Cystic Interstitial Lung Diseases: A Pictorial Review and a Practical Guide for the Radiologist

 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
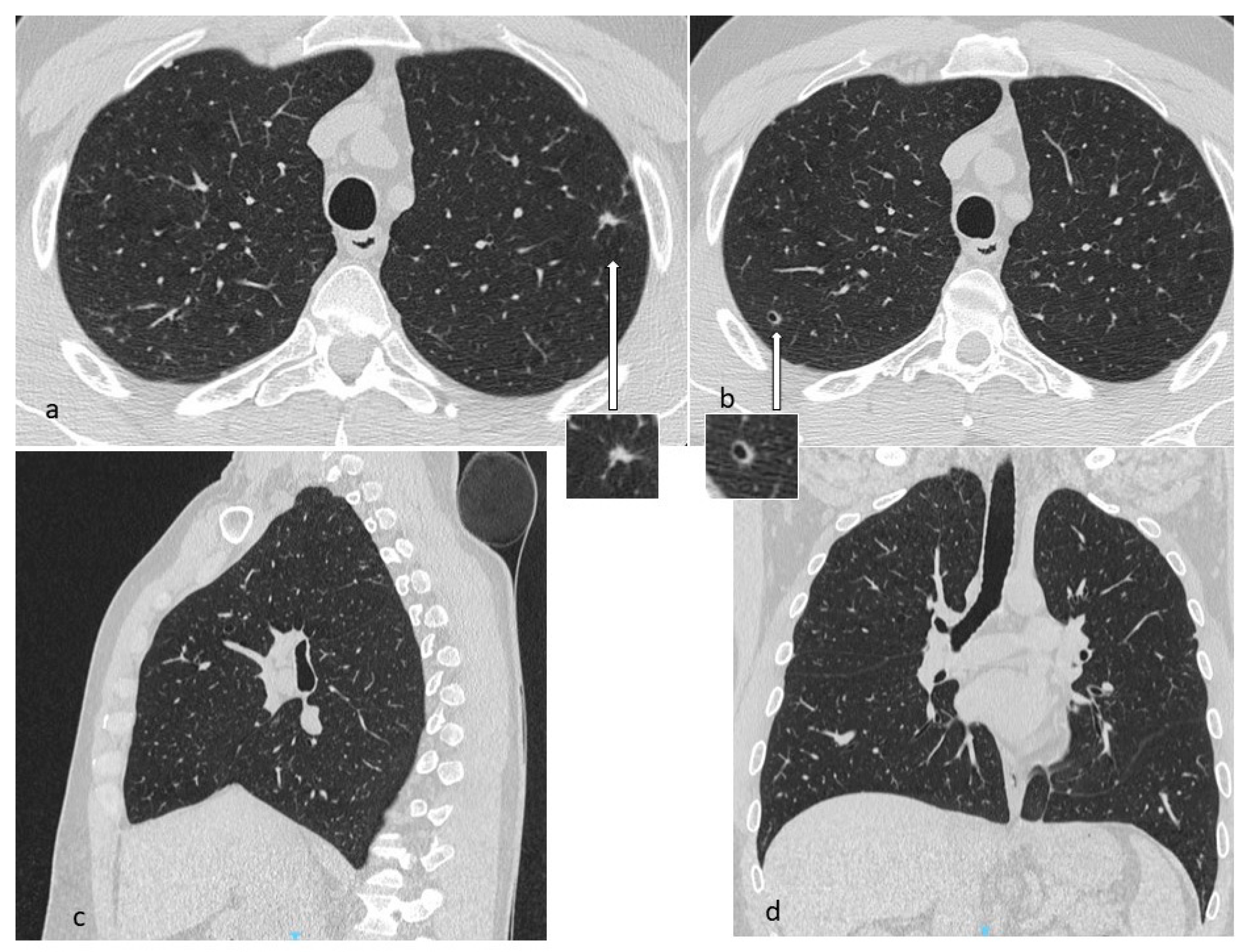
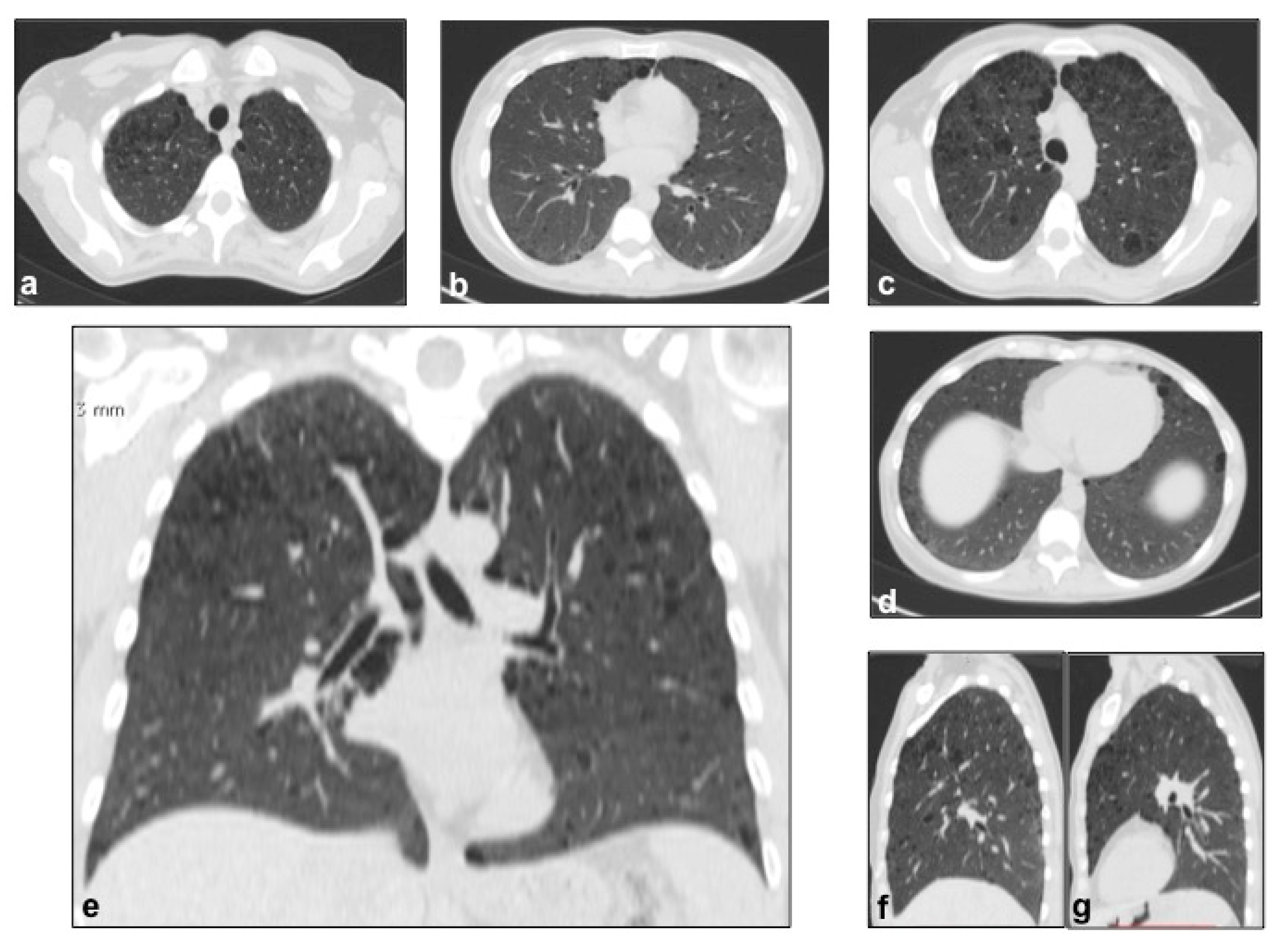
4.1. Histiocytosis X or Pulmonary Langerhans Cell Histiocytosis (LCH)
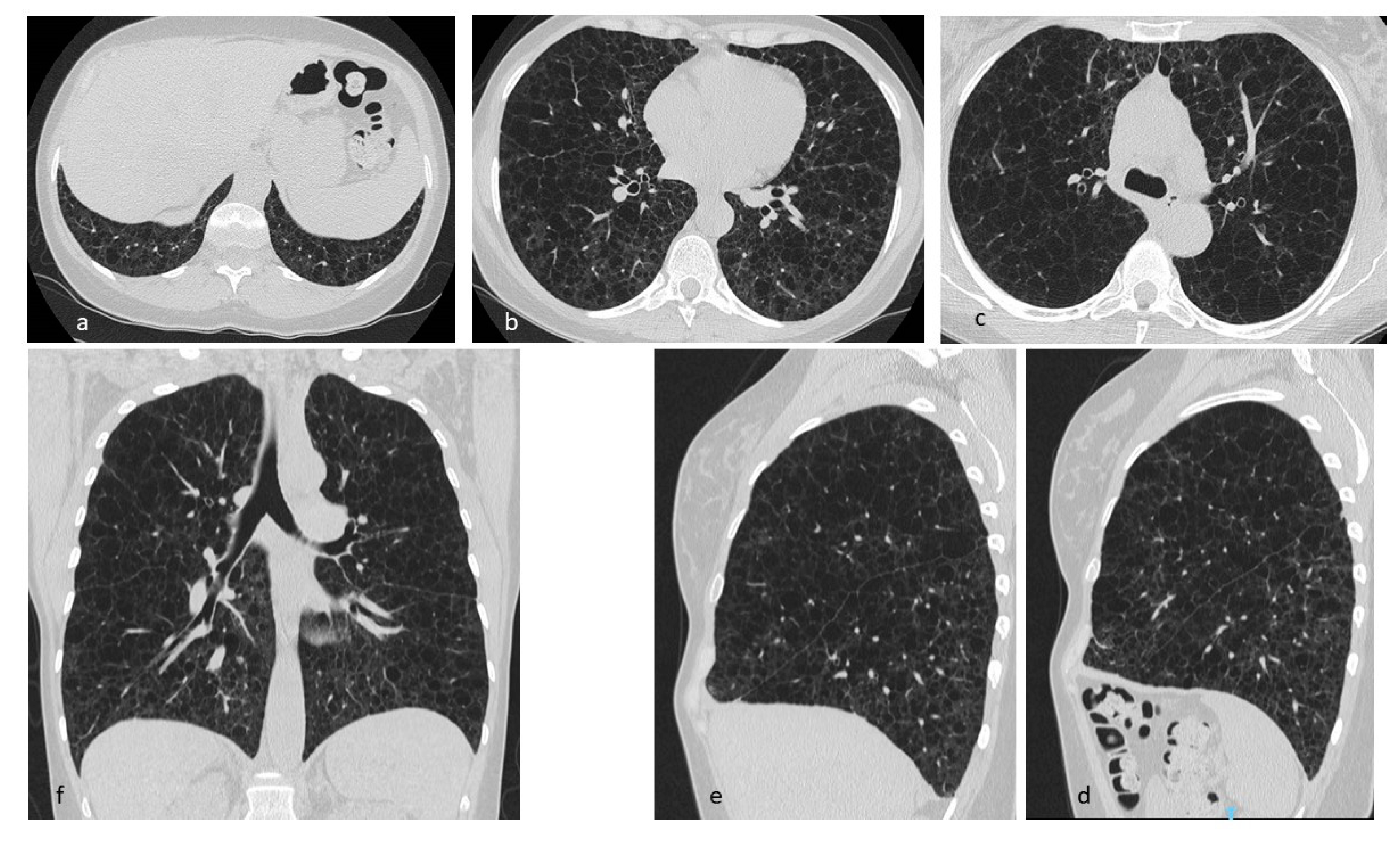
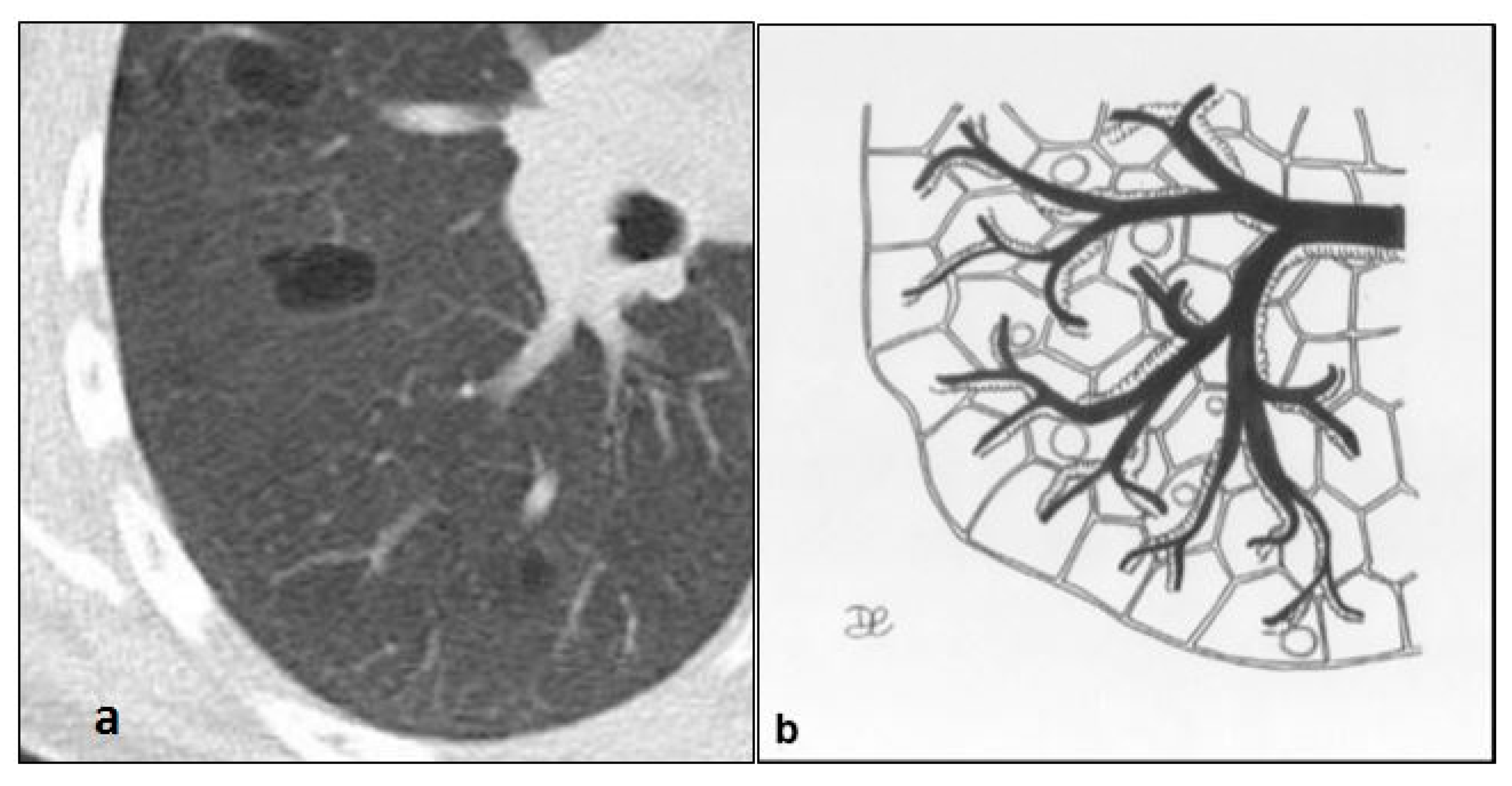
4.2. Lymphangioleiomiomatosis (LAM)
- Tuberous Sclerosis complex;
- Chylous effusions;
- Angiomyolipomas;
- Lymphatic involvement;
- Serum VEFG-D >800 pg/mL (VEGF-D is a lymphangiogenic growth factor that is increased in the serum of patients with LAM);
- Histological findings.
4.3. Lymphocytic Interstitial Pneumonia (LIP)
4.4. Desquamative Interstitial Pneumonia (DIP)
4.5. Neurofibromatosis (NF)
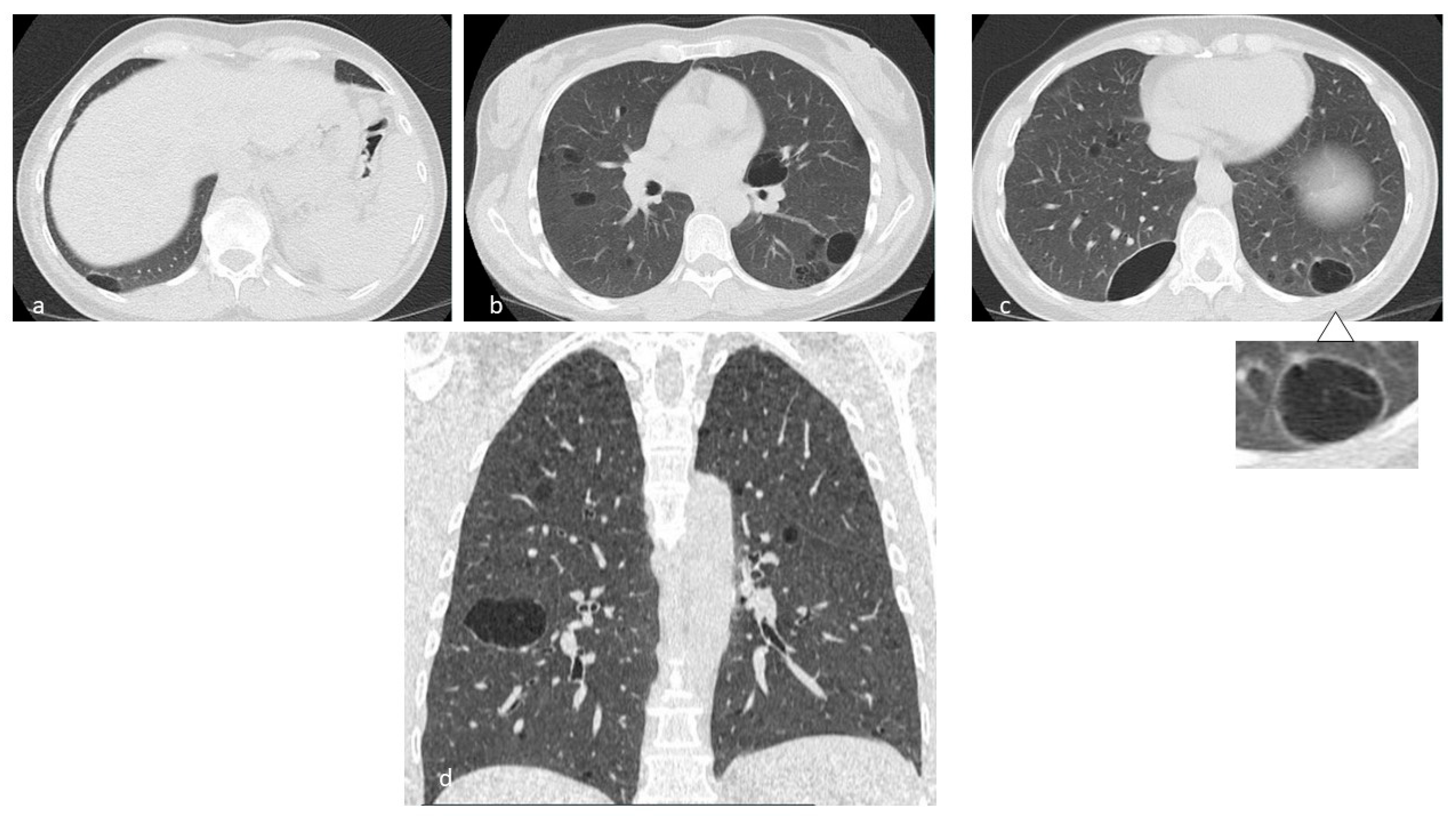
4.6. Birt–Hogg–Dubè (BHD) Syndrome
4.7. Infectious Pneumatocele, Recurrent Respiratory Papillomatosis (RRP) and Paragonimiasis
4.8. Cystic Fibrosis (CF)
4.9. Aging Lung Cysts
4.10. Cystic Metastasis
4.11. Amyloidosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Genereux, G.P. The end-stage lung: Pathogenesis, pathology, and radiology. Radiology 1975, 116, 279–289. [Google Scholar] [CrossRef]
- Caltabiano, D.C.; Costanzo, V.; Mammino, L.; Vindigni, V.; Torrisi, S.; Rosso, R.; Mauro, L.A.; Vancheri, C.; Palmucci, S. Cystic Pattern in Lung Diseases: A Simplified HRCT Guide Based on Free-Hand Drawings. 2017. Available online: https://epos.myesr.org/esr/viewing/index.php?module=viewing_poster&task=viewsection&pi=138603&ti=474530&si=1641&searchkey (accessed on 20 September 2019).
- Basset, F.; Corrin, B.; Spencer, H.; Lacronique, J.; Roth, C.; Soler, P.; Battesti, J.P.; Georges, R.; Chrétien, J. Pulmonary histiocytosis X. Am. Rev. Respir. Dis. 1978, 118, 811–820. [Google Scholar] [PubMed]
- Xaubet, A.; Agustí, C.; Picado, C.; Gueréquiz, S.; Martos, J.; Carrión, M.; Agustí-Vidal, A. Bronchoalveolar lavage analysis with anti-T6 monoclonal antibody in the evaluation of diffuse lung diseases. Respiration 1989, 56, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Francisco, F.A.F.; Souza, A.S.; Zanetti, G.; Marchiori, E. Multiple cystic lung disease. Eur. Respir. Rev. 2015, 24, 552–564. [Google Scholar] [CrossRef] [PubMed]
- DeMartino, E.; Go, R.S.; Vassallo, R. Langerhans cell hystiocytosis and other histiocytic diseases of the lung. Clin. Chest Med. 2016, 37, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, G.P.; Frankel, S.K.; Brown, K.K. Challenges in pulmonary fibrosis. 3: Cystic lung disease. Thorax 2007, 62, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.F.; Rosado-de-Christenson, M.L.; Franks, T.J.; Franzier, A.A.; Galvin, J.R. From the archives of AFIP: Pulmonary Langerhans cell histiocytosis. RadioGraphics 2004, 24, 821–841. [Google Scholar] [CrossRef]
- Colby, T.V.; Lombard, C. Histiocytosis X in the lung. Hum. Pathol. 1983, 14, 847–856. [Google Scholar] [CrossRef]
- Jawad, H.; Walker, C.M.; Wu, C.C.; Chung, J.H. Cystic Interstitial Lung Diseases: Recognizing the Common and Uncommon Entities. Curr. Probl. Diagn. Radiol. 2014, 43, 115–127. [Google Scholar] [CrossRef]
- Juvet, S.C.; Hwang, D.; Downey, G.P. Rare lung diseases III: Pulmonary Langerhans’ cell histiocytosis. Can. Respir. J. 2010, 17, e55–e62. [Google Scholar] [CrossRef]
- Vassallo, R.; Ryu, J.H.; Schroeder, D.R.; Decker, P.A.; Limper, A.H. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N. Engl. J. Med. 2002, 346, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Baqir, M.; Vassallo, R.; Maldonado, F.; Yi, E.S.; Ryu, J.H. Utility of bronchoscopy in pulmonary Langerhans cell histiocytosis. J. Bronchol. Interv. Pulmonol. 2013, 20, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, J.M.; Grossman, R.F.; Chamberlain, D.; Tremblay, C.E. Pulmonary manifestations of tuberous sclerosis in first degree relatives. Thorax 1989, 44, 212–214. [Google Scholar] [CrossRef]
- Ho, T.B.; Hull, J.H.; Hughes, N.C. An 86-year-old female with lymphangioleiomyomatosis. Eur. Respir. J. 2006, 28, 1065. [Google Scholar] [CrossRef]
- Gu, X.; Yu, J.J.; Ilter, D.; Blenis, N.; Henske, E.P.; Blenis, J. Integration of mTOR and estrogen-ERK2 signaling in lymphangioleiomyomatosis pathogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 14960–14965. [Google Scholar] [CrossRef]
- Cottin, V.; Harari, S.; Humbert, M.; Mal, H.; Dorfmu, P.; Jaïs, X.; Reynaud-Gaubert, M.; Prevot, G.; Lazor, R.; Taille, C.; et al. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Pulmonary hypertension in lymphangioleiomyomatosis: Characteristics in 20 patients. Eur. Respir. J. 2012, 40, 630–640. [Google Scholar] [CrossRef]
- Rappaport, D.; Weisbrod, G.; Herman, S.; Chamberlain, D. Pulmonary lymphangioleiomyomatosis: High-resolution CT findings in four cases. AJR Am. J. Roentgenol. 1989, 152, 961–964. [Google Scholar] [CrossRef]
- Cui, H.; Tian, X.; Wang, H.; Zhao, J.; Huang, H.; Zhang, W.; Lo, B.H.; Xu, K.-F.; Feng, R. Diffuse Cystic Lung Disease: Diagnostic Considerations. Semin. Respir. Crit. Care Med. 2016, 37, 457–467. [Google Scholar] [CrossRef]
- McCormack, F.X.; Gupta, N.; Finlay, G.R.; Young, L.R.; Taveira-DaSilva, A.M.; Glasgow, C.G.; Steagall, W.K.; Johnson, S.R.; Sahn, S.A.; Ryu, J.H.; et al. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2016, 194, 748–761. [Google Scholar] [CrossRef]
- Anton, E.; Casanova, A.; Xaubet, A.; Román, A.; Villena, V.; Montero, M.C.; Molina-Molina, M.; Pérez-Sánchez, E.; Sueiro, A.; Morell, F.; et al. Lymphangioleiomyomatosis: A study of 72 patients from the Spanish registry. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 85–91. [Google Scholar]
- Koss, M.N.; Hochholzer, L.; Langloss, J.M.; Wehunt, W.D.; Lazarus, A.A. Lymphoid interstitial pneumonia: Clinicopathological and immunopathologic findings in 18 cases. Pathology 1987, 19, 178–185. [Google Scholar] [CrossRef]
- Liebow, A.; Carrington, C.B. The interstitial pneumonias. In Frontiers of Pulmonary Radiology, 1st ed.; Simon, M., Potchen, E.J., LeMay, M., Eds.; Grune & Stratton: New York, NY, USA, 1969; pp. 102–141. [Google Scholar]
- Liebow, A.; Carrington, C. Diffuse pulmonary lymphoreticular infiltrations associated with dysproteinemia. Med. Clin. N. Am. 1973, 57, 809–843. [Google Scholar] [CrossRef]
- Cha, S.I.; Fessler, M.B.; Cool, C.D.; Schwarz, M.I.; Brown, K.K. Lymphoid interstitial pneumonia: Clinical features, associations and prognosis. Eur. Respir. J. 2006, 28, 364–369. [Google Scholar] [CrossRef]
- Silva, C.I.; Flint, J.D.; Levy, R.D.; Muller, N.L. Diffuse lung cysts in lymphoid interstitial pneumonia: High-resolution CT and pathologic findings. J. Thorac. Imaging 2006, 21, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Johkoh, T.; Honda, O.; Tsubamoto, M.; Kozuka, T.; Tomiyama, N.; Hamada, S.; Nakamura, H.; Akira, M.; Ichikado, K.; et al. Chronic cystic lung disease: Diagnostic accuracy of high-resolution CT in 92 patients. AJR Am. J. Roentgenol. 2003, 180, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Johkoh, T.; Muller, N.L.; Pickford, H.A.; Hartman, T.E.; Ichikado, K.; Akira, M.; Honda, O.; Nakamura, H. Lymphocytic interstitial pneumonia: Thin-section CT findings in 22 patients. Radiology 1999, 212, 567–572. [Google Scholar] [CrossRef]
- Nicholson, A.G. Lymphocytic interstitial pneumonia and other lymphoproliferative disorders in the lung. Semin. Respir. Crit. Care Med. 2001, 22, 409–422. [Google Scholar] [CrossRef]
- Attili, A.K.; Kazerooni, E.A.; Gross, B.H.; Flaherty, K.R.; Myers, J.L.; Martinez, F.J. Smoking-related interstitial lung disease: Radiologic-clinical-pathologic correlation. Radiographics 2008, 28, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Doan, M.L.; Guillerman, R.P.; Dishop, M.K.; Nogee, L.M.; Langston, C.; Mallory, G.B.; Sockrider, M.M.; Fan, L.L. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax 2008, 63, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Craig, P.J.; Wells, A.U.; Doffman, S.; Rassl, D.; Colby, T.V.; Hansell, D.; Bois, R.M.D.; Nicholson, A.G. Desquamative interstitial pneumonia, respiratory bronchiolitis and their relationship to smoking. Histopathology 2004, 45, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Colby, T.; Hartman, T.E.; Vassallo, R. Smoking-related interstitial lung diseases: A concise review. Eur. Respir. J. 2001, 17, 122–132. [Google Scholar] [CrossRef]
- Bone, R.C.; Wolfe, J.; Sobonya, R.E.; Kerby, G.R.; Stechschulte, D.; Ruth, W.E.; Welch, M. Desquamative interstitial pneumonia following long-term nitrofurantoin therapy. Am. J. Med. 1976, 60, 697–701. [Google Scholar] [CrossRef]
- Rao, R.N.; Goodman, L.R.; Tomashefski, J.F. Smoking-related interstitial lung disease. Ann. Diagn. Pathol. 2008, 12, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Johkoh, T.; Müller, N.L.; Cartier, Y.; Kavanagh, P.V.; Hartman, T.E.; Akira, M.; Ichikado, K.; Ando, M.; Nakamura, H. Idiopathic interstitial pneumonias: Diagnostic accuracy of thin-section CT in 129 patients. Radiology 1999, 211, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.E.; Primack, S.L.; Swensen, S.J.; Hansell, D.; McGuinness, G.; Muller, N.L. Desquamative interstitial pneumonia: Thin-section CT findings in 22 patients. Radiology 1993, 187, 787–790. [Google Scholar] [CrossRef]
- Desai, S.R.; Ryan, S.M.; Colby, T.V. Smoking-related interstitial lung diseases: Histopathological and imaging perspectives. Clin. Radiol. 2003, 58, 259–268. [Google Scholar] [CrossRef]
- Lee, K.-H.; Lee, J.S.; Lynch, D.A.; Song, K.-S.; Lim, T.-H. The radiologic differential diagnosis of diffuse lung diseases characterized by multiple cysts or cavities. J. Comput. Assist. Tomogr. 2002, 26, 5–12. [Google Scholar] [CrossRef]
- Akira, M.; Yamamoto, S.; Hara, H.; Sakatani, M.; Ueda, E. Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax 1997, 52, 333–337. [Google Scholar] [CrossRef]
- Hartman, T.E.; Primack, S.L.; Kang, E.Y.; Swensen, S.J.; Hansell, D.M.; McGuinness, G.; Müller, N.L. Disease progression in usual interstitial pneumonia compared with desquamative interstitial pneumonia: Assessment with serial CT. Chest 1996, 110, 378–382. [Google Scholar] [CrossRef]
- Riccardi, V.M. Von Recklinghausen neurofibromatosis. N. Engl. J. Med. 1981, 305, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Browning, G.G.P.; Nawroz, I.; Campbell, I.W. Von Recklinghausen’s neurofibromatosis: Neurofibromatosis type 1. Lancet Lond. Engl. 2003, 361, 1552–1554. [Google Scholar] [CrossRef]
- Zamora, A.C.; Collard, H.R.; Wolters, P.J.; Webb, W.R.; King, T.E. Neurofibromatosis-associated lung disease: A case series and literature review. Eur. Respir. J. 2007, 29, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Nardecchia, E.; Perfetti, L.; Castiglioni, M.; Di Natale, D.; Imperatori, A.; Rotolo, N. Bullous lung disease and neurofibromatosis type-1. Monaldi Arch. Chest Dis. 2012, 77, 105–107. [Google Scholar] [CrossRef][Green Version]
- Tate, J.M.; Gyorffy, J.B.; Colburn, J.A. The importance of pheochromocytoma case detection in patients with neurofibromatosis type 1: A case report and review of literature. SAGE Open Med. Case Rep. 2017, 5, 2050313X17741016. [Google Scholar] [CrossRef]
- Souza, C.A.; Finley, R.; Müller, N.L. Birt-Hogg-Dubé syndrome: A rare cause of pulmonary cysts. AJR Am. J. Roentgenol. 2005, 185, 1237–1239. [Google Scholar] [CrossRef]
- Gupta, N.; Sunwoo, B.Y.; Kotloff, R.M. Birt-Hogg-Dubè Syndrome. Clin. Chest Med. 2016, 37, 475–486. [Google Scholar] [CrossRef]
- Donabedian, H.; Gallin, J.I. The hyperimmunoglobulin E recurrent-infection (Job’s) syndrome: A review of the NIH experience and the literature. Medicine (Baltimore) 1983, 62, 195–208. [Google Scholar] [CrossRef]
- Moore, F.A.; Moore, E.E.; Haenel, J.B.; Waring, B.J.; Parsons, P.E. Post-traumatic pulmonary pseudocyst in the adult: Pathophysiology, recognition, and selective management. J. Trauma 1989, 29, 1380–1385. [Google Scholar] [CrossRef]
- Quigley, M.J.; Fraser, R.S. Pulmonary pneumatocele: Pathology and pathogenesis. AJR Am. J. Roentgenol. 1988, 150, 1275–1277. [Google Scholar] [CrossRef]
- Gentina, T.; Tillie-Leblond, I.; Birolleau, S.; Saidi, F.; Saelens, T.; Boudoux, L.; Vervloet, D.; Delaval, P.; Tonnel, A.B.; Faycal, S. Fire-eater’s lung: Seventeen cases and a review of the literature. Medicine (Baltimore) 2001, 80, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Cordier, J.-F.; Cottin, V.; Khouatra, C.; Giraud, S.; Lazor, R. Multiple cystic lung diseases. Eur. Respir. Mon. 2011, 54, 46–83. [Google Scholar]
- Ruan, S.; Chen, K.; Yang, P. Recurrent respiratory papillomatosis with pulmonary involvement: A case report and review of the literature. Respirology 2009, 14, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Schraff, S.; Derkay, C.S.; Burke, B.; Lawson, M.L. American Society of Pediatric Otolaryngology members’ experience with recurrent respiratory papillomatosis and the use of adjuvant therapy. Arch. Otolaryngol. Head Neck Surg. 2004, 130, 1039–1042. [Google Scholar] [CrossRef]
- Kuroki, M.; Hatabu, H.; Nakata, H.; Hashiguchi, N.; Shimizu, T.; Uchino, N.; Tamura, S. High-resolution computed tomography findings of P. Westermani. J. Thorac. Imaging 2005, 20, 210–213. [Google Scholar] [CrossRef]
- Im, J.G.; Whang, H.Y.; Kim, W.S.; Han, M.C.; Shim, Y.S.; Cho, S.Y. Pleuropulmonary paragonimiasis: Radiologic findings in 71 patients. AJR Am. J. Roentgenol. 1992, 159, 39–43. [Google Scholar] [CrossRef]
- Amodio, J.B.; Berdon, W.E.; Abramson, S.B.; Baker, D. Cystic fibrosis in childhood: Pulmonary, paranasal sinus, and skeletal manifestations. Semin. Roentgenol. 1987, 22, 125–135. [Google Scholar] [CrossRef]
- Tager, A.M.; Wu, J.; Vermeulen, M.W. The effect of chloride concentration on human neutrophil functions: Potential relevance to cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 1998, 19, 643–652. [Google Scholar] [CrossRef]
- Arakawa, H.; Webb, W.R. Air trapping on expiratory high-resolution CT scans in the absence of inspiratory scan abnormalities: Correlation with pulmonary function tests and differential diagnosis. AJR Am. J. Roentgenol. 1998, 170, 1349–1353. [Google Scholar] [CrossRef]
- Robinson, T.E.; Leung, A.N.; Northway, W.H.; Blankenberg, F.G.; Bloch, D.A.; Oehlert, J.W.; Al-Dabbagh, H.; Hubli, S.; Moss, R.B. Spirometer-triggered high-resolution computed tomography and pulmonary function measurements during an acute exacerbation in patients with cystic fibrosis. J. Pediatr. 2001, 138, 553–559. [Google Scholar] [CrossRef]
- Copley, S.J.; Wells, A.U.; Hawtin, K.E.; Gibson, D.J.; Hodson, J.M.; Jacques, A.E.T.; Hansell, D.M. Lung morphology in the elderly: Comparative CT study of subjects over 75 years old versus those under 55 years old. Radiology 2009, 251, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Ateishi, U.; Hasegawa, T.; Kusumoto, M.; Yamazaki, N.; Iinuma, G.; Muramatsu, Y.; Moriyama, N. Metastatic Angiosarcoma of the Lung: Spectrum of CT Findings. Am. J. Roentgenol. 2003, 180, 1671–1674. [Google Scholar] [CrossRef] [PubMed]
- Yogi, A.; Miyara, T.; Ogawa, K.; Iraha, S.; Matori, S.; Haranaga, S.; Murayama, S. Pulmonary metastases from angiosarcoma: A spectrum of CT findings. Acta Radiol. 2016, 57, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Czeyda-Pommersheim, F.; Hwang, M.; Chen, S.S.; Strollo, D.; Fuhrman, C.; Bhalla, S. Amyloidosis: Modern cross-sectional imaging. Radiographics 2015, 35, 1381–1392. [Google Scholar] [CrossRef]
- Martinez, A.C.Z.; White, D.B.; Sykes, A.-M.G.; Hoskote, S.S.; Moua, T.; Yi, E.S.; Ryu, J.H. Amyloid-associated cystic lung disease. Chest 2016, 149, 1223–1233. [Google Scholar]
- Georgiades, C.S.; Neyman, E.G.; Barish, M.; Fishman, E.K. Amyloidosis: Review and CT manifestations. Radiographics 2004, 24, 405–416. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Hrct Features | Wall | Distribution | Other Features | |
|---|---|---|---|---|---|
| Diffused | LCH | Variable shape, “bizzarre” variable size Thin walls | thick | Wide spreading, with sparing of costo-phrenic angles | Centrilobular nodules at early stages |
| LAM | Round and regular shape Uniform size (10–20 mm) Thick walls | thin | Wide spreading No zonal sparing | No nodules | |
| LIP | Regular shape Uniform size Thin walls | thin | Peri-broncho-vascular regions | Ground-glass opacities, septa thickening, centrilobular nodules | |
| DIP | Regular shape Uniform size Thin walls | thin | Peripheral regions | Ground-glass opacities, linear opacities | |
| BHD | Variable shape, irregular, septated or round | thin | Lung bases paramediastinal areas | Pneumothorax | |
| Other | NF1 | Irregular shape and small size | thick | Predominant in the upper lobes | Ground-glass areas with reticular basal opacities |
| Pneumatocele | Regular shape and variable size | thick/thin | Focal or multi-focal predominant at upper lobes | Ground-glass opacities in peri-hilar regions | |
| Cystic Fibrosis | Regular shape and variable size bronchiectasis | thick | Predominant in upper lobes and dorsal segments of lower lobes | Air trapping areas, impaired perfusions areas, enlarged mediastinal and hylar lymphnodes | |
| Aging Lung Cysts | Regular shape and variable size | thin | Wide | Linear irregular opacities and reticular subpleural opacities |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aquilina, G.; Caltabiano, D.C.; Galioto, F.; Cancemi, G.; Pino, F.; Vancheri, A.; Vancheri, C.; Foti, P.V.; Mauro, L.A.; Basile, A. Cystic Interstitial Lung Diseases: A Pictorial Review and a Practical Guide for the Radiologist. Diagnostics 2020, 10, 346. https://doi.org/10.3390/diagnostics10060346
Aquilina G, Caltabiano DC, Galioto F, Cancemi G, Pino F, Vancheri A, Vancheri C, Foti PV, Mauro LA, Basile A. Cystic Interstitial Lung Diseases: A Pictorial Review and a Practical Guide for the Radiologist. Diagnostics. 2020; 10(6):346. https://doi.org/10.3390/diagnostics10060346
Chicago/Turabian StyleAquilina, Giulia, Daniele Carmelo Caltabiano, Federica Galioto, Giovanna Cancemi, Fabio Pino, Ada Vancheri, Carlo Vancheri, Pietro Valerio Foti, Letizia Antonella Mauro, and Antonio Basile. 2020. "Cystic Interstitial Lung Diseases: A Pictorial Review and a Practical Guide for the Radiologist" Diagnostics 10, no. 6: 346. https://doi.org/10.3390/diagnostics10060346
APA StyleAquilina, G., Caltabiano, D. C., Galioto, F., Cancemi, G., Pino, F., Vancheri, A., Vancheri, C., Foti, P. V., Mauro, L. A., & Basile, A. (2020). Cystic Interstitial Lung Diseases: A Pictorial Review and a Practical Guide for the Radiologist. Diagnostics, 10(6), 346. https://doi.org/10.3390/diagnostics10060346