The Influence of Methylating Mutations on Acute Myeloid Leukemia: Preliminary Analysis on 56 Patients



 , ,
, ,
Abstract
1. Introduction
2. Material and Methods
TCGA Analysis
3. Results
3.1. Patient Selection
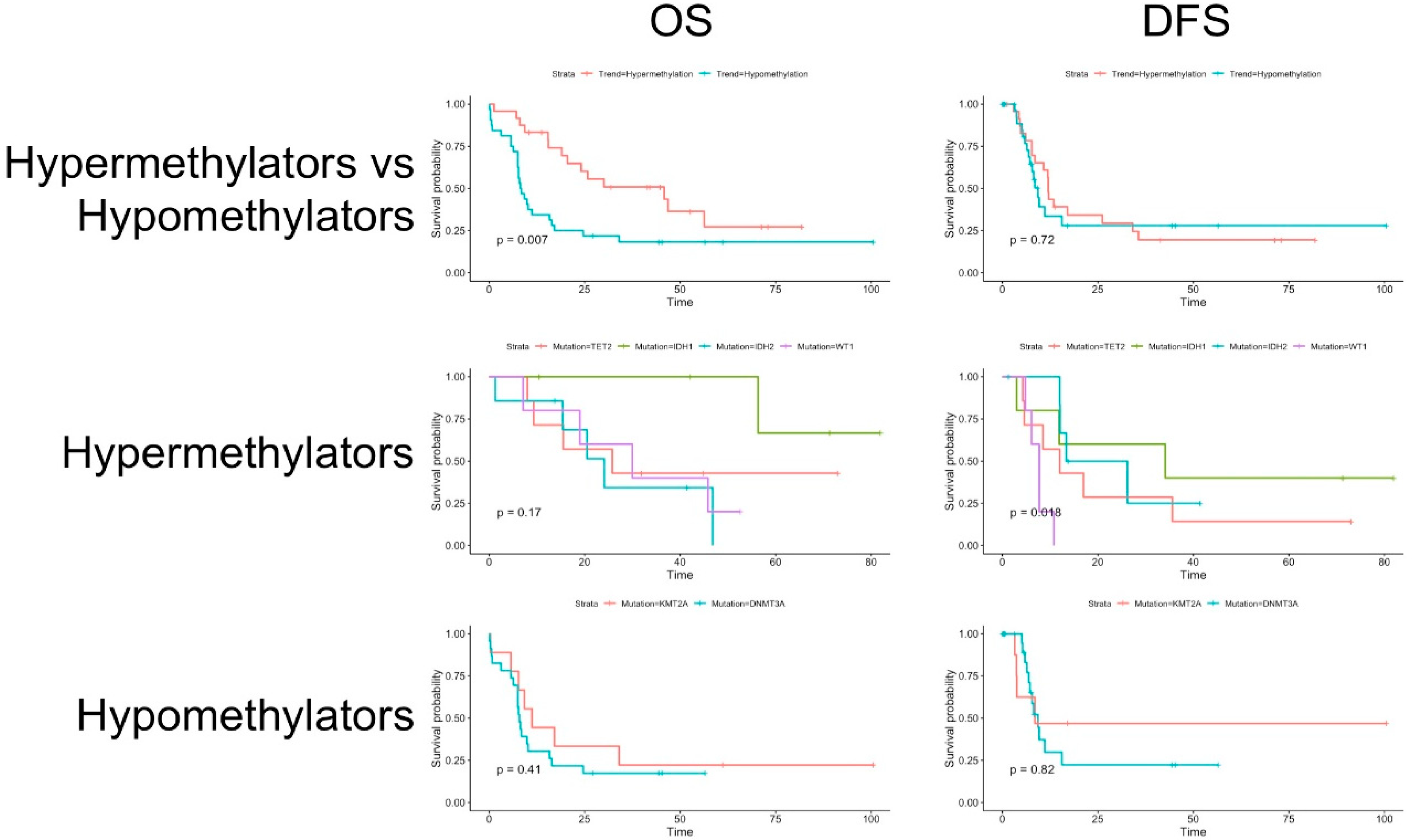
3.2. Clinical Data and Survival Analysis
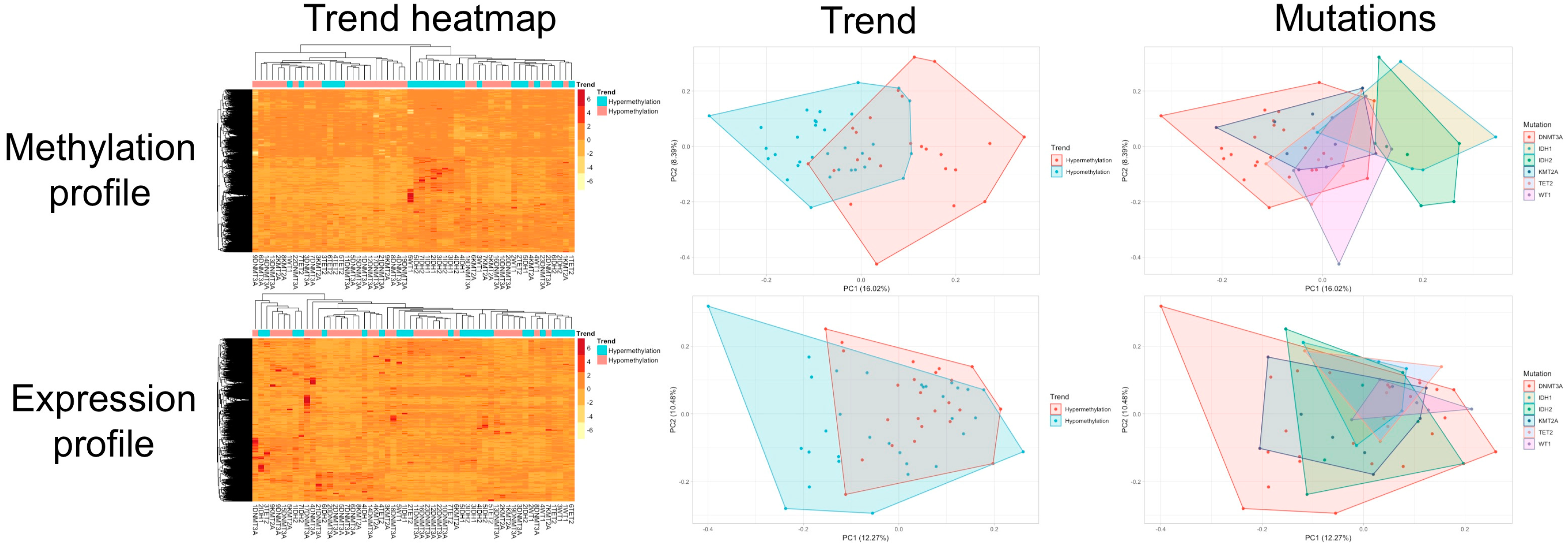
3.3. Clustering and Principal Component Analysis
3.4. Supervised Machine Learning
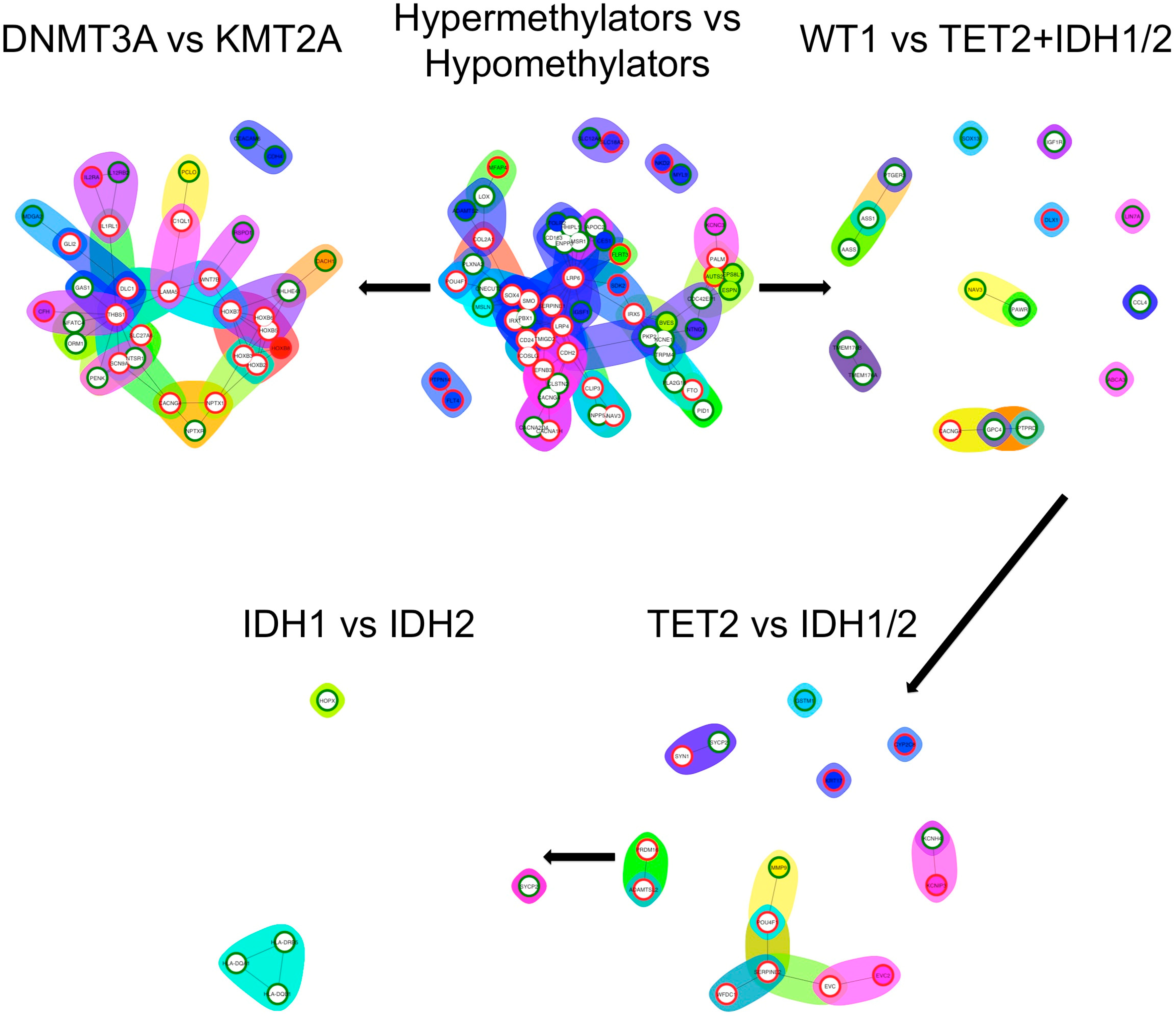
3.5. Differential Expression Analysis and Functional Enrichment Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khwaja, A.; Bjorkholm, M.; Gale, R.E.; Levine, R.L.; Jordan, C.T.; Ehninger, G.; Bloomfield, C.D.; Estey, E.; Burnett, A.; Cornelissen, J.J.; et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers 2016, 2, 16010. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Basheer, F.; Giotopoulos, G.; Meduri, E.; Yun, H.; Mazan, M.; Sasca, D.; Gallipoli, P.; Marando, L.; Gozdecka, M.; Asby, R.; et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J. Exp. Med. 2019, 216, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-H.; Liu, Y.; Gao, S.-Y. Correlation between IL-7 genomic protein methylation level and acute myeloid leukemia. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1196–1202. [Google Scholar] [CrossRef]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef]
- Stengel, A.; Kern, W.; Meggendorfer, M.; Nadarajah, N.; Perglerovà, K.; Haferlach, T.; Haferlach, C. Number of RUNX1 mutations, wild-type allele loss and additional mutations impact on prognosis in adult RUNX1-mutated AML. Leukemia 2018, 32, 295–302. [Google Scholar] [CrossRef]
- Kelly, A.D.; Kroeger, H.; Yamazaki, J.; Taby, R.; Neumann, F.; Yu, S.; Lee, J.T.; Patel, B.; Li, Y.; He, R.; et al. A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia 2017, 31, 2011–2019. [Google Scholar] [CrossRef]
- Sellers, Z.P.; Bolkun, L.; Kloczko, J.; Wojtaszewska, M.L.; Lewandowski, K.; Moniuszko, M.; Ratajczak, M.Z.; Schneider, G. Increased methylation upstream of the MEG3 promotor is observed in acute myeloid leukemia patients with better overall survival. Clin. Epigenetics 2019, 11, 50. [Google Scholar] [CrossRef]
- Lavallée, V.-P.; Lemieux, S.; Boucher, G.; Gendron, P.; Boivin, I.; Armstrong, R.N.; Sauvageau, G.; Hébert, J. RNA-sequencing analysis of core binding factor AML identifies recurrent ZBTB7A mutations and defines RUNX1-CBFA2T3 fusion signature. Blood 2016, 127, 2498–2501. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Shen, Y.; Chen, H.; Wang, X.; Zhang, R.; Peng, Y.; Lei, X.; Liu, T.; Liu, J.; Gu, L.; et al. Expression profile analysis of long non-coding RNA in acute myeloid leukemia by microarray and bioinformatics. Cancer Sci. 2018, 109, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.E.; Huang, Y.-H.; Muise, A.; Goodell, M.A.; Reich, N.O. Mutations in the DNMT3A DNA methyltransferase in acute myeloid leukemia patients cause both loss and gain of function and differential regulation by protein partners. J. Biol. Chem. 2019, 294, 4898–4910. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Ma, L.; Merker, J.D.; Gotlib, J.R.; Schrijver, I.; Zehnder, J.L.; Arber, D.A. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod. Pathol. 2015, 28, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Lemonnier, F.; Mak, T.W. Roles of IDH1/2 and TET2 mutations in myeloid disorders. Int. J. Hematol. 2016, 103, 627–633. [Google Scholar] [CrossRef]
- Petrova, L.; Vrbacky, F.; Lanska, M.; Zavrelova, A.; Zak, P.; Hrochova, K. IDH1 and IDH2 mutations in patients with acute myeloid leukemia: Suitable targets for minimal residual disease monitoring? Clin. Biochem. 2018, 61, 34–39. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA Hydroxymethylation Profiling Reveals that WT1 Mutations Result in Loss of TET2 Function in Acute Myeloid Leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T.L.; Vijayvargiya, P.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer J. 2016, 6, e385. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Lin, S.-J.; Shih, H.-Y.; Chou, C.-H.; Chu, H.-H.; Chiu, C.-C.; Yuh, C.-H.; Yeh, T.-H.; Cheng, Y.-C. Epigenetic regulation of NOTCH1 and NOTCH3 by KMT2A inhibits glioma proliferation. Oncotarget 2017, 8, 63110–63120. [Google Scholar] [CrossRef][Green Version]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Birke, M.; Schreiner, S.; García-Cuéllar, M.-P.; Mahr, K.; Titgemeyer, F.; Slany, R.K. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002, 30, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Aibar, S.; Fontanillo, C.; Droste, C.; De Las Rivas, J. Functional Gene Networks: R/Bioc package to generate and analyse gene networks derived from functional enrichment and clustering. Bioinformatics 2015, 31, 1686–1688. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Bera, R.; Chiu, M.-C.; Huang, Y.-J.; Liang, D.-C.; Lee, Y.-S.; Shih, L.-Y. Genetic and Epigenetic Perturbations by DNMT3A-R882 Mutants Impaired Apoptosis through Augmentation of PRDX2 in Myeloid Leukemia Cells. Neoplasia 2018, 20, 1106–1120. [Google Scholar] [CrossRef]
- Mrózek, K.; Marcucci, G.; Paschka, P.; Whitman, S.P.; Bloomfield, C.D. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: Are we ready for a prognostically prioritized molecular classification? Blood 2007, 109, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Gong, M.; Gao, L.; Cai, X.; Zhang, H.; Ma, Y. RASSF1A hypermethylation is associated with ASXL1 mutation and indicates an adverse outcome in non-M3 acute myeloid leukemia. OncoTargets Ther. 2017, 10, 4143–4151. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Smeitink, J. Metabolism, gliomas, and IDH1. N. Engl. J. Med. 2010, 362, 1144–1145. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Abbas, S.; Erpelinck-Verschueren, C.A.J.; Goudswaard, C.S.; Löwenberg, B.; Valk, P.J.M. Mutant Wilms’ tumor 1 (WT1) mRNA with premature termination codons in acute myeloid leukemia (AML) is sensitive to nonsense-mediated RNA decay (NMD). Leukemia 2010, 24, 660–663. [Google Scholar] [CrossRef]
- Miyagi, T.; Ahuja, H.; Kubota, T.; Kubonishi, I.; Koeffler, H.P.; Miyoshi, I. Expression of the candidate Wilm’s tumor gene, WT1, in human leukemia cells. Leukemia 1993, 7, 970–977. [Google Scholar]
- Yan, X.-J.; Xu, J.; Gu, Z.-H.; Pan, C.-M.; Lu, G.; Shen, Y.; Shi, J.-Y.; Zhu, Y.-M.; Tang, L.; Zhang, X.-W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef]
- Cierpicki, T.; Risner, L.E.; Grembecka, J.; Lukasik, S.M.; Popovic, R.; Omonkowska, M.; Shultis, D.D.; Zeleznik-Le, N.J.; Bushweller, J.H. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 2010, 17, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Erfurth, F.E.; Popovic, R.; Grembecka, J.; Cierpicki, T.; Theisler, C.; Xia, Z.-B.; Stuart, T.; Diaz, M.O.; Bushweller, J.H.; Zeleznik-Le, N.J. MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc. Natl. Acad. Sci. USA 2008, 105, 7517–7522. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Lin, C.; Guest, E.; Shilatifard, A. Licensed to elongate: A molecular mechanism for MLL-based leukaemogenesis. Nat. Rev. Cancer 2010, 10, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-H.; Li, H.-Y.; Fan, S.-C.; Yuan, T.-H.; Chen, M.; Hsu, Y.-H.; Yang, Y.-H.; Li, L.-Y.; Yeh, S.-P.; Bai, L.-Y.; et al. A targeted next-generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: Implications for clinical practice. Cancer Med. 2017, 6, 349–360. [Google Scholar] [CrossRef]
- Sato, T.; Issa, J.-P.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef]
- Derissen, E.J.B.; Beijnen, J.H.; Schellens, J.H.M. Concise Drug Review: Azacitidine and Decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef]
- Müller-Tidow, C.; Tschanter, P.; Röllig, C.; Thiede, C.; Koschmieder, A.; Stelljes, M.; Koschmieder, S.; Dugas, M.; Gerss, J.; Study Alliance Leukemia Group; et al. Azacitidine in combination with intensive induction chemotherapy in older patients with acute myeloid leukemia: The AML-AZA trial of the study alliance leukemia. Leukemia 2016, 30, 555–561. [Google Scholar] [CrossRef]
- Garz, A.-K.; Wolf, S.; Grath, S.; Gaidzik, V.; Habringer, S.; Vick, B.; Rudelius, M.; Ziegenhain, C.; Herold, S.; Weickert, M.-T.; et al. Azacitidine combined with the selective FLT3 kinase inhibitor crenolanib disrupts stromal protection and inhibits expansion of residual leukemia-initiating cells in FLT3-ITD AML with concurrent epigenetic mutations. Oncotarget 2017, 8. [Google Scholar] [CrossRef]
- Döhner, H.; Dolnik, A.; Tang, L.; Seymour, J.F.; Minden, M.D.; Stone, R.M.; del Castillo, T.B.; Al-Ali, H.K.; Santini, V.; Vyas, P.; et al. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia 2018, 32, 2546–2557. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 inhibition in AML: An unexpected bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; DiNardo, C.D. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018, 14, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Nassereddine, S.; Lap, C.J.; Tabbara, I.A. Evaluating ivosidenib for the treatment of relapsed/refractory AML: Design, development, and place in therapy. OncoTargets Ther. 2018, 12, 303–308. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Krönke, J.; Bullinger, L.; Späth, D.; Kayser, S.; Zucknick, M.; Götze, K.; et al. IDH1 and IDH2 Mutations Are Frequent Genetic Alterations in Acute Myeloid Leukemia and Confer Adverse Prognosis in Cytogenetically Normal Acute Myeloid Leukemia with NPM1 Mutation Without FLT3 Internal Tandem Duplication. JCO 2010, 28, 3636–3643. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypermethylating | Hypomethylating | p Value | ||
|---|---|---|---|---|
| (n = 24) | (n = 32) | |||
| Sex | Female | 14 (58%) | 15 (47%) | 0.430 |
| Male | 10 (42%) | 17 (53%) | ||
| Median age (quartile 1, quartile 3) | 61 (38, 67) | 58 (48, 66) | 0.684 | |
| WHO NOS | AML with minimal maturation | 3 (13%) | 2 (6%) | 0.00271 |
| AML without maturation | 10 (42%) | 4 (13%) | ||
| AML with maturation | 6 (25%) | 5 (16%) | ||
| Acute myelomonocytic leukemia | 5 (21%) | 9 (28%) | ||
| Acute monoblastic/monocytic leukemia | 0 (0%) | 11 (34%) | ||
| Acute megakaryocytoblastic leukemia | 0 (0%) | 1 (3%) | ||
| Median WBC (quartile 1, quartile 3) (/μL) | 16.95 (5.40, 60.33) | 42.70 (8.13, 91.20) | 0.224 | |
| Median bone marrow blast percentage (quartile 1, quartile 3) | 76 (59, 87) | 76 (57, 86) | 0.734 | |
| Median peripheral blood blast percentage (quartile 1, quartile 3) | 51 (17, 86) | 10 (4, 58) | 0.0048 | |
| Cytogenetic risk | Good | 2 (8%) | 0 (0%) | 0.388 |
| Intermediate | 20 (83%) | 27 (84%) | ||
| Poor | 1 (4%) | 4 (13%) | ||
| Not determined | 1 (4%) | 1 (3%) | ||
| TET2 | IDH1 | IDH2 | WT1 | p Value | ||
|---|---|---|---|---|---|---|
| (n = 7) | (n = 5) | (n = 7) | (n = 5) | |||
| Gender | Female | 5 (71%) | 2 (40%) | 5 (71%) | 2 (40%) | 0.525 |
| Male | 2 (29%) | 3 (60%) | 2 (29%) | 3 (60%) | ||
| Median age (quartile 1, quartile 3) | 61 (49, 72) | 32 (27, 38) | 67 (62, 70) | 57 (53, 61) | 0.0118 | |
| WHO NOS | AML with minimal maturation | 0 (0%) | 0 (0%) | 2 (29%) | 1 (20%) | 0.153 |
| AML without maturation | 3 (43%) | 5 (100%) | 1 (14%) | 1 (20%) | ||
| AML with maturation | 3 (43%) | 0 (0%) | 2 (29%) | 1 (20%) | ||
| Acute myelomonocytic leukemia | 1 (14%) | 0 (0%) | 2 (29%) | 2 (40%) | ||
| Acute monoblastic/monocytic leukemia | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||
| Acute megakaryoblastic leukemia | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||
| Median WBC (quartile 1, quartile 3) (/μL) | 9.80 (3.95, 30.75) | 39.80 (8.20, 63.70) | 11.50 (3.80, 38.40) | 27.70 (27.10, 61.60) | 0.339 | |
| Median bone marrow blast percentage (quartile 1, quartile 3) | 63 (51, 86) | 86 (85, 91) | 72 (57, 81) | 72 (61, 86) | 0.255 | |
| Median peripheral blood blast percentage (quartile 1, quartile 3) | 32 (15, 60) | 85 (83, 88) | 43 (14, 68) | 52 (49, 63) | 0.164 | |
| Cytogenetic risk | Good | 1 (14%) | 0 (0%) | 0 (0%) | 1 (20%) | 0.7187 |
| Intermediate | 6 (84%) | 4 (80%) | 6 (84%) | 4 (80%) | ||
| Poor | 0 (0%) | 0 (0%) | 1 (14%) | 0 (0%) | ||
| Not determined | 0 (0%) | 1 (20%) | 0 (0%) | 0 (0%) | ||
| DNMT3A | KMT2A | p Value | ||
|---|---|---|---|---|
| (n = 23) | (n = 9) | |||
| Gender | Female | 13 (57%) | 2 (22%) | 0.122 |
| Male | 10 (43%) | 7 (78%) | ||
| Median age (quartile 1, quartile 3) | 58 (50, 71) | 54 (45, 64) | 0.571 | |
| WHO NOS | AML with minimal maturation | 0 (0%) | 2 (22%) | |
| AML without maturation | 4 (17%) | 0 (0%) | ||
| AML with maturation | 4 (17%) | 1 (11%) | ||
| Acute myelomonocytic leukemia | 6 (26%) | 3 (33%) | ||
| Acute monoblastic/monocytic leukemia | 8 (35%) | 3 (33%) | ||
| Acute megakaryoblastic leukemia | 1 (4%) | 0 (0%) | ||
| Median WBC (quartile 1, quartile 3) (/μL) | 75.20 (15.15, 98.70) | 8.40 (2.30, 25.90) | 0.00497 | |
| Median bone marrow blast percentage (quartile 1, quartile 3) | 76 (55, 86) | 75 (67, 83) | 0.949 | |
| Median peripheral blood blast percentage (quartile 1, quartile 3) | 11 (6, 76) | 0 (0, 14) | 0.0477 | |
| Cytogenetic risk | Good | 0 (0%) | 0 (0%) | 0.0572 |
| Intermediate | 21 (91%) | 6 (66%) | ||
| Poor | 1 (4%) | 3 (33%) | ||
| Not determined | 1 (4%) | 0 (0%) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasca, S.; Turcas, C.; Jurj, A.; Teodorescu, P.; Iluta, S.; Hotea, I.; Bojan, A.; Selicean, C.; Fetica, B.; Petrushev, B.; et al. The Influence of Methylating Mutations on Acute Myeloid Leukemia: Preliminary Analysis on 56 Patients. Diagnostics 2020, 10, 263. https://doi.org/10.3390/diagnostics10050263
Pasca S, Turcas C, Jurj A, Teodorescu P, Iluta S, Hotea I, Bojan A, Selicean C, Fetica B, Petrushev B, et al. The Influence of Methylating Mutations on Acute Myeloid Leukemia: Preliminary Analysis on 56 Patients. Diagnostics. 2020; 10(5):263. https://doi.org/10.3390/diagnostics10050263
Chicago/Turabian StylePasca, Sergiu, Cristina Turcas, Ancuta Jurj, Patric Teodorescu, Sabina Iluta, Ionut Hotea, Anca Bojan, Cristina Selicean, Bogdan Fetica, Bobe Petrushev, and et al. 2020. "The Influence of Methylating Mutations on Acute Myeloid Leukemia: Preliminary Analysis on 56 Patients" Diagnostics 10, no. 5: 263. https://doi.org/10.3390/diagnostics10050263
APA StylePasca, S., Turcas, C., Jurj, A., Teodorescu, P., Iluta, S., Hotea, I., Bojan, A., Selicean, C., Fetica, B., Petrushev, B., Moisoiu, V., Zimta, A.-A., Sas, V., Constantinescu, C., Zdrenghea, M., Dima, D., & Tomuleasa, C. (2020). The Influence of Methylating Mutations on Acute Myeloid Leukemia: Preliminary Analysis on 56 Patients. Diagnostics, 10(5), 263. https://doi.org/10.3390/diagnostics10050263