Microbiome Profile of Deep Endometriosis Patients: Comparison of Vaginal Fluid, Endometrium and Lesion

 , , and
, , and 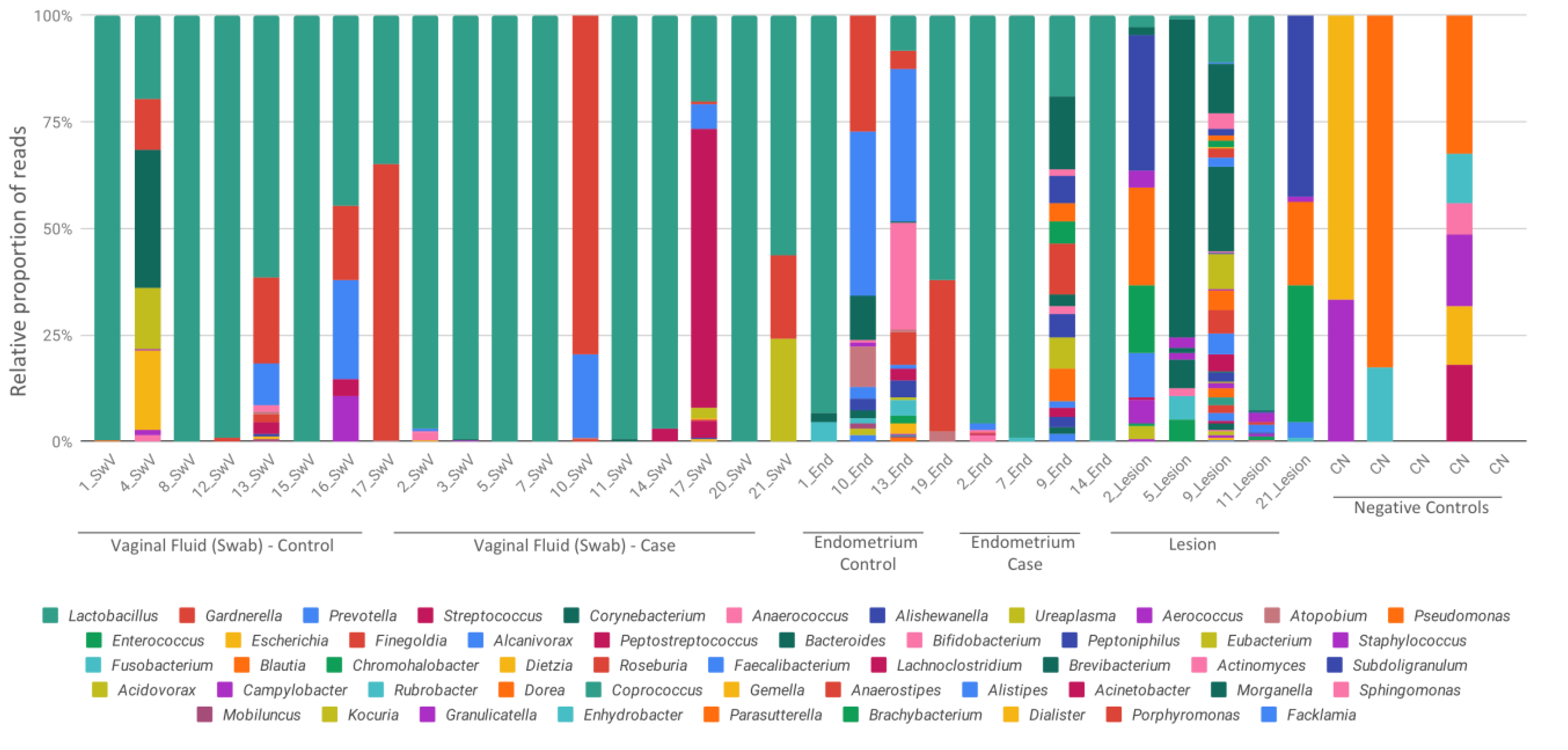{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and 16S rRNA Amplicon Sequencing
2.3. Microbiome Profile Evaluation Through Bioinformatics Analysis
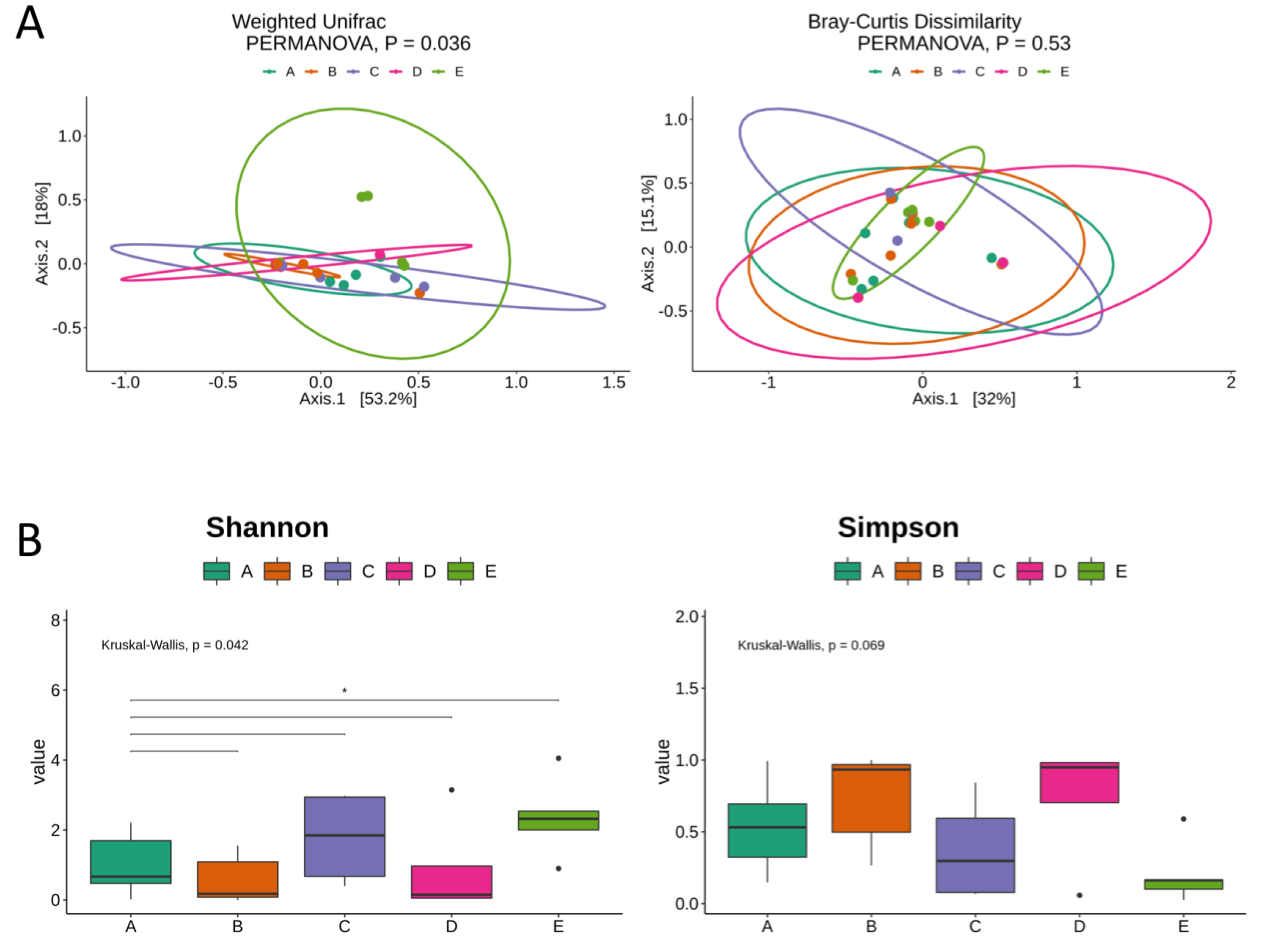
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Paula Andres, M.; Lopes, L.A.; Baracat, E.C.; Podgaec, S. Dienogest in the treatment of endometriosis: Systematic review. Arch. Gynecol. Obstet. 2015, 292, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Parasar, P.; Ozcan, P.; Terry, K.L. Endometriosis: Epidemiology, Diagnosis and Clinical Management. Curr. Obstet. Gynecol. Rep. 2017, 6, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Nisolle, M.; Donnez, J. Peritoneal endometriosis, ovarian endometriosis, and adenomyotic nodules of the rectovaginal septum are three different entities. Fertil. Steril. 1997, 68, 585–596. [Google Scholar] [CrossRef]
- Bassi, M.A.; Arias, V.; D’Amico, N.; Gueuvoghlanian-Silva, B.Y.; Abrao, M.S.; Podgaec, S. Deep Invasive Endometriosis Lesions of the Rectosigmoid May Be Related to Alterations in Cell Kinetics. Reprod Sci. 2015, 22, 1122–1128. [Google Scholar] [CrossRef]
- Santulli, P.; Tran, C.; Gayet, V.; Bourdon, M.; Maignien, L.; Pocate-Cheriet, K.; Chapron, C.; de Ziegler, D. Oligo-anovulation is not a rarer feature in women with documented endometriosis. Fertil. Steril. 2018, 110, 941–948. [Google Scholar] [CrossRef]
- Klemmt, P.A.B.; Starzinski-Powitz, A. Molecular and Cellular Pathogenesis of Endometriosis. Curr. Womens Health Rev. 2018, 14, 106–116. [Google Scholar] [CrossRef]
- Sampson, J.A. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110. [Google Scholar]
- Silvae, J.C.R.; da Fortunato, G.G.; Barbosa, C.P. Aspectos Gerais da Etiopatogenia da Endometriose. In Coleção Febrasgo Endometriose, 1st ed.; Podgaec, S., de Janeiro, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 27–34. [Google Scholar]
- Zhang, T.; De Carolis, C.; Man, G.C.W.; Wang, C.C. The link between immunity, autoimmunity and endometriosis: A literature update. Autoimmun. Rev. 2018, 17, 945–955. [Google Scholar] [CrossRef]
- Podgaec, S.; junior, J.A.D.; Chapron, C.; de Oliveira, R.M.; Baracat, E.; Abrao, M.S. Th1 and Th2 immune responses related to pelvic endometriosis. Rev. Assoc. Med. Bras. 2010, 56, 92–98. [Google Scholar] [CrossRef]
- Podgaec, S.; Rizzo, L.V.; Fernandes, L.F.; Baracat, E.C.; Abrao, M.S. CD4+ CD25high Foxp3+ cells increased in the peritoneal fluid of patients with endometriosis. Am. J. Reprod Immunol. 2012, 68, 301–308. [Google Scholar] [CrossRef]
- Sourial, S.; Tempest, N.; Hapangama, D.K. Theories on the pathogenesis of endometriosis. Int. J. Reprod. Med. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; Fujishita, A.; Hiraki, K.; Kitajima, M.; Nakashima, M.; Fushiki, S.; Kitawaki, J. Bacterial contamination hypothesis: A new concept in endometriosis. Reprod. Med. Biol. 2018, 18, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Gueuvoghlanian-Silva, B.Y.; Bellelis, P.; Barbeiro, F.D.; Hernandes, C.; Podgaec, S. Treg and NK cells related cytokines are associated with deep rectosigmoid endometriosis and clinical symptoms related to the disease. J. Reprod. Immunol. 2018, 126, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Bellelis, P.; Barbeiro, D.F.; Gueuvoghlanian-Silva, B.Y.; Kalil, J.; Abrão, M.S.; Podgaec, S. Interleukin-15 and Interleukin-7 are the Major Cytokines to Maintain Endometriosis. Gynecol. Obstet. Invest. 2019, 84, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Nishioka, K.; Khan, K.N.; Tanaka, Y.; Mori, T.; Nakaya, T.; Kitawaki, J. Molecular detection of microbial colonization in cervical mucus of women with and without endometriosis. Am. J. Reprod. Immunol. 2019, 82, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Manuel Navarro, J. Inflammasome as a key pathogenic mechanism in endometriosis. Curr. Drug Targets 2017, 18, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Chadchan, S.B.; Cheng, M.; Parnell, L.A.; Yin, Y.; Schriefer, A.; Mysorekar, I.U.; Kommagani, R. Antibiotic therapy with metronidazole reduces endometriosis disease progression in mice: A potential role for gut microbiota. Human Reprod. 2019, 34, 1106–1116. [Google Scholar] [CrossRef]
- Campos, G.B.; Marques, L.M.; Rezende, I.S. Mycoplasma genitalium can modulate the local immune response in patients with endometriosis. Fertil. Steril. 2018, 109, 549–560. [Google Scholar] [CrossRef]
- Khan, K.N.; Kitajima, M.; Hiraki, K.; Yamaguchi, N.; Katamine, S.; Matsuyama, T.; Nakashima, M.; Fujishita, A.; Ishimaru, T.; Masuzaki, H. Escherichia coli contamination of menstrual blood and effect of bacterial endotoxin on endometriosis. Fertil. Steril. 2010, 94, 860–863. [Google Scholar] [CrossRef]
- Khan, K.N.; Fujishita, A.; Kitajima, M.; Hiraki, K.; Nakashima, M.; Masuzaki, H. Intra-uterine microbial colonization and occurrence of endometritis in women with endometriosis. Human Reprod. 2014, 29, 2446–2456. [Google Scholar] [CrossRef]
- Khan, K.N.; Fujishita, A.; Masumoto, H.; Muto, H.; Kitajima, M.; Masuzaki, H.; Kitawaki, J. Molecular detection of intrauterine microbial colonization in women with endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 199, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 17, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Ata, B.; Yildiz, S.; Turkgeldi, E.; Brocal, V.P.; Dinleyici, E.C.; Moya, A.; Urman, B. The Endobiota Study: Comparison of Vaginal, Cervical and Gut Microbiota between Women with Stage 3/4 Endometriosis and Healthy Controls. Sci. Rep. 2019, 9, 2204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, P. Conservative Fragments in Bacterial 16S rRNA Genes and Primer Design for 16S Ribosomal DNA Amplicons in Metagenomic Studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Christian, L.L.; Walters, W.A.; Berg-lyons, D.; Huntley, J.; Fierer, N.; Owens, S.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Christoff, A.P.; Cruz, G.F.N.; Sereia, A.F.R.; Yamanaka, L.E.; Silveira, P.P.; Oliveira, L.F.V. End-to-end assessment of fecal bacteriome analysis: From sample processing to DNA sequencing and bioinformatics results. bioRxiv 2019, 646349. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Xu, Z.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. Peerj 2016, 4, e2584. [Google Scholar] [CrossRef]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 2, 635–645. [Google Scholar] [CrossRef]
- McMurdie, P.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Dehal, P.; Arkin, A. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2015, 71, 8228. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef]
- McMurdie, P.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Hothorn, T.; Hornik, K.; Wiel, M.A.; Zeileis, A. Implementing a class of permutation tests: The coin package. J. Stat. Softw. 2008, 28, 23. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Hernandes, C.; Gueuvoghlanian-Silva, B.Y.; Monnaka, V.U.; Ribeiro, N.M.; Pereira, W.O.; Podgaec, S. Regulatory T cells isolated from endometriotic peritoneal fluid express a different number of Toll-like receptors expressed. Einstein (São Paulo) 2020, 18, eAO5294. [Google Scholar]
- Tai, F.W.; Chang, C.Y.; Chiang, J.H.; Lin, W.C.; Wan, L. Association of Pelvic Inflammatory Disease with Risk of Endometriosis: A Nationwide Cohort Study Involving 141,460 Individuals. J. Clin. Med. 2018, 7, 379. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 15, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Goodall, J.C. New concepts in Chlamydia induced inflammasome responses. Microb. Infect. 2018, 20, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Caramalho, I.; Lopes-Carvalho, T.; Ostler, D.; Zelenay, S.; Haury, M.; Demengeot, J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J. Exp. Med. 2003, 197, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Sutmuller, R.; Hermann, C.; Van der Graaf, C.A.; Van der Meer, J.W.; Van Krieken, J.H.; Hartung, T.; Adema, G.; Kullberg, B.J. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J. Immunol. 2004, 172, 3712–3718. [Google Scholar] [CrossRef]
- Peng, G.; Guo, Z.; Kiniwa, Y.; Voo, K.S.; Peng, W.; Fu, T.; Wang, D.Y.; Li, Y.; Wang, H.Y. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 2005, 309, 1380–1384. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, Y. Toll-like receptors and immune regulation: Their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology 2007, 122, 149–156. [Google Scholar] [CrossRef]
- Van Maren, W.W.; Jacobs, J.F.; de Vries, I.J.; Nierkens, S.; Adema, G.J. Toll-like receptor signalling on Tregs: To suppress or not to suppress? Immunology 2008, 124, 445–452. [Google Scholar] [CrossRef]
- Olivier, A.; Dong, H.; Sparwasser, T.; Majilessi, L.; Leclerc, C. The adjuvant effect of TLR agonists on CD4+ effector T cells is under the indirect control of regulatory T cells. Eur. J. Immunol. 2011, 41, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- de Barros, I.B.L.; Malvezzi, H.; Gueuvoghlanian-Silva, B.Y.; Piccinato, C.A.; Rizzo, L.V.; Podgaec, S. Corrigendum to “What do we know about regulatory T cells and endometriosis? A systematic review”. J. Reprod. Immunol. 2017, 121, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, A.; Cottone, L.; Monno, A.; Manfredi, A.A.; Rovere-Querini, P. The peritoneum: Healing, immunity, and diseases. J. Pathol. 2017, 243, 137–147. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandes, C.; Silveira, P.; Rodrigues Sereia, A.F.; Christoff, A.P.; Mendes, H.; Valter de Oliveira, L.F.; Podgaec, S. Microbiome Profile of Deep Endometriosis Patients: Comparison of Vaginal Fluid, Endometrium and Lesion. Diagnostics 2020, 10, 163. https://doi.org/10.3390/diagnostics10030163
Hernandes C, Silveira P, Rodrigues Sereia AF, Christoff AP, Mendes H, Valter de Oliveira LF, Podgaec S. Microbiome Profile of Deep Endometriosis Patients: Comparison of Vaginal Fluid, Endometrium and Lesion. Diagnostics. 2020; 10(3):163. https://doi.org/10.3390/diagnostics10030163
Chicago/Turabian StyleHernandes, Camila, Paola Silveira, Aline Fernanda Rodrigues Sereia, Ana Paula Christoff, Helen Mendes, Luiz Felipe Valter de Oliveira, and Sergio Podgaec. 2020. "Microbiome Profile of Deep Endometriosis Patients: Comparison of Vaginal Fluid, Endometrium and Lesion" Diagnostics 10, no. 3: 163. https://doi.org/10.3390/diagnostics10030163
APA StyleHernandes, C., Silveira, P., Rodrigues Sereia, A. F., Christoff, A. P., Mendes, H., Valter de Oliveira, L. F., & Podgaec, S. (2020). Microbiome Profile of Deep Endometriosis Patients: Comparison of Vaginal Fluid, Endometrium and Lesion. Diagnostics, 10(3), 163. https://doi.org/10.3390/diagnostics10030163