The Role of Innate Lymphoid Cells in the Regulation of Immune Homeostasis in Sepsis-Mediated Lung Inflammation

 ,
, 
 , , ,
, , ,  ,
,
Abstract
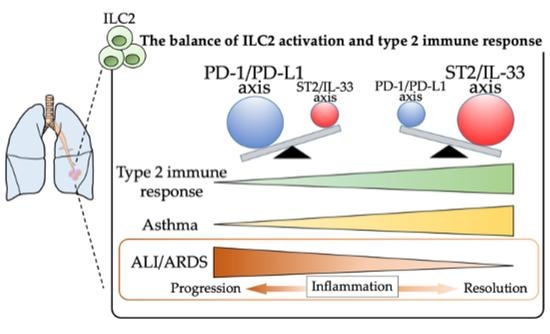
1. Introduction
2. Innate Lymphoid Cells Protect against Pathogens and Contribute to Tissue Repair
3. A Pathogenic Role of ILC2s and Lung ILC2s
4. ILC2s in Sepsis-Induced Lung Injury
5. ILC2 Subsets in the Septic Lung
6. The Mechanism That Drives IL-33/ST2 Signaling Stimulates ILC2s
7. The Mechanism Underlying PD-1/PD-L1 Signaling Inhibits ILC2s
8. The Functional Dynamics of Lung ILC2s during Sepsis
9. PD-1 on ILC2s as a Target for PD-1 Blocking Therapy in the Septic Lung
10. Summary and Future Challenges
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Abe, T.; on behalf of JAAM FORECAST group; Ogura, H.; Shiraishi, A.; Kushimoto, S.; Saitoh, D.; Fujishima, S.; Mayumi, T.; Shiino, Y.; Nakada, T.-A.; et al. Characteristics, management, and in-hospital mortality among patients with severe sepsis in intensive care units in Japan: The FORECAST study. Crit. Care 2018, 22, 322. [Google Scholar] [CrossRef]
- Vincent, J.-L.; Jones, G.; David, S.; Olariu, E.; Cadwell, K.K. Frequency and mortality of septic shock in Europe and North America: A systematic review and meta-analysis. Crit. Care 2019, 23, 196. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells — a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Spits, H.; Di Santo, J.P. The expanding family of innate lymphoid cells: Regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 2010, 12, 21–27. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Dutton, E.E.; Camelo, A.; Sleeman, M.; Herbst, R.; Carlesso, G.; Belz, G.T.; Withers, D.R. Characterisation of innate lymphoid cell populations at different sites in mice with defective T cell immunity. Wellcome Open Res. 2018, 2, 117. [Google Scholar] [CrossRef]
- Lai, D.; Tang, J.; Chen, L.; Fan, E.K.; Scott, M.J.; Li, Y.; Billiar, T.R.; Wilson, M.A.; Fang, X.; Shu, Q.; et al. Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Krishack, P.A.; Louviere, T.J.; Decker, T.S.; Kuzel, T.G.; Greenberg, J.A.; Camacho, D.F.; Hrusch, C.L.; Sperling, A.I.; Verhoef, P.A. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; Velde, A.A.T.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; E Heinsbroek, S.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.M.; Chaix, J.; Rupp, L.J.; Wu, J.; Madera, S.; Sun, J.C.; Lindsten, T.; Reiner, S.L. The Transcription Factors T-bet and Eomes Control Key Checkpoints of Natural Killer Cell Maturation. Immunity 2012, 36, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Weizman, O.-E.; Adams, N.M.; Schuster, I.S.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 2017, 171, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.-I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nat. Cell Biol. 2009, 463, 540–544. [Google Scholar] [CrossRef]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nat. Cell Biol. 2010, 464, 1367–1370. [Google Scholar] [CrossRef]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T.; et al. MHCII-Mediated Dialog between Group 2 Innate Lymphoid Cells and CD4+ T Cells Potentiates Type 2 Immunity and Promotes Parasitic Helminth Expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Kato, T.; Motomura, Y.; Kageyama, T.; Taguchi-Atarashi, N.; Kinoshita-Daitoku, R.; Kuroda, E.; Di Santo, J.P.; Mimuro, H.; Moro, K.; et al. Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection through Induction of IgA. Immunity 2020, 52, 635–649. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Vosshenrich, C.A.J.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.-J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial Flora Drives Interleukin 22 Production in Intestinal NKp46+ Cells that Provide Innate Mucosal Immune Defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.M.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nat. Cell Biol. 2008, 457, 722–725. [Google Scholar] [CrossRef]
- Cupedo, T. (Tom); Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue–inducer cells are interleukin 17–producing precursors to RORC+ CD127+ natural killer–like cells. Nat. Immunol. 2008, 10, 66–74. [Google Scholar] [CrossRef]
- Van Maele, L.; Carnoy, C.; Cayet, D.; Ivanov, S.; Porte, R.; Deruy, E.; Chabalgoity, J.A.; Renauld, J.-C.; Eberl, G.; Benecke, A.G.; et al. Activation of Type 3 Innate Lymphoid Cells and Interleukin 22 Secretion in the Lungs During Streptococcus pneumoniae Infection. J. Infect. Dis. 2014, 210, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Abt, M.C.; Lewis, B.B.; Caballero, S.; Xiong, H.; Carter, R.A.; Sušac, B.; Ling, L.; Leiner, I.; Pamer, E.G. Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium difficile Infection. Cell Host Microbe 2015, 18, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Nabekura, T.; Riggan, L.; Hildreth, A.D.; O’Sullivan, T.E.; Shibuya, A. Type 1 Innate Lymphoid Cells Protect Mice from Acute Liver Injury via Interferon-γ Secretion for Upregulating Bcl-xL Expression in Hepatocytes. Immunity 2020, 52, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging Functions of Amphiregulin in Orchestrating Immunity, Inflammation, and Tissue Repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin–EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef]
- Li, Z.; Hodgkinson, T.; Gothard, E.J.; Boroumand, S.; Lamb, R.; Cummins, I.; Narang, P.; Sawtell, A.; Coles, J.; Leonov, G.; et al. Epidermal Notch1 recruits RORγ+ group 3 innate lymphoid cells to orchestrate normal skin repair. Nat. Commun. 2016, 7, 11394. [Google Scholar] [CrossRef]
- Starkey, M.R.; McKenzie, A.N.; Belz, G.T.; Hansbro, P.M. Pulmonary group 2 innate lymphoid cells: Surprises and challenges. Mucosal Immunol. 2019, 12, 299–311. [Google Scholar] [CrossRef]
- McKenzie, A.N.J. Type-2 Innate Lymphoid Cells in Asthma and Allergy. Ann. Am. Thorac. Soc. 2014, 11, S263–S270. [Google Scholar] [CrossRef]
- Helfrich, S.; Mindt, B.C.; Fritz, J.H.; Duerr, C. Group 2 Innate Lymphoid Cells in Respiratory Allergic Inflammation. Front. Immunol. 2019, 10, 930. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2014, 16, 45–56. [Google Scholar] [CrossRef]
- Smith, S.G.; Chen, R.; Kjarsgaard, M.; Huang, C.; Oliveria, J.-P.; O’Byrne, P.M.; Gauvreau, G.M.; Boulet, L.; Lemière, C.; Martin, J.; et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J. Allergy Clin. Immunol. 2016, 137, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Bartemes, K.R.; Kephart, G.M.; Fox, S.J.; Kita, H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J. Allergy Clin. Immunol. 2014, 134, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Christianson, C.A.; Goplen, N.P.; Zafar, I.; Irvin, C.; Good, J.T.; Rollins, D.R.; Gorentla, B.; Liu, W.; Gorska, M.M.; Chu, H.; et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J. Allergy Clin. Immunol. 2015, 136, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, S.; Brestoff, J.R.; Flamar, A.-L.; Moeller, J.B.; Klose, C.S.N.; Rankin, L.C.; Yudanin, N.A.; Monticelli, L.A.; Putzel, G.G.; Rodewald, H.-R.; et al. β2-adrenergic receptor–mediated negative regulation of group 2 innate lymphoid cell responses. Science 2018, 359, 1056–1061. [Google Scholar] [CrossRef]
- Kabata, H.; Moro, K.; Fukunaga, K.; Suzuki, Y.; Miyata, J.; Masaki, K.; Betsuyaku, T.; Koyasu, S.; Asano, K. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat. Commun. 2013, 4, 2675. [Google Scholar] [CrossRef]
- Von Moltke, J.; O’Leary, C.E.; Barrett, N.A.; Kanaoka, Y.; Austen, K.F.; Locksley, R.M. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J. Exp. Med. 2016, 214, 27–37. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; O’Byrne, P.M.; Boulet, L.-P.; Wang, Y.; Cockcroft, D.; Bigler, J.; Fitzgerald, J.M.; Boedigheimer, M.; Davis, B.E.; Dias, C.; et al. Effects of an Anti-TSLP Antibody on Allergen-Induced Asthmatic Responses. N. Engl. J. Med. 2014, 370, 2102–2110. [Google Scholar] [CrossRef]
- Erpenbeck, V.J.; Popov, T.A.; Miller, D.; Weinstein, S.F.; Spector, S.; Magnusson, B.; Osuntokun, W.; Goldsmith, P.; Weiss, H.M.; Beier, J. The oral CRTh2 antagonist QAW039 (fevipiprant): A phase II study in uncontrolled allergic asthma. Pulm. Pharmacol. Ther. 2016, 39, 54–63. [Google Scholar] [CrossRef]
- Halim, T.Y.F.; MacLaren, A.; Romanish, M.T.; Gold, M.; McNagny, K.M.; Takei, F. Retinoic-Acid-Receptor-Related Orphan Nuclear Receptor Alpha Is Required for Natural Helper Cell Development and Allergic Inflammation. Immunity 2012, 37, 463–474. [Google Scholar] [CrossRef]
- Halim, T.Y.; Steer, C.A.; Mathä, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.; Takei, F. Group 2 Innate Lymphoid Cells Are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef]
- Gold, M.; Antignano, F.; Halim, T.Y.; Hirota, J.A.; Blanchet, M.-R.; Zaph, C.; Takei, F.; McNagny, K.M. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J. Allergy Clin. Immunol. 2014, 133, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.F.; Hwang, Y.Y.; Scanlon, S.T.; Zaghouani, H.; Garbi, N.; Fallon, P.G.; McKenzie, A.N.J. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat. Immunol. 2015, 17, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Guardado, J.; Hoffman, R.; Xu, H.; Namas, R.; Vodovotz, Y.; Xu, L.; Ramadan, M.; Brown, J.; Turnquist, H.R.; et al. IL33-mediated ILC2 activation and neutrophil IL5 production in the lung response after severe trauma: A reverse translation study from a human cohort to a mouse trauma model. PLoS Med. 2017, 14, e1002365. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Adachi, T.; Koida, A.; Nakanishi, K. Nematode-Infected Mice Acquire Resistance to Subsequent Infection With Unrelated Nematode by Inducing Highly Responsive Group 2 Innate Lymphoid Cells in the Lung. Front. Immunol. 2018, 9, 2132. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.F.; Rana, B.M.; Walker, J.A.; Kerscher, B.; Knolle, M.D.; Jolin, H.E.; Serrao, E.M.; Haim-Vilmovsky, L.; Teichmann, S.A.; Rodewald, H.-R.; et al. Tissue-Restricted Adaptive Type 2 Immunity Is Orchestrated by Expression of the Costimulatory Molecule OX40L on Group 2 Innate Lymphoid Cells. Immunity 2018, 48, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Donovan, C.; Starkey, M.R.; Kim, R.Y.; Rana, B.M.J.; Barlow, J.L.; Jones, B.; Haw, T.J.; Nair, P.M.; Budden, K.; Cameron, G.J.; et al. Roles for T/B lymphocytes and ILC2s in experimental chronic obstructive pulmonary disease. J. Leukoc. Biol. 2018, 105, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.D.; Steinberg, K.P. Epidemiology of acute lung injury and ARDS. Chest 1999, 116, 74S–82S. [Google Scholar] [CrossRef]
- Mikkelsen, M.E.; Shah, C.V.; Meyer, N.J.; Gaieski, D.F.; Lyon, S.; Miltiades, A.N.; Goyal, M.; Fuchs, B.D.; Bellamy, S.L.; Christie, J.D. The Epidemiology of Acute Respiratory Distress Syndrome in Patients Presenting to the Emergency Department with Severe Sepsis. Shock 2013, 40, 375–381. [Google Scholar] [CrossRef]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 562–572. [Google Scholar] [CrossRef]
- Linch, S.N.; Danielson, E.T.; Kelly, A.M.; Tamakawa, R.A.; Lee, J.J.; Gold, J.A. Interleukin 5 Is Protective during Sepsis in an Eosinophil-Independent Manner. Am. J. Respir. Crit. Care Med. 2012, 186, 246–254. [Google Scholar] [CrossRef]
- Hrusch, C.L.; Manns, S.T.; Bryazka, D.; Casaos, J.; Bonham, C.A.; Jaffery, M.R.; Blaine, K.M.; Mills, K.A.; Verhoef, P.A.; Adegunsoye, A.; et al. ICOS protects against mortality from acute lung injury through activation of IL-5+ ILC2s. Mucosal Immunol. 2017, 11, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Hübner, M.P.; Layland, L.E.; Hoerauf, A. Helminths and their implication in sepsis - a new branch of their immunomodulatory behaviour? Pathog. Dis. 2013, 69, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.-E.; Morrison, P.J.; Wilhelm, C.; Wilson, M.; Ahlfors, H.; Renauld, J.-C.; Panzer, U.; Helmby, H.; Stockinger, B. IL-9–mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 2013, 210, 2951–2965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Jin, Y.; Lai, D.; Wang, J.; Wang, Y.; Wu, X.; Scott, M.; Li, Y.; Hou, J.; Billiar, T.; et al. RAGE-induced ILC2 expansion in acute lung injury due to haemorrhagic shock. Thorax 2020, 75, 209–219. [Google Scholar] [CrossRef]
- Nascimento, D.C.; Melo, P.H.; Piñeros, A.R.; Ferreira, R.G.; Colón, D.F.; Donate, P.B.; Castanheira, F.V.; Gozzi, A.; Czaikoski, P.G.; Niedbala, W.; et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat. Commun. 2017, 8, 14919. [Google Scholar] [CrossRef]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 Is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33–LacZ Gene Trap Reporter Strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of Interleukin-33 Bioactivity through Proteolysis by Apoptotic Caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R., Jr.; Urban, J.F.; Paul, W.E. IL-25-responsive, lineage-negative KLRG1hi cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 2014, 16, 161–169. [Google Scholar] [CrossRef]
- Huang, Y.; Mao, K.; Chen, X.; Sun, M.-A.; Kawabe, T.; Li, W.; Usher, N.; Zhu, J.; Urban, J.F.; Paul, W.E.; et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. [Google Scholar] [CrossRef]
- Huang, Y.; Paul, W.E. Inflammatory group 2 innate lymphoid cells. Int. Immunol. 2015, 28, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Akama, Y.; Park, E.J.; Satoh-Takayama, N.; Gaowa, A.; Ito, A.; Kawamoto, E.; Darkwah, S.; Appiah, M.G.; Myint, P.K.; Ohno, H.; et al. Sepsis Induces Deregulation of IL-13 Production and PD-1 Expression in Lung Group 2 Innate Lymphoid Cells. Shock 2020. [Google Scholar] [CrossRef] [PubMed]
- Closa, D.; Folch-Puy, E. Oxygen Free Radicals and the Systemic Inflammatory Response. IUBMB Life 2004, 56, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Löhning, M.; Stroehmann, A.; Coyle, A.J.; Grogan, J.L.; Lin, S.; Gutierrez-Ramos, J.-C.; Levinson, D.; Radbruch, A.; Kamradt, T. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc. Natl. Acad. Sci. USA 1998, 95, 6930–6935. [Google Scholar] [CrossRef] [PubMed]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nat. Cell Biol. 2014, 513, 564–568. [Google Scholar] [CrossRef]
- Moritz, D.R.; Rodewald, H.R.; Gheyselinck, J.; Klemenz, R. The IL-1 receptor-related T1 antigen is expressed on immature and mature mast cells and on fetal blood mast cell progenitors. J. Immunol. 1998, 161, 4866–4874. [Google Scholar] [PubMed]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; Van Rooijen, N.; et al. IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef]
- Cherry, W.B.; Yoon, J.; Bartemes, K.R.; Iijima, K.; Kita, H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J. Allergy Clin. Immunol. 2008, 121, 1484–1490. [Google Scholar] [CrossRef]
- Stier, M.T.; Zhang, J.; Goleniewska, K.; Cephus, J.Y.; Rusznak, M.; Wu, L.; Van Kaer, L.; Zhou, B.; Newcomb, D.C.; Peebles, R.S. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J. Exp. Med. 2017, 215, 263–281. [Google Scholar] [CrossRef]
- Wang, L.; Tang, J.; Yang, X.; Zanvit, P.; Cui, K.; Ku, W.L.; Jin, W.; Zhang, D.; Goldberg, N.; Cain, A.; et al. TGF-β induces ST2 and programs ILC2 development. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, W.; Moon, U.J.; Kim, H.J.; Choi, H.-J.; Sin, J.-I.; Park, N.-H.; Cho, H.R.; Kwon, B. Intratumorally Establishing Type 2 Innate Lymphoid Cells Blocks Tumor Growth. J. Immunol. 2016, 196, 2410–2423. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.; Chesné, J.; Ribeiro, H.; García-Cassani, B.; Carvalho, T.; Bouchery, T.; Shah, K.; Barbosa-Morais, N.L.; Harris, N.L.; Veiga-Fernandes, H. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nat. Cell Biol. 2017, 549, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2017, 18, 153–167. [Google Scholar] [CrossRef]
- Schwartz, C.; Khan, A.R.; Floudas, A.; Saunders, S.P.; Hams, E.; Rodewald, H.-R.; McKenzie, A.N.; Fallon, P.G. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J. Exp. Med. 2017, 214, 2507–2521. [Google Scholar] [CrossRef]
- Nishimura, H.; Agata, Y.; Kawasaki, A.; Sato, M.; Imamura, S.; Minato, N.; Yagita, H.; Nakano, T.; Honjo, T. Developmentally regulated expression of the PD-1 protein on the surface of double-negative(CD4–CD8–) thymocytes. Int. Immunol. 1996, 8, 773–780. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Oldenhove, G.; Boucquey, E.; Taquin, A.; Acolty, V.; Bonetti, L.; Ryffel, B.; Le Bert, M.; Englebert, K.; Boon, L.; Moser, M. PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep. 2018, 25, 2053–2060. [Google Scholar] [CrossRef]
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.-A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S.; et al. PD-1 regulates KLRG1+ group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013, 13, 260–268. [Google Scholar] [CrossRef]
- Patil, N.K.; Guo, Y.; Luan, L.; Sherwood, E.R. Targeting Immune Cell Checkpoints during Sepsis. Int. J. Mol. Sci. 2017, 18, 2413. [Google Scholar] [CrossRef]
- Semler, M.W.; Wheeler, A.P.; Thompson, B.T.; Bernard, G.R.; Wiedemann, H.P.; Rice, T.W. Impact of Initial Central Venous Pressure on Outcomes of Conservative Versus Liberal Fluid Management in Acute Respiratory Distress Syndrome. Crit. Care Med. 2016, 44, 782–789. [Google Scholar] [CrossRef] [PubMed]
- National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network; Wiedemann, H.P.; Wheeler, A.P.; Bernard, G.R.; Thompson, B.T.; Hayden, D.; Deboisblanc, B.; Connors, A.F.; Hite, R.D.; Harabin, A.L. Comparison of Two Fluid-Management Strategies in Acute Lung Injury. N. Engl. J. Med. 2006, 354, 2564–2575. [Google Scholar] [CrossRef]
- Network, A.R.D.S.; Brower, R.G.; A Matthay, M.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef]
- Matsukawa, A.; Hogaboam, C.M.; Lukacs, N.W.; Lincoln, P.M.; Evanoff, H.L.; Strieter, R.M.; Kunkel, S.L. Expression and Contribution of Endogenous IL-13 in an Experimental Model of Sepsis. J. Immunol. 2000, 164, 2738–2744. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chang, Y.; Bae, B.; Sohn, K.-H.; Cho, S.-H.; Chung, O.H.; Kang, H.-R.; Kim, H.Y. Innate immune crosstalk in asthmatic airways: Innate lymphoid cells coordinate polarization of lung macrophages. J. Allergy Clin. Immunol. 2019, 143, 1769–1782. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, H.; Wang, C.; Li, H.; Zhang, Q.; Bai, J. M2A and M2C Macrophage Subsets Ameliorate Inflammation and Fibroproliferation in Acute Lung Injury Through Interleukin 10 Pathway. Shock 2017, 48, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Brahmamdam, P.; Inoue, S.; Unsinger, J.; Chang, K.C.; McDunn, J.E.; Hotchkiss, R.S. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J. Leukoc. Biol. 2010, 88, 233–240. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Lou, J.; Zhou, Y.; Bo, L.; Zhu, J.; Zhu, K.; Wan, X.; Cai, Z.; Deng, X. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit. Care 2011, 15, R70. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.T.; Chung, C.-S.; Fallon, E.A.; Hutchins, N.A.; Clarke, E.; Rossi, A.-L.; Cioffi, W.G.; Heffernan, D.S.; Ayala, A. Group 2 Innate Lymphoid Cells (ILC2s) Are Key Mediators of the Inflammatory Response in Polymicrobial Sepsis. Am. J. Pathol. 2018, 188, 2097–2108. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rorasg/sg BMT Mice | ||
| Disease Model | Roles of ILC2s in the Lung | References |
| Papain-induced asthma model | ILC2s play a crucial role in the inflammatory response to allergens, even in the presence of Th2 cells. | [39] |
| Papain-induced asthma model | ILC2-derived IL-13 primes naive T cells to differentiate into Th2 cells by promoting the migration of DCs to LNs. | [40] |
| House dust mite-induced asthma model | BAL eosinophils are significantly decreased in ILC2-deficient mice. | [41] |
| Papain-induced asthma model | ILC2s induce DCs, thereby promoting memory Th2 function. | [42] |
| Hemorrhagic shock model | ILC2-derived IL-5 promotes IL-5 production of neutrophils. | [43] |
| Helminth-infected mouse model | ILC2-deficiency leads to a reduction in eosinophil accumulation. | [44] |
| Rorafl/sgIl7rCre Mice | ||
| Disease Model | Roles of ILC2s in the Lung | References |
| Papain-induced asthma model | ILC2s induce DCs, thereby promoting memory Th2 functionality. (The authors used both Rorasg/sg BMT and Rorafl/sgIl7rCre mice). | [42] |
| Papain-induced asthma model Helminth-infected mouse model | ILC2s promote increases in Th2 cells in mLN and IgE concentrations in the lung. | [45] |
| Cigarette smoke exposure model | ILC2s help induce collagen deposition following cigarette exposure. | [46] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akama, Y.; Satoh-Takayama, N.; Kawamoto, E.; Ito, A.; Gaowa, A.; Park, E.J.; Imai, H.; Shimaoka, M. The Role of Innate Lymphoid Cells in the Regulation of Immune Homeostasis in Sepsis-Mediated Lung Inflammation. Diagnostics 2020, 10, 808. https://doi.org/10.3390/diagnostics10100808
Akama Y, Satoh-Takayama N, Kawamoto E, Ito A, Gaowa A, Park EJ, Imai H, Shimaoka M. The Role of Innate Lymphoid Cells in the Regulation of Immune Homeostasis in Sepsis-Mediated Lung Inflammation. Diagnostics. 2020; 10(10):808. https://doi.org/10.3390/diagnostics10100808
Chicago/Turabian StyleAkama, Yuichi, Naoko Satoh-Takayama, Eiji Kawamoto, Atsushi Ito, Arong Gaowa, Eun Jeong Park, Hiroshi Imai, and Motomu Shimaoka. 2020. "The Role of Innate Lymphoid Cells in the Regulation of Immune Homeostasis in Sepsis-Mediated Lung Inflammation" Diagnostics 10, no. 10: 808. https://doi.org/10.3390/diagnostics10100808
APA StyleAkama, Y., Satoh-Takayama, N., Kawamoto, E., Ito, A., Gaowa, A., Park, E. J., Imai, H., & Shimaoka, M. (2020). The Role of Innate Lymphoid Cells in the Regulation of Immune Homeostasis in Sepsis-Mediated Lung Inflammation. Diagnostics, 10(10), 808. https://doi.org/10.3390/diagnostics10100808