Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects

Abstract
1. Introduction
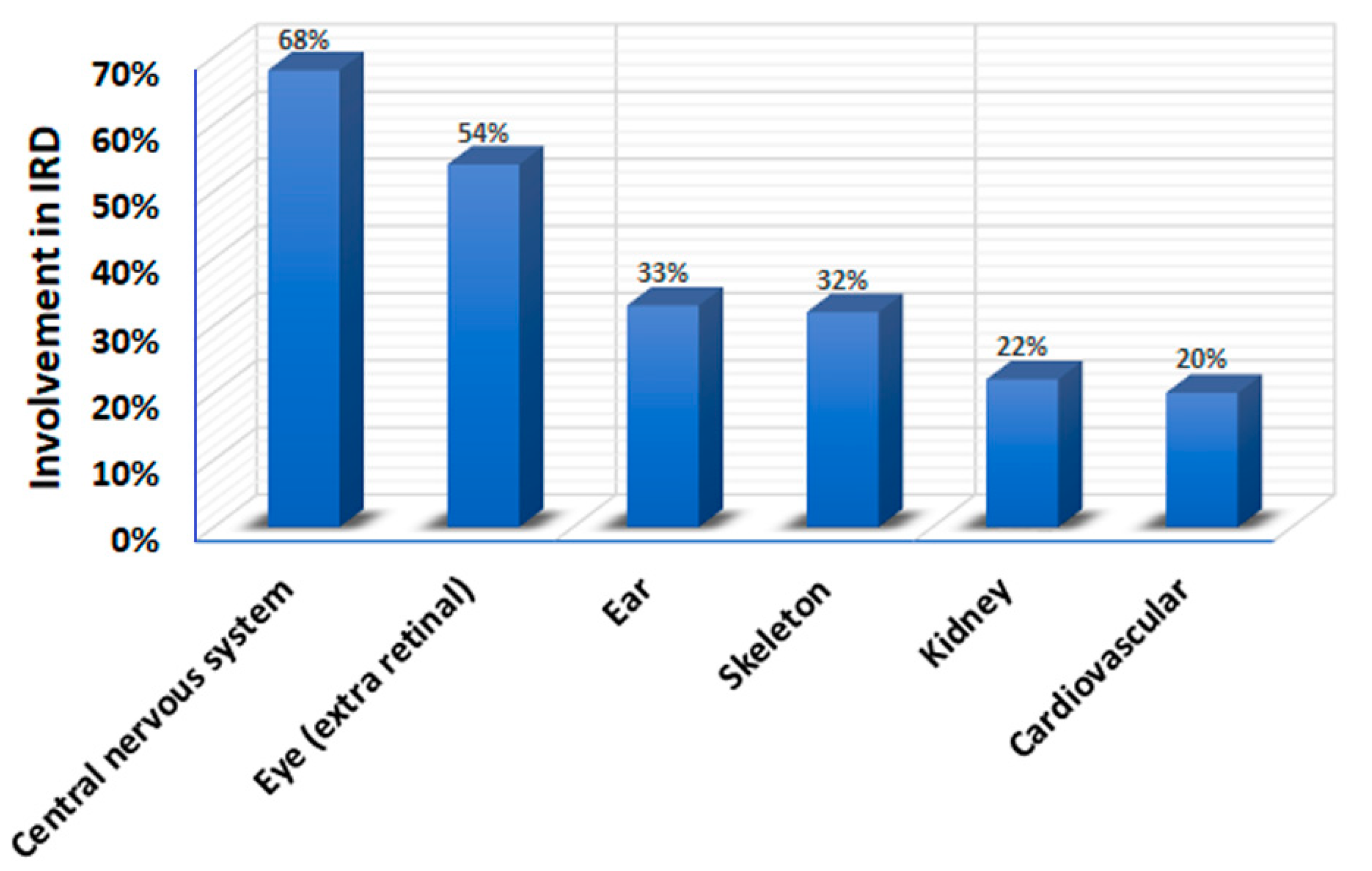
2. Syndromic IRD Types
3. Genetic Heterogeneity in Syndromic IRDs
4. Phenotypic Overlap in Syndromic IRDs
4.1. Phenotypic Overlap between Different IRD Syndromes
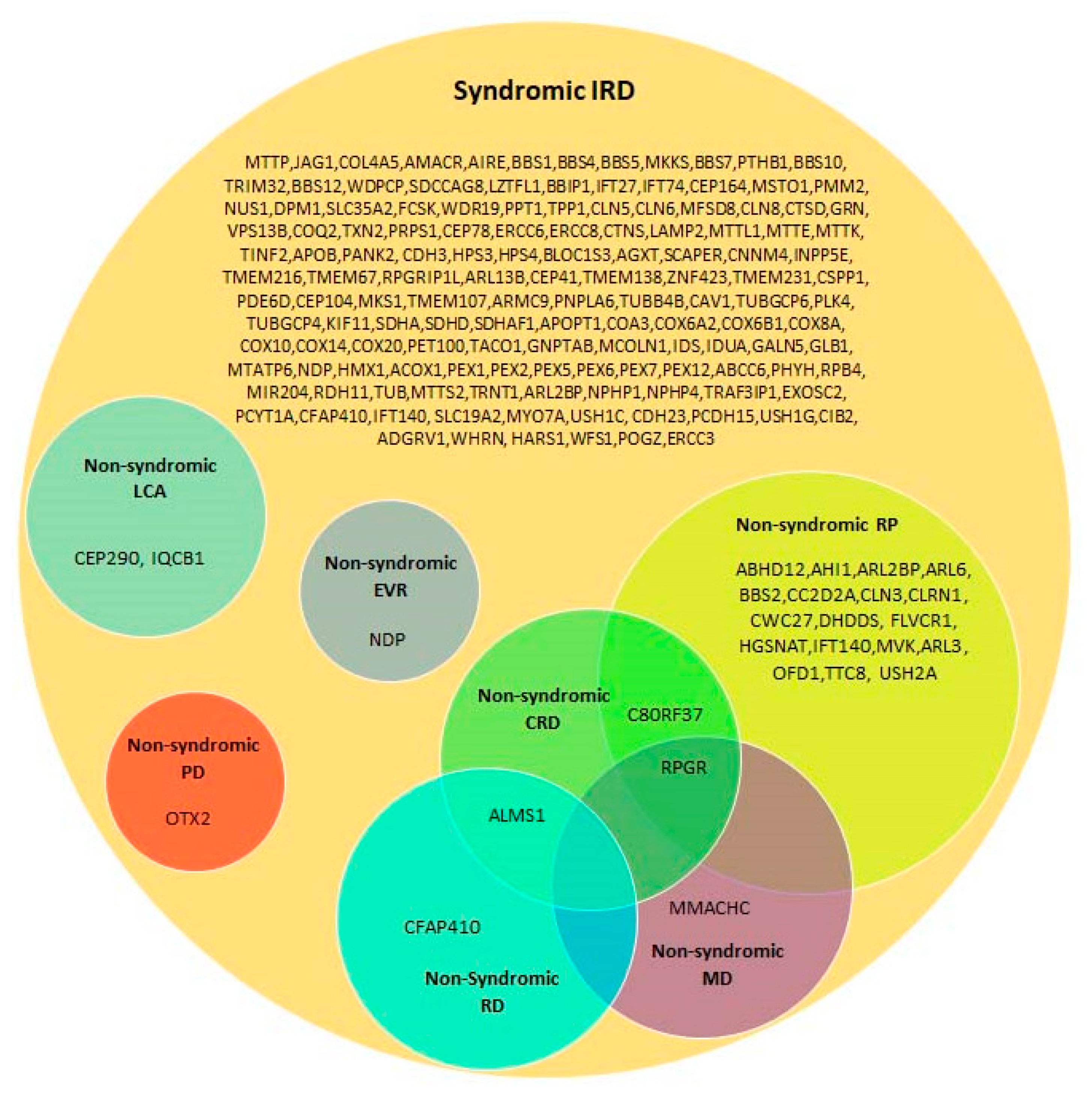
4.2. Syndromic Versus Non-Syndromic IRD Caused by the Same Genes
4.3. Co-Existence of Non-Syndromic IRD and Additional Non-Ocular Diseases
5. Diagnostic Challenges
6. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.; Phan, T.M.; Zekveld-Vroon, R.C.; Leroy, B.P.; van den Born, L.I.; Hoyng, C.B.; Klaver, C.C.; Roosing, S.; Pott, J.W.; van Schooneveld, M.J.; et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 2012, 119, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Sharma, T. Rod Monochromatism (Achromatopsia). Adv. Exp. Med. Biol. 2018, 1085, 119–123. [Google Scholar]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Shaheen, R.; Ewida, N.; Bubshait, D.K.; Alkuraya, H.; Almardawi, E.; Howaidi, A.; Sabr, Y.; Abdalla, E.M.; Alfaifi, A.Y.; et al. The morbid genome of ciliopathies: An update. Genet. Med. 2020, 22, 1051–1060. [Google Scholar] [CrossRef]
- Iwama, K.; Takaori, T.; Fukushima, A.; Tohyama, J.; Ishiyama, A.; Ohba, C.; Mitsuhashi, S.; Miyatake, S.; Takata, A.; Miyake, N.; et al. Novel recessive mutations in MSTO1 cause cerebellar atrophy with pigmentary retinopathy. J. Hum. Genet. 2018, 63, 263–270. [Google Scholar] [CrossRef]
- Ferreira, C.R.; van Karnebeek, C.D.M. Inborn errors of metabolism. Handb. Clin. Neurol. 2019, 162, 449–481. [Google Scholar]
- Freeze, H.H.; Schachter, H.; Kinoshita, T. Genetic Disorders of Glycosylation. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2017; Chapter 45. [Google Scholar]
- Nita, D.A.; Mole, S.E.; Minassian, B.A. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016, 18, 73–88. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford) 2011, 50 (Suppl. 5), v4–v12. [Google Scholar] [CrossRef]
- Imanaka, T. Biogenesis and Function of Peroxisomes in Human Disease with a Focus on the ABC Transporter. Biol. Pharm. Bull. 2019, 42, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, V.; Norris, D.P. Cilia and development. Curr. Opin. Genet. Dev. 2019, 56, 15–21. [Google Scholar] [CrossRef] [PubMed]
- May-Simera, H.; Nagel-Wolfrum, K.; Wolfrum, U. Cilia—The sensory antennae in the eye. Prog. Retin. Eye Res. 2017, 60, 144–180. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Bardet-Biedl Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 171–174. [Google Scholar] [PubMed]
- Valente, E.M.; Dallapiccola, B.; Bertini, E. Joubert syndrome and related disorders. Handb. Clin. Neurol. 2013, 113, 1879–1888. [Google Scholar] [PubMed]
- Geleoc, G.G.S.; El-Amraoui, A. Disease mechanisms and gene therapy for Usher syndrome. Hear. Res. 2020, 394, 107932. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Senior-Loken Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 175–178. [Google Scholar] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Alstrom Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 179–180. [Google Scholar] [PubMed]
- Petriman, N.A.; Lorentzen, E. Moving proteins along in the cilium. Elife 2020, 9, e55254. [Google Scholar] [CrossRef]
- Hsu, Y.; Garrison, J.E.; Kim, G.; Schmitz, A.R.; Searby, C.C.; Zhang, Q.; Datta, P.; Nishimura, D.Y.; Seo, S.; Sheffield, V.C. BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet. 2017, 13, e1007057. [Google Scholar] [CrossRef]
- Hsu, Y.; Garrison, J.E.; Seo, S.; Sheffield, V.C. The absence of BBSome function decreases synaptogenesis and causes ectopic synapse formation in the retina. Sci. Rep. 2020, 10, 8321. [Google Scholar] [CrossRef]
- Wang, S.F.; Kowal, T.J.; Ning, K.; Koo, E.B.; Wu, A.Y.; Mahajan, V.B.; Sun, Y. Review of Ocular Manifestations of Joubert Syndrome. Genes 2018, 9, 605. [Google Scholar] [CrossRef] [PubMed]
- El-Amraoui, A.; Petit, C. The retinal phenotype of Usher syndrome: Pathophysiological insights from animal models. Comptes Rendus Biol. 2014, 337, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Benson, M.D.; MacDonald, I.M.; Innes, A.M. A diagnostic approach to syndromic retinal dystrophies with intellectual disability. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 538–570. [Google Scholar] [CrossRef] [PubMed]
- Namburi, P.; Ratnapriya, R.; Khateb, S.; Lazar, C.H.; Kinarty, Y.; Obolensky, A.; Erdinest, I.; Marks-Ohana, D.; Pras, E.; Ben-Yosef, T.; et al. Bi-allelic Truncating Mutations in CEP78, Encoding Centrosomal Protein 78, Cause Cone-Rod Degeneration with Sensorineural Hearing Loss. Am. J. Hum. Genet. 2016, 99, 777–784. [Google Scholar] [CrossRef]
- Luscan, R.; Mechaussier, S.; Paul, A.; Tian, G.; Gerard, X.; Defoort-Dellhemmes, S.; Loundon, N.; Audo, I.; Bonnin, S.; LeGargasson, J.F.; et al. Mutations in TUBB4B Cause a Distinctive Sensorineural Disease. Am. J. Hum. Genet. 2017, 101, 1006–1012. [Google Scholar] [CrossRef]
- Sims, K.B. NDP-Related Retinopathies. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2014. [Google Scholar]
- Tsang, S.H.; Sharma, T. Inborn Errors of Metabolism: Refsum Disease. Adv. Exp. Med. Biol. 2018, 1085, 191–192. [Google Scholar]
- Raas-Rothschild, A.; Wanders, R.J.; Mooijer, P.A.; Gootjes, J.; Waterham, H.R.; Gutman, A.; Suzuki, Y.; Shimozawa, N.; Kondo, N.; Eshel, G.; et al. A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am. J. Hum. Genet. 2002, 70, 1062–1068. [Google Scholar] [CrossRef]
- Smith, C.E.; Poulter, J.A.; Levin, A.V.; Capasso, J.E.; Price, S.; Ben-Yosef, T.; Sharony, R.; Newman, W.G.; Shore, R.C.; Brookes, S.J.; et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 2016, 24, 1565–1571. [Google Scholar] [CrossRef]
- Le Quesne Stabej, P.; Saihan, Z.; Rangesh, N.; Steele-Stallard, H.B.; Ambrose, J.; Coffey, A.; Emmerson, J.; Haralambous, E.; Hughes, Y.; Steel, K.P.; et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 2012, 49, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2019, 41, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Heon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Pierrache, L.H.; Hartel, B.P.; van Wijk, E.; Meester-Smoor, M.A.; Cremers, F.P.; de Baere, E.; de Zaeytijd, J.; van Schooneveld, M.J.; Cremers, C.W.; Dagnelie, G.; et al. Visual Prognosis in USH2A-Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type IIa Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2016, 123, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Cabral, T.; Schuerch, K.; Duong, J.; Lee, W.; Boudreault, K.; Xu, Y.; Justus, S.; Sparrow, J.R.; Mahajan, V.B.; et al. Electroretinography Reveals Difference in Cone Function between Syndromic and Nonsyndromic USH2A Patients. Sci. Rep. 2017, 7, 11170. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Avila-Fernandez, A.; van Huet, R.A.; Corton, M.; Perez-Carro, R.; Martin-Garrido, E.; Lopez-Molina, M.I.; Blanco-Kelly, F.; Hoefsloot, L.H.; van Zelst-Stams, W.A.; et al. Exome sequencing extends the phenotypic spectrum for ABHD12 mutations: From syndromic to nonsyndromic retinal degeneration. Ophthalmology 2014, 121, 1620–1627. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Hull, S.; Roepman, R.; van den Born, L.I.; Oud, M.M.; de Vrieze, E.; Hetterschijt, L.; Letteboer, S.J.F.; van Beersum, S.E.C.; Blokland, E.A.; et al. Missense mutations in the WD40 domain of AHI1 cause non-syndromic retinitis pigmentosa. J. Med. Genet. 2017, 54, 624–632. [Google Scholar] [CrossRef]
- Aldrees, A.; Abdelkader, E.; Al-Habboubi, H.; Alrwebah, H.; Rahbeeni, Z.; Schatz, P. Non-syndromic retinal dystrophy associated with homozygous mutations in the ALMS1 gene. Ophthalmic Genet. 2019, 40, 77–79. [Google Scholar] [CrossRef]
- Audo, I.; El Shamieh, S.; Mejecase, C.; Michiels, C.; Demontant, V.; Antonio, A.; Condroyer, C.; Boyard, F.; Letexier, M.; Saraiva, J.P.; et al. ARL2BP mutations account for 0.1% of autosomal recessive rod-cone dystrophies with the report of a novel splice variant. Clin. Genet. 2017, 92, 109–111. [Google Scholar] [PubMed]
- Davidson, A.E.; Schwarz, N.; Zelinger, L.; Stern-Schneider, G.; Shoemark, A.; Spitzbarth, B.; Gross, M.; Laxer, U.; Sosna, J.; Sergouniotis, P.I.; et al. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2013, 93, 321–329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holtan, J.P.; Teigen, K.; Aukrust, I.; Bragadottir, R.; Houge, G. Dominant ARL3-related retinitis pigmentosa. Ophthalmic Genet. 2019, 40, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Safieh, L.A.; Alkuraya, H.; Al-Rajhi, A.; Shamseldin, H.; Hashem, M.; Alzahrani, F.; Khan, A.O.; Alqahtani, F.; Rahbeeni, Z.; et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol. Vis. 2009, 15, 2464–2469. [Google Scholar]
- Shevach, E.; Ali, M.; Mizrahi-Meissonnier, L.; McKibbin, M.; El-Asrag, M.; Watson, C.M.; Inglehearn, C.F.; Ben-Yosef, T.; Blumenfeld, A.; Jalas, C.; et al. Association Between Missense Mutations in the BBS2 Gene and Nonsyndromic Retinitis Pigmentosa. JAMA Ophthalmol. 2015, 133, 312–318. [Google Scholar] [CrossRef]
- Khan, A.O.; Decker, E.; Bachmann, N.; Bolz, H.J.; Bergmann, C. C8orf37 is mutated in Bardet-Biedl syndrome and constitutes a locus allelic to non-syndromic retinal dystrophies. Ophthalmic Genet. 2016, 37, 290–293. [Google Scholar] [CrossRef]
- Mejecase, C.; Hummel, A.; Mohand-Said, S.; Andrieu, C.; El Shamieh, S.; Antonio, A.; Condroyer, C.; Boyard, F.; Foussard, M.; Blanchard, S.; et al. Whole exome sequencing resolves complex phenotype and identifies CC2D2A mutations underlying non-syndromic rod-cone dystrophy. Clin. Genet. 2019, 95, 329–333. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef]
- Khan, A.O.; Eisenberger, T.; Nagel-Wolfrum, K.; Wolfrum, U.; Bolz, H.J. C21orf2 is mutated in recessive early-onset retinal dystrophy with macular staphyloma and encodes a protein that localises to the photoreceptor primary cilium. Br. J. Ophthalmol. 2015, 99, 1725–1731. [Google Scholar] [CrossRef]
- Suga, A.; Mizota, A.; Kato, M.; Kuniyoshi, K.; Yoshitake, K.; Sultan, W.; Yamazaki, M.; Shimomura, Y.; Ikeo, K.; Tsunoda, K.; et al. Identification of Novel Mutations in the LRR-Cap Domain of C21orf2 in Japanese Patients With Retinitis Pigmentosa and Cone-Rod Dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4255–4263. [Google Scholar] [CrossRef]
- Ku, C.A.; Hull, S.; Arno, G.; Vincent, A.; Carss, K.; Kayton, R.; Weeks, D.; Anderson, G.W.; Geraets, R.; Parker, C.; et al. Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol. 2017, 135, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Kersten, F.F.; Azam, M.; Collin, R.W.; Hussain, A.; Shah, S.T.; Keunen, J.E.; Kremer, H.; Cremers, F.P.; Qamar, R.; et al. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology 2011, 118, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xie, Y.A.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Zuchner, S.L.; Dallman, J.; Wen, R.; Alfonso, E.C.; Vance, J.M.; Pericak-Vance, M.A. Mutation K42E in dehydrodolichol diphosphate synthase (DHDDS) causes recessive retinitis pigmentosa. Adv. Exp. Med. Biol. 2014, 801, 165–170. [Google Scholar]
- Zelinger, L.; Banin, E.; Obolensky, A.; Mizrahi-Meissonnier, L.; Beryozkin, A.; Bandah-Rozenfeld, D.; Frenkel, S.; Ben-Yosef, T.; Merin, S.; Schwartz, S.B.; et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 2011, 88, 207–215. [Google Scholar] [CrossRef]
- Kuehlewein, L.; Schols, L.; Llavona, P.; Grimm, A.; Biskup, S.; Zrenner, E.; Kohl, S. Phenotypic spectrum of autosomal recessive retinitis pigmentosa without posterior column ataxia caused by mutations in the FLVCR1 gene. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 629–638. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; Newman, H.; Leibu, R.; Bax, N.M.; Baris, H.N.; Rizel, L.; Banin, E.; Massarweh, A.; Roosing, S.; Lefeber, D.J.; et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. Genet. 2015, 24, 3742–3751. [Google Scholar] [CrossRef]
- Xu, M.; Yang, L.; Wang, F.; Li, H.; Wang, X.; Wang, W.; Ge, Z.; Wang, K.; Zhao, L.; Li, H.; et al. Mutations in human IFT140 cause non-syndromic retinal degeneration. Hum. Genet. 2015, 134, 1069–1078. [Google Scholar] [CrossRef]
- Stone, E.M.; Cideciyan, A.V.; Aleman, T.S.; Scheetz, T.E.; Sumaroka, A.; Ehlinger, M.A.; Schwartz, S.B.; Fishman, G.A.; Traboulsi, E.I.; Lam, B.L.; et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch. Ophthalmol. 2011, 129, 81–87. [Google Scholar] [CrossRef]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef]
- Collison, F.T.; Xie, Y.A.; Gambin, T.; Jhangiani, S.; Muzny, D.; Gibbs, R.; Lupski, J.R.; Fishman, G.A.; Allikmets, R. Whole Exome Sequencing Identifies an Adult-Onset Case of Methylmalonic Aciduria and Homocystinuria Type C (cblC) with Non-Syndromic Bull’s Eye Maculopathy. Ophthalmic Genet. 2015, 36, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Siemiatkowska, A.M.; van den Born, L.I.; van Hagen, P.M.; Stoffels, M.; Neveling, K.; Henkes, A.; Kipping-Geertsema, M.; Hoefsloot, L.H.; Hoyng, C.B.; Simon, A.; et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology 2013, 120, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Battinelli, E.M.; Fielder, A.; Bundey, S.; Sims, K.; Breakefield, X.O.; Craig, I.W. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 1993, 5, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.R.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E.; Zito, I.; Thiselton, D.L.; Ressa, J.H.; Apergi, M.; et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef]
- Vincent, A.; Forster, N.; Maynes, J.T.; Paton, T.A.; Billingsley, G.; Roslin, N.M.; Ali, A.; Sutherland, J.; Wright, T.; Westall, C.A.; et al. OTX2 mutations cause autosomal dominant pattern dystrophy of the retinal pigment epithelium. J. Med. Genet. 2014, 51, 797–805. [Google Scholar] [CrossRef]
- Tee, J.J.; Smith, A.J.; Hardcastle, A.J.; Michaelides, M. RPGR-associated retinopathy: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2016, 100, 1022–1027. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Iqbal, M.; Wang, Y.; Masuda, T.; Chen, Y.; Bowne, S.; Sullivan, L.S.; Waseem, N.H.; Bhattacharya, S.; Daiger, S.P.; et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet. 2010, 86, 805–812. [Google Scholar] [CrossRef]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef]
- Ehrenberg, M.; Weiss, S.; Orenstein, N.; Goldenberg-Cohen, N.; Ben-Yosef, T. The co-occurrence of rare non-ocular phenotypes in patients with inherited retinal degenerations. Mol. Vis. 2019, 25, 691–702. [Google Scholar]
- Ku, C.A.; Pennesi, M.E. The new landscape of retinal gene therapy. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 846–859. [Google Scholar] [CrossRef]
- Bach, G.; Webb, M.B.; Bargal, R.; Zeigler, M.; Ekstein, J. The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of 3 novel MCOLN1 mutations. Hum. Mutat. 2005, 26, 591. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yosef, T.; Ness, S.L.; Madeo, A.C.; Bar-Lev, A.; Wolfman, J.H.; Ahmed, Z.M.; Desnick, R.J.; Willner, J.P.; Avraham, K.B.; Ostrer, H.; et al. A mutation of PCDH15 among Ashkenazi Jews with the type 1 Usher syndrome. N. Engl. J. Med. 2003, 348, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Fedick, A.; Jalas, C.; Abeliovich, D.; Krakinovsky, Y.; Ekstein, J.; Ekstein, A.; Treff, N.R. Carrier frequency of two BBS2 mutations in the Ashkenazi population. Clin. Genet. 2014, 85, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Ness, S.L.; Ben-Yosef, T.; Bar-Lev, A.; Madeo, A.C.; Brewer, C.C.; Avraham, K.B.; Kornreich, R.; Desnick, R.J.; Willner, J.P.; Friedman, T.B.; et al. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J. Med. Genet. 2003, 40, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, T.; Hamalainen, R.; Yuan, B.; Johnson, C.; Tegelberg, S.; Gasparini, P.; Zelante, L.; Pirvola, U.; Pakarinen, L.; Lehesjoki, A.E.; et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 2001, 69, 673–684. [Google Scholar] [CrossRef]
- Kyttala, M.; Tallila, J.; Salonen, R.; Kopra, O.; Kohlschmidt, N.; Paavola-Sakki, P.; Peltonen, L.; Kestila, M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006, 38, 155–157. [Google Scholar] [CrossRef]
- Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, K.T. Genetic testing for inherited retinal degenerations: Triumphs and tribulations. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 571–577. [Google Scholar] [CrossRef]
- Mansfield, B.C.; Yerxa, B.R.; Branham, K.H. Implementation of a registry and open access genetic testing program for inherited retinal diseases within a non-profit foundation. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 838–845. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Syndrome (MIM/Reference) | Gene | Inheritance * | Main Ocular Phenotypes # | Main Extra-Ocular Phenotypes ¶ |
|---|---|---|---|---|
| Abetalipoproteinemia; ABL (#200100) | MTTP | AR | RP | Fat malabsorption, neurodegeneration, acanthocytosis |
| Aicardi Syndrome; AIC (#304050) | Xp22 abnormalities | XLD | Chorioretinopathy, OA, microphthalmia, optic nerve coloboma, cataract | Callosal agenesis, PGR, microcephaly, ID, skeletal anomalies, neoplasia |
| Alagille Syndrome 1; ALGS1 (#118450) | JAG1 | AD | Iris stromal hypoplasia, posterior embryotoxon, microcornea, anomalous optic disc, peripapillary retinal depigmentation, chorioretinopathy | Liver disease, skeletal and renal involvement, characteristic facial features, ID, FTT |
| Alport Syndrome 1; ATS1 (#3010150) | COL4A5 | XLD | Fleck retinopathy, cataract, myopia, corneal abnormalities | HL, renal disease |
| Alstrom Syndrome; ALMS (#203800) | ALMS1 | AR | CRD, MD, cataract | DD, SS, obesity, HL, cardiac, skeletal, hepatic, renal and endocrine involvement |
| Alpha-Methylacyl-CoA Racemase Deficiency; AMACRD (#614307) | AMACR | AR | RP | Neurodegeneration |
| Autoimmune Polyendocrine Syndrome, Type I, with or without Reversible Metaphyseal Dysplasia; APS1 (#240300) | AIRE | AD, AR | RP, keratopathy, keratoconjunctivitis | Multiple autoantibodies, anemia, hepatic, gastrointestinal, dental, skin, hair and endocrine involvement, hypogonadism |
| Bardet–Biedl Syndrome; BBS (#209900, #615981, #600151, #615982, #615983, #605231, #615984, #615985, #615986, #615987, #615988, #615989, #615990, #615991, 615992, #615993, #615994, #615995, #615996, #617119, #617406) [7] | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, PTHB1, BBS10, TRIM32, BBS12, MKS1, CEP290, WDPCP, SDCCAG8, LZTFL1, BBIP1, IFT27, IFT74, C8ORF37, CEP164 | AR | RP, strabismus, cataract | ID, SS, obesity, hypogonadism, renal disease, polydactyly |
| Cerebellar Atrophy with Pigmentary Retinopathy [8] | MSTO1 | AR | RD | Cerebellar atrophy, ID, PGR |
| Congenital Disorder of Glycosylation; CDG (#212065, #617082, #613861, #608799, #300896) | PMM2, NUS1, DHDDS, DPM1, SLC35A2 | AR | RP | FTT, microcephaly, ID, neurodegeneration, cardiac, hepatic, gastrointestinal, renal and hematological involvement |
| Congenital Disorder of Glycosylation with Defective Fucosylation 2; CDGF2 (#618324) | FCSK | AR | MD, OA, strabismus | FTT, ID, hypotonia, neurodegeneration, gastrointestinal anomalies |
| Cranioectodermal Dysplasia 4; CED4 (#614378) | WDR19 | AR | RP | Skeletal anomalies, SS, respiratory, hepatic and renal involvement |
| Ceroid Lipofuscinosis, Neuronal; CLN (#256730, #204500, #204200, #256731, #601780, #610951, #600143, #610127, #614706) | PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, GRN | AR | RP, CRD, OA | Microcephaly, ID, neurodegeneration |
| Cohen Syndrome; COH1 (#216550) | VPS13B | AR | RD, OA, strabismus, high myopia | ID, DD, microcephaly, SS, obesity, skeletal, cardiac, hematological and endocrine involvement |
| Coenzyme Q10 Deficiency, Primary, 1; COQ10D1 (#607426) | COQ2 | AR | RP | ID, cerebellar atrophy, HL, cardiac, hepatic, renal and muscular involvement |
| Combined Oxidative Phosphorylation Deficiency 29; COXPD29 (#616811) | TXN2 | AR | RD, OA | Microcephaly, hypotonia, DD, ID, neurodegeneration |
| Charcot–Marie–Tooth Disease, X-linked recessive, 5; CMTX5 (#311070) | PRPS1 | XLR | RP, OA | Peripheral neuropathy, HL |
| Cone–Rod Dystrophy and Hearing Loss 1; CRDHL1 (#617236) | CEP78 | AR | CRD | HL |
| Cockayne Syndrome; CS (#216400, #133540) | ERCC8, ERCC6 | AR | RD, OA, cataract, strabismus | IUGR, PGR, microcephaly, ID, neurodegeneration, HL, renal, skeletal and skin involvement |
| Cystinosis, Nephropathic; CTNS (#219800, #219900) | CTNS | AR | RD, corneal crystals | Renal disease, neurodegeneration, skeletal and endocrine anomalies |
| Danon Disease (#300257) | LAMP2 | XLD | RD | Cardiac disease, myopathy, ID |
| Diabetes and Deafness, Maternally Inherited; MIDD (#520000) | MTTL1, MTTE, MTTK, mitochondrial DNA rearrangements | Mi | RD, MD, ophthalmoplegia | HL, cardiac and neurological anomalies, diabetes mellitus |
| Dyskeratosis Congenita, Autosomal Dominant 3; DKCA3 (#613990) | TINF2 | AD | RD, blockage of lacrimal ducts | IUGR, SS, microcephaly, ID, HL, respiratory, skin, skeletal and hematological involvement, neoplasia |
| Hypobetalipoproteinemia, Familial, 1; FHBL1 (#615558) | APOB | AR | RP | Fat malabsorption, neurodegeneration, acanthocytosis |
| Hypobetalipoproteinemia, Acanthocytosis, Retinitis Pigmentosa and Pallidal Degeneration; HARP (#607236) | PANK2 | AR | RP | Fat malabsorption, neurodegeneration, acanthocytosis |
| Hypotrichosis, Congenital, with Juvenile Macular Dystrophy; HJMD (#601553) | CDH3 | AR | MD | Hypotrichosis |
| Hermansky–Pudlak Syndrome; HPS (#614072, #614073, #614077) | HPS3, HPS4, BLOC1S3 | AR | Hypopigmentation of retina and choroid, foveal hypoplasia, nystagmus, iris transillumination | Skin and hair hypopigmentation, bleeding diathesis |
| Hyper-IgD Syndrome; HIDS (#260920) | MVK | AR | RP | Hematological anomalies, gastrointestinal and skeletal involvement, periodic fever |
| Hyperoxaluria, Primary, Type I; HP1 (#259900) | AGXT | AR | RD, OA | Renal disease, dental, cardiovascular and skin involvement, peripheral neuropathy |
| Intellectual Developmental Disorder and Retinitis Pigmentosa; IDDRP (#618195) | SCAPER | AR | RP, MD, cataract | ID, skeletal abnormalities, male sterility |
| Jalili Syndrome (#217080) | CNNM4 | AR | CRD | Amelogenesis imperfecta |
| Joubert Syndrome; JBTS (#213300, #608091, #608629, #610188, #610688, #611560, #612291, #612285, #614464, #614465, #614844, #614970, #615636, #615665, #616781, #617121, #617562, #617622, #618161, #300804) | INPP5E, TMEM216, AHI1, CEP290, TMEM67, RPGRIP1L, ARL13B, CC2D2A, CEP41, TMEM138, ZNF423, TMEM231, CSPP1, PDE6D, CEP104, MKS1, TMEM107, ARMC9, ARL3 | AR | RD, chorioretinal coloboma, optic nerve coloboma, microphthalmia, oculomotor apraxia, esotropia, ptosis | Brain structural anomalies, FTT, macrocephaly, ID, neurodegeneration, genitourinary, hepatic, respiratory and skeletal involvement |
| OFD1 | XLR | |||
| Kearns–Sayre Syndrome; KSS (#530000) | Mitochondrial DNA deletions | Mi | RD, ophthalmoplegia | SS, microcephaly, neurodegeneration, cardiac, renal and endocrine involvement |
| Laurence–Moon Syndrome; LNMS (#245800) | PNPLA6 | AR | Chorioretinal degeneration | ID, neurodegeneration, genitourinary abnormalities |
| Leber Congenital Amaurosis with Early-Onset Deafness; LCAEOD (#617879) | TUBB4B | AD | LCA | HL |
| Lipodystrophy, familial partial, type7; FPLD7 (#606721) | CAV1 | AD | RD, cataract | Lack of facial fat, orthostatic hypotension, neurological and skin involvement |
| Methylmalonic Aciduria and Homocystinuria, cblC type; MAHCC (#277400) | MMACHC | AR | RP, CRD | FTT, microcephaly, ID, neurodegeneration, renal and hematological involvement |
| Mevalonic Aciduria; MEVA (#610377) | MVK | AR | RP, OA, cataract | FTT, DD, neurodegeneration, spleen, hepatic, skeletal, skin and hematological involvement |
| Microcephaly and Chorioretinopathy, autosomal recessive; MCCRP (#251270, #616171, #616335) | TUBGCP6, PLK4, TUBGCP4 | AR | Chorioretinopathy, OA, microphthalmia, microcornea, cataract | IUGR, microcephaly, brain structural anomalies, DD, ID, neurodegeneration, SS |
| Microcephaly with or without Chorioretinopathy, Lymphedema or Mental Retardation; MCLMR (#152950) | KIF11 | AD | Chorioretinopathy, myopia, hypermetropia, corneal opacity, microcornea, microphthalmia, cataract | Microcephaly, ID, neurodegeneration, lymphedema |
| Microphthalmia, Syndromic 5; MCOPS5 (#610125) | OTX2 | AD | RD, microphthalmia, anophthalmia, optic nerve hypoplasia or agenesis, microcornea, cataract | Brain structural anomalies, hypotonia, pituitary dysfunction, DD, SS, cleft palate, abnormal genitalia, joint laxity |
| Mitochondrial Complex II Deficiency (#252011) | SDHA, SDHD, SDHAF1 | AR | RD, OA, ptosis, ophthalmoplegia | SS, cardiac, skeletal, muscular and neurological involvement |
| Mitochondrial Complex IV Deficiency (#220110) | APOPT1, COA3, COX6A2, COX6B1, COX8A, COX10, COX14, COX20, PET100, TACO1 | AR | RD, OA, ptosis | FTT, brain structural anomalies, ID, HL, cardiac, respiratory, hepatic, renal and muscular involvement |
| Mucolipidosis III alpha/beta; MLIII A/B (#252600) | GNPTAB | AR | RD, corneal clouding | Neurodegeneration, ID, SS, coarse facies, skeletal, cardiac and skin involvement |
| Mucolipidosis IV; ML4 (#252650) | MCOLN1 | AR | RD, OA, corneal disease, strabismus | Microcephaly, ID, neurodegeneration |
| Mucopolysaccharidosis; MPS (#309900, #252930, #607014, #253000, #253010) | IDS | XLR | RP, ptosis, corneal clouding | Neurodegeneration, ID, SS, coarse facies, HL, skeletal, cardiac, respiratory, hepatic, gastrointestinal and skin involvement |
| HGSNAT, IDUA, GALN5, GLB1 | AR | |||
| Nephronophthisis 15; NPHP15 (#614845) | CEP164 | AR | LCA | Renal disease |
| Neurodegeneration with Brain Iron Accumulation 1; NBIA1 (#234200) | PANK2 | AR | RD, OA, eyelid apraxia | Neurodegeneration, gastrointestinal, skeletal, skin and muscular involvement |
| Neuropathy, Ataxia and Retinitis Pigmentosa; NARP (#551500) | MTATP6 | Mi | RP | Neurodegeneration, ataxia |
| Norrie Disease; ND (#310600) | NDP | XLR | Retinal dysgenesis, retinal dysplasia, OA, microphthalmia, vitreous atrophy, corneal opacities, iris atrophy, cataract | HL, ID, neurodegeneration |
| Oculoauricular Syndrome; OCACS (#612109) | HMX1 | AR | RP, microphthalmia, microcornea, cataract, microphakia, sclerocornea, increased intraocular pressure | External ear abnormalities |
| Orofaciodigital Syndrome XVI; OFD16 (#617563) | TMEM107 | AR | RD, oculomotor apraxia, ptosis | Facial anomalies, breathing abnormalities, polydactyly, hypotonia, ID, neurological anomalies |
| Oliver–McFarlane Syndrome; OMCS (#275400) | PNPLA6 | AR | Chorioretinopathy, OA | SS, ID, neurodegeneration, obesity, male external genitalia abnormalities, endocrine anomalies |
| Peroxisomal Acyl-CoA Oxidase Deficiency (#264470) | ACOX1 | AR | RD, OA, strabismus | Neurodegeneration, ID, HL, liver disease |
| Peroxisome Biogenesis Disorder; PBD (#214100, #614866, #601539, #234580, #614879, #266510) | PEX1, PEX2, PEX5, PEX6, PEX7, PEX12 | AR | RD, OA, corneal clouding, cataract | FTT, neurodegeneration, ID, HL, dental, cardiac, hepatic, genitourinary and skeletal involvement |
| Posterior Column Ataxia with Retinitis Pigmentosa; AXPC1 (#609033) | FLVCR1 | AR | RP, OA | Posterior column ataxia, neurodegeneration, gastrointestinal and skeletal involvement |
| Polyneuropathy, Hearing Loss, Ataxia, Retinitis Pigmentosa and Cataract; PHARC (#612674) | ABHD12 | AR | RP, OA, cataract | Ataxia, neurodegeneration, HL |
| Pseudoxanthoma Elasticum; PXE (#264800) | ABCC6 | AR | RD, MD, choroidal neovascularization | Skin lesions, cardiovascular disease, gastrointestinal and genitourinary involvement |
| Refsum Disease, classic (#266500) | PHYH | AR | RP | Neurodegeneration, ataxia, HL, anosmia, cardiac, skeletal and skin involvement |
| Retinal Dystrophy, Iris Coloboma and Comedogenic Acne Syndrome; RDCCAS (#615147) | RPB4 | AR | RD, coloboma of the iris, displacement of the pupil, microcornea, cataract | Comedogenic acne |
| Retinal Dystrophy and Iris Coloboma with or without Cataract; RDICC (#616722) | MIR204 | AD | RD, coloboma of the iris, congenital cataract | |
| Retinal Dystrophy, Juvenile Cataracts and Short Stature Syndrome; RDJCSS (#616108) | RDH11 | AR | RD, juvenile cataracts | SS, DD, ID, dental anomalies |
| Retinal Dystrophy and Obesity; RDOB (#616188) | TUB | AR | RD | Obesity |
| Revesz Syndrome (#268130) | TINF2 | AD | RD | IUGR, brain structural anomalies, neurodegeneration, ID, aplastic anemia, skin, hair and nail abnormalities |
| Retinitis Pigmentosa–Deafness Syndrome (#500004) | MTTS2 | Mi | RP | HL |
| Retinitis Pigmentosa and Erythrocytic Microcytosis; RPEM (#616959) | TRNT1 | AR | RP | Erythrocytic microcytosis and additional hematologic abnormalities |
| Retinitis Pigmentosa, Hypopituitarism, Nephronophtisis and mild Skeletal Dysplasia; RHYNS (#602152) | TMEM67 | AR | RP | Hypopituitarism, renal disease, skeletal anomalies, HL |
| Retinitis Pigmentosa 82 with or without Situs Inversus; RP82 (#615434) | ARL2BP | AR | RP | Situs inversus, male infertility |
| Retinitis Pigmentosa with or without Skeletal Anomalies; RPSKA (#250410) | CWC27 | AR | RP | SS, skeletal anomalies, ID |
| Retinitis Pigmentosa, X-linked and Sinorespiratory Infections, with or without Deafness (#300455) | RPGR | XL | RP | Recurrent respiratory infections, HL |
| Senior–Løken Syndrome; SLSN (#266900, #606996, #609254, #610189, #613615, #616307, #616629) | NPHP1, NPHP4, IQCB1, CEP290, SDCCAG8, WDR19, TRAF3IP1 | AR | RP, LCA | Renal disease |
| Short Stature, Hearing Loss, Retinitis Pigmentosa and Distinctive Facies; SHRF (#617763) | EXOSC2 | AR | RP, corneal dystrophy, glaucoma, strabismus | SS, facial anomalies, HL, neurodegeneration, DD, ID |
| Sideroblastic Anemia with B-cell Immunodeficiency, Periodic Fevers and Developmental Delay; SIFD (#616084) | TRNT1 | AR | RP | Sideroblastic anemia, immunodeficiency, growth retardation, DD, periodic fever, HL, neurological, cardiac and renal involvement |
| Spondylometaphyseal Dysplasia with Cone–Rod Dystrophy; SMDCRD (#608940) | PCYT1A | AR | CRD | Skeletal anomalies, PGR |
| Spondylometaphyseal Dysplasia, Axial; SMDAX (#602271) | CFAP410 | AR | RP, CRD, OA | Skeletal anomalies, respiratory disease, reduced sperm motility |
| Short-Rib Thoracic Dysplasia 9 with or without Polydactyly; SRTD9 (#266920) | IFT140 | AR | RP | Skeletal anomalies, renal disease, ID |
| Thiamine-Responsive Megaloblastic Anemia Syndrome; TRMA (#249270) | SLC19A2 | AR | OA, RD | Megaloblastic anemia, diabetes mellitus, HL |
| Usher Syndrome; USH (#276900, #276904, #601067, #602083, #606943, #614869, #276901, #605472, #611383, #276902, #614504) | MYO7A, USH1C, CDH23, PCDH15, USH1G, CIB2, USH2A, ADGRV1, WHRN, CLRN1, HARS1 | AR | RP | HL, vestibular dysfunction |
| Wolfram Syndrome 1, WFS1 (#222300) | WFS1 | AR | OA, RD | Diabetes mellitus, diabetes insipidus, HL, neurodegeneration, genitourinary and neurologic involvement |
| White–Sutton Syndrome, WHSUS (#616364) | POGZ | AD | RP, OA, cortical blindness | DD, characteristic facial features, hypotonia, HL, joint laxity, gastrointestinal anomalies |
| Xeroderma Pigmentosum, group B; XPB (#610651) | ERCC3 | AR | RD, OA, micropathalmia | Neoplasia, skin anomalies, SS, microcephaly, HL, ID, brain structural anomalies, neurodegeneration |
| Gene | Syndromic IRD | Non-Syndromic IRD (MIM) | Reference |
|---|---|---|---|
| ABHD12 | PHARC | arRP | [40] |
| AHI1 | JBTS3 | arRP | [41] |
| ALMS1 | ALMS | arCRD, arEORD | [42] |
| ARL2BP | RP with situs inversus | arRP (#615434) | [43,44] |
| ARL3 | JBTS35 | adRP (#618173) | [45] |
| ARL6 | BBS3 | arRP (#613575) | [46] |
| BBS2 | BBS2 | arRP (#616562) | [47] |
| C8ORF37 | BBS21 | arCRD, arRP (#614500) | [48] |
| CC2D2A | JBTS9, MKS6 | arRP | [49] |
| CEP290 | BBS14, JBTS5, MKS4, SLSN6 | arLCA (#611755) | [50] |
| CFAP410 | SMDAX | arRD with or without macular staphyloma (#617547) | [51,52] |
| CLN3 | CLN3 | arRP | [53] |
| CLRN1 | USH3A | arRP (#614180) | [54] |
| CWC27 | RPSKA | arRP (#250410) | [55] |
| DHDDS | CDG1BB | arRP (#613861) | [56,57] |
| FLVCR1 | PCARP | arRP | [58] |
| HGSNAT | MPS3C | arRP (#616544) | [59] |
| IFT140 | SRTD9 with/without polydactyly | arRP (#617781) | [60] |
| IQCB1 | SLSN5 | arLCA | [61] |
| MFSD8 | CLN7 | arMD (#616170), arRD | [62] |
| MMACHC | MAHCC | arMD | [63] |
| MVK | HIDS, MEVA | arRP | [64] |
| NDP | ND | XL EVR (#305390) | [65] |
| OFD1 | JBTS10 | XL RP (#300424) | [66] |
| OTX2 | RD with pituitary dysfunction | adPD (#610125) | [67] |
| RPGR | RP, sinorespiratory infections and deafness | XL CRD (#304020), XL MD (#300834), XL RP (#300029) | [68] |
| TTC8 | BBS8 | arRP (#613464) | [69] |
| USH2A | USH2A | arRP (#613809) | [70] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. https://doi.org/10.3390/diagnostics10100779
Tatour Y, Ben-Yosef T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics. 2020; 10(10):779. https://doi.org/10.3390/diagnostics10100779
Chicago/Turabian StyleTatour, Yasmin, and Tamar Ben-Yosef. 2020. "Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects" Diagnostics 10, no. 10: 779. https://doi.org/10.3390/diagnostics10100779
APA StyleTatour, Y., & Ben-Yosef, T. (2020). Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics, 10(10), 779. https://doi.org/10.3390/diagnostics10100779