Whole Exome Sequencing as a Diagnostic Tool for Unidentified Muscular Dystrophy in a Vietnamese Family

 ,
,
Abstract
:1. Introduction
2. Case Presentation
2.1. Patients
2.2. Whole Exome Sequencing, Variant Calling and Annotation
2.3. Filtration of Variants
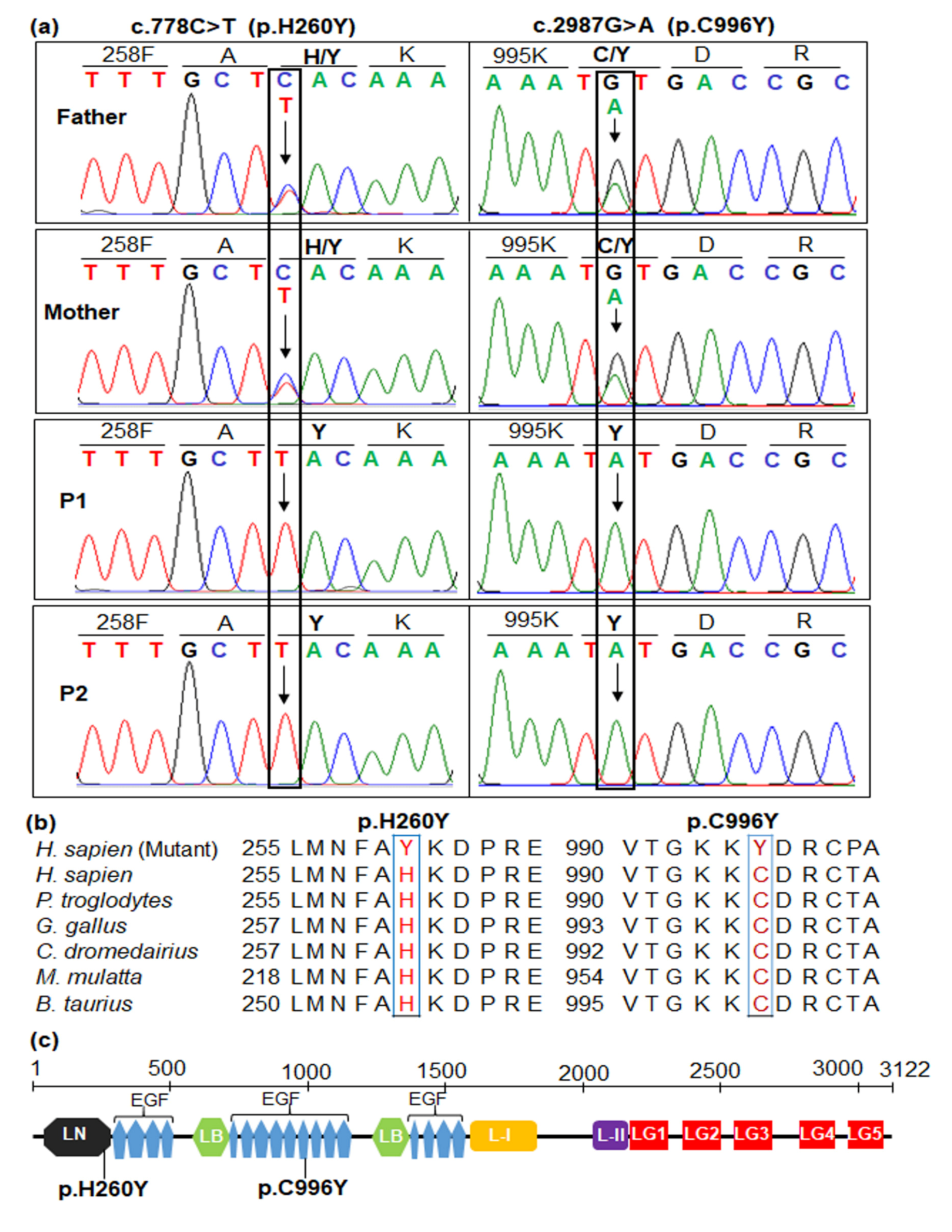
2.4. Sanger Sequencing for Variant Confirmation and Familial Segregation Analysis
2.5. In Silico Analyses and Pathogenic Interpretation
2.6. Amino Acid Sequence Alignment and Protein Analyses
3. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kanagawa, M.; Toda, T. The genetic and molecular basis of muscular dystrophy: Roles of cell–matrix linkage in the pathogenesis. J. Hum. Genet. 2006, 51, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef]
- El-Tallawy, H.N.; Khedr, E.M.; Qayed, M.H.; Helliwell, T.R.; Kamel, N.F. Epidemiological study of muscular disorders in Assiut, Egypt. Neuroepidemiology 2005, 25, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Falsaperla, R.; Praticò, A.D.; Ruggieri, M.; Parano, E.; Rizzo, R.; Corsello, G.; Vitaliti, G.; Pavone, P. Congenital muscular dystrophy: From muscle to brain. Ital. J. Paediatr. 2016, 42, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, E.; D’Amico, A.; Gualandi, F.; Petrini, S. Congenital muscular dystrophies: A brief review. Semin. Pediatr. Neurol. 2011, 18, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Grady, G.L.; Lek, M.; Lamande, S.R.; Waddell, L.; Oates, E.C.; Punetha, J.; Ghaoui, R.; Sandaradura, S.A.; Best, H.; Kaur, S.; et al. Diagnosis and etiology of congenital muscular dystrophy: We are halfway there. Ann. Neurol. 2016, 80, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2019, 29, 980–1018. [Google Scholar] [CrossRef] [PubMed]
- Graziano, A.; Bianco, F.; D’Amico, A.; Moroni, I.; Messina, S.; Bruno, C.; Pegoraro, E.; Mora, M.; Astrea, G.; Magri, F.; et al. Prevalence of congenital muscular dystrophy in Italy. Neurology 2015, 84, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Zhang, C.; Wang, Z.; Chan, S.H.S.; Zhu, W.; Han, C.; Zhang, X.; Zheng, H.; Wu, L.; Jin, B.; et al. Congenital muscular dystrophies in China. Clin. Genet. 2019, 96, 207–215. [Google Scholar] [CrossRef]
- Geranmayeh, F.; Clement, E.; Feng, L.H.; Sewry, C.; Pagan, J.; Mein, R.; Abbs, S.; Brueton, L.; Childs, A.-M.; Jungbluth, H.; et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 2010, 20, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Allamand, V.; Guicheney, P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for α2 chain of laminin). Eur. J. Hum. Genet. 2002, 10, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sframeli, M.; Sarkozy, A.; Bertoli, M.; Astrea, G.; Hudson, J.; Scoto, M.; Mein, R.; Yau, M.; Phadke, R.; Feng, L.; et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul. Disord. 2017, 27, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Løkken, N.; Born, A.P.; Duno, M.; Vissing, J. LAMA2-related myopathy: Frequency among congenital and limb-girdle muscular dystrophies. Muscle Nerve 2015, 52, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Abdel Aleem, A.; Elsaid, M.F.; Chalhoub, N.; Chakroun, A.; Mohamed, K.A.S.; AlShami, R.; Kuzu, O.; Mohamed, R.B.; Ibrahim, K.; AlMudheki, N.; et al. Clinical and genomic characteristics of LAMA2 related congenital muscular dystrophy in a patients’ cohort from Qatar. A population specific founder variant. Neuromuscul. Disord. 2020, 30, 457–471. [Google Scholar] [CrossRef]
- Meinen, S.; Barzaghi, P.; Lin, S.; Lochmüller, H.; Ruegg, M.A. Linker molecules between laminins and dystroglycan ameliorate laminin-α2–deficient muscular dystrophy at all disease stages. J. Cell. Biol. 2007, 176, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Gawlik, K.I.; Durbeej, M. Skeletal muscle laminin and MDC1A: Pathogenesis and treatment strategies. Skelet. Muscle 2011, 1, 9. [Google Scholar] [CrossRef]
- Quijano-Roy, S.; Sparks, S.E.; Rutkowski, A. LAMA2-related muscular dystrophy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2012. [Google Scholar]
- He, Z.; Luo, X.; Liang, L.; Li, P.; Li, D.; Zhe, M. Merosin-deficient congenital muscular dystrophy type 1A: A case report. Exp. Ther. Med. 2013, 6, 1233–1236. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tan, J.; Ma, D.; Zhang, J.; Cheng, J.; Luo, C.; Liu, G.; Wang, Y.; Xu, Z. Identification of two novel LAMA2 mutations in a Chinese patient with congenital muscular dystrophy. Front. Genet. 2018, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Saredi, S.; Gibertini, S.; Matalonga, L.; Farina, L.; Ardissone, A.; Moroni, I.; Mora, M. Exome sequencing detects compound heterozygous nonsense LAMA2 mutations in two siblings with atypical phenotype and nearly normal brain MRI. Neuromuscul. Disord. 2019, 29, 376–380. [Google Scholar] [CrossRef]
- Cohn, R.D.; Campbell, K.P. Molecular basis of muscular dystrophies. Muscle Nerve 2000, 23, 1456–1471. [Google Scholar] [CrossRef] [PubMed]
- Gaina, G.; Budisteanu, M.; Manole, E.; Ionica, E. Clinical and molecular diagnosis in muscular dystrophies. In Muscular Dystrophies; Sakuma, K., Ed.; IntechOpen: London, UK, 2019; pp. 4–27. [Google Scholar] [CrossRef] [Green Version]
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Gruber, A.; Cardoso, M.; Taipa, R.; Fineza, I.; Gonçalves, A.; Laner, A.; Winder, T.L.; Schroeder, J.; Rath, J.; et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum. Mutat. 2018, 39, 1314–1337. [Google Scholar] [CrossRef] [PubMed]
- Madej-Pilarczyk, A.; Niezgoda, A.; Janus, M.; Wojnicz, R.; Marchel, M.; Fidziańska, A.; Grajek, S.; Hausmanowa-Petrusewicz, I. Limb-girdle muscular dystrophy with severe heart failure overlapping with lipodystrophy in a patient with LMNA mutation p.Ser334del. J. Appl. Genet. 2017, 58, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Reeuwijk, J.; Janssen, M.; van den Elzen, C.; de Bernabé, D.B.-V.; Sabatelli, P.; Merlini, L.; Boon, M.; Scheffer, H.; Brockington, M.; Muntoni, F.; et al. POMT2 mutations cause α-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005, 42, 907–912. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; D’Amico, A.; Tessa, A.; Berardinelli, A.; Pane, M.; Messina, S.; van Reeuwijk, J.; Bertini, E.; Muntoni, F.; Santorelli, F.M. POMT2 mutation in a patient with ‘MEB-like’ phenotype. Neuromuscul. Disord. 2006, 16, 446–448. [Google Scholar] [CrossRef]
- Biancheri, R.; Falace, A.; Tessa, A.; Pedemonte, M.; Scapolan, S.; Cassandrini, D.; Aiello, C.; Rossi, A.; Broda, P.; Zara, F.; et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem. Biophys. Res. Commun. 2007, 363, 1033–1037. [Google Scholar] [CrossRef]
- Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve 2020, 61, 436–448. [Google Scholar] [CrossRef]
- Taghizadeh, E.; Rezaee, M.; Barreto, G.E.; Sahebkar, A. Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: A review. J. Cell. Physiol. 2019, 234, 7874–7884. [Google Scholar] [CrossRef]
- Dardas, Z.; Swedan, S.; Al-Sheikh Qassem, A.; Azab, B. The impact of exome sequencing on the diagnostic yield of muscular dystrophies in consanguineous families. Eur. J. Med. Genet. 2020, 63, 103845. [Google Scholar] [CrossRef]
- Efthymiou, S.; Manole, A.; Houlden, H. Next generation sequencing in neuromuscular diseases. Curr. Opin. Neurol. 2016, 29, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichna, J.P.; Macias, A.; Piechota, M.; Korostyński, M.; Potulska-Chromik, A.; Redowicz, M.J.; Zekanowski, C. Whole-exome sequencing identifies novel pathogenic mutations and putative phenotype-influencing variants in Polish limb-girdle muscular dystrophy patients. Hum. Genom. 2018, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaoui, R.; Cooper, S.T.; Lek, M.; Jones, K.; Corbett, A.; Reddel, S.W.; Needham, M.; Liang, C.; Waddell, L.B.; Nicholson, G.; et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: Outcomes and lessons learned. JAMA Neurol. 2015, 72, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Reddy, H.M.; Cho, K.-A.; Lek, M.; Estrella, E.; Valkanas, E.; Jones, M.D.; Mitsuhashi, S.; Darras, B.T.; Amato, A.A.; Lidov, H.G.; et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J. Hum. Genet. 2017, 62, 243–252. [Google Scholar] [CrossRef]
- Bonne, G.; Rivier, F.; Hamroun, D. The 2019 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2018, 28, 1031–1063. [Google Scholar] [CrossRef]
- Nguyen, N.-L.; Thi Bich Ngoc, C.; Dung Vu, C.; Van Tung, N.; Hoang Nguyen, H. A novel frameshift PHKA2 mutation in a family with glycogen storage disease type IXa: A first report in Vietnam and review of literature. Clin. Chim. Acta 2020, 508, 9–15. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Tran, K.T.; Le, V.S.; Vu, C.D.; Nguyen, L.T. A novel de novo variant of LAMA2 contributes to merosin deficient congenital muscular dystrophy type 1A: Case report. Biomed. Rep. 2020, 12, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Nelson, I.; Stojkovic, T.; Allamand, V.; Leturcq, F.; Bécane, H.-M.; Babuty, D.; Toutain, A.; Béroud, C.; Richard, P.; Romero, N.B.; et al. Laminin α2 deficiency-related muscular dystrophy mimicking Emery-Dreifuss and collagen VI related diseases. J. Neuromuscul. Dis. 2015, 2, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Tan, D.; Wang, S.; Song, S.; Yang, H.; Gao, K.; Liu, A.; Jiao, H.; Mao, B.; Ding, J.; et al. Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin. Genet. 2015, 87, 233–243. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; McKee, K.K. Linker protein repair of LAMA2 dystrophic neuromuscular basement membranes. Front. Mol. Neurosci. 2019, 12, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, M.; Rashid, S.; Kupsky, W.; Moore, S.A.; Jiang, H. Child Neurology: LAMA2 muscular dystrophy without contractures. Neurology 2017, 88, e199–e203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissinen, M.; Helbling-Leclerc, A.; Zhang, X.; Evangelista, T.; Topaloglu, H.; Cruaud, C.; Weissenbach, J.; Fardeau, M.; Tomé, F.M.; Schwartz, K.; et al. Substitution of a conserved cysteine-996 in a cysteine-rich motif of the laminin alpha2-chain in congenital muscular dystrophy with partial deficiency of the protein. Am. J. Hum. Genet. 1996, 58, 1177–1184. [Google Scholar]
- Kubota, A.; Ishiura, H.; Mitsui, J.; Sakuishi, K.; Iwata, A.; Yamamoto, T.; Nishino, I.; Tsuji, S.; Shimizu, J. A homozygous LAMA2 mutation of c.818G>A caused partial merosin deficiency in a Japanese patient. Intern. Med. 2018, 57, 877–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi-Gorji, F.; Yassaee, V.R.; Dashti, P.; Miryounesi, M. Novel LAMA2 gene mutations associated with merosin-deficient congenital muscular dystrophy. Iran Biomed. J. 2018, 22, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Wang, X.; Zeng, Y.; Lu, Z.; Cai, S.; Gao, M.; Zhu, W.; Luo, S.; Zhao, C.; Xiao, Z. Missense mutations in LAMA2 causing a new phenotype of mild cognitive impairment, proximal myopathy, seizure, and severe leukoencephalopathy: A case report and protein analysis. Clin. Neuropathol. 2019, 38, 100–108. [Google Scholar] [CrossRef]
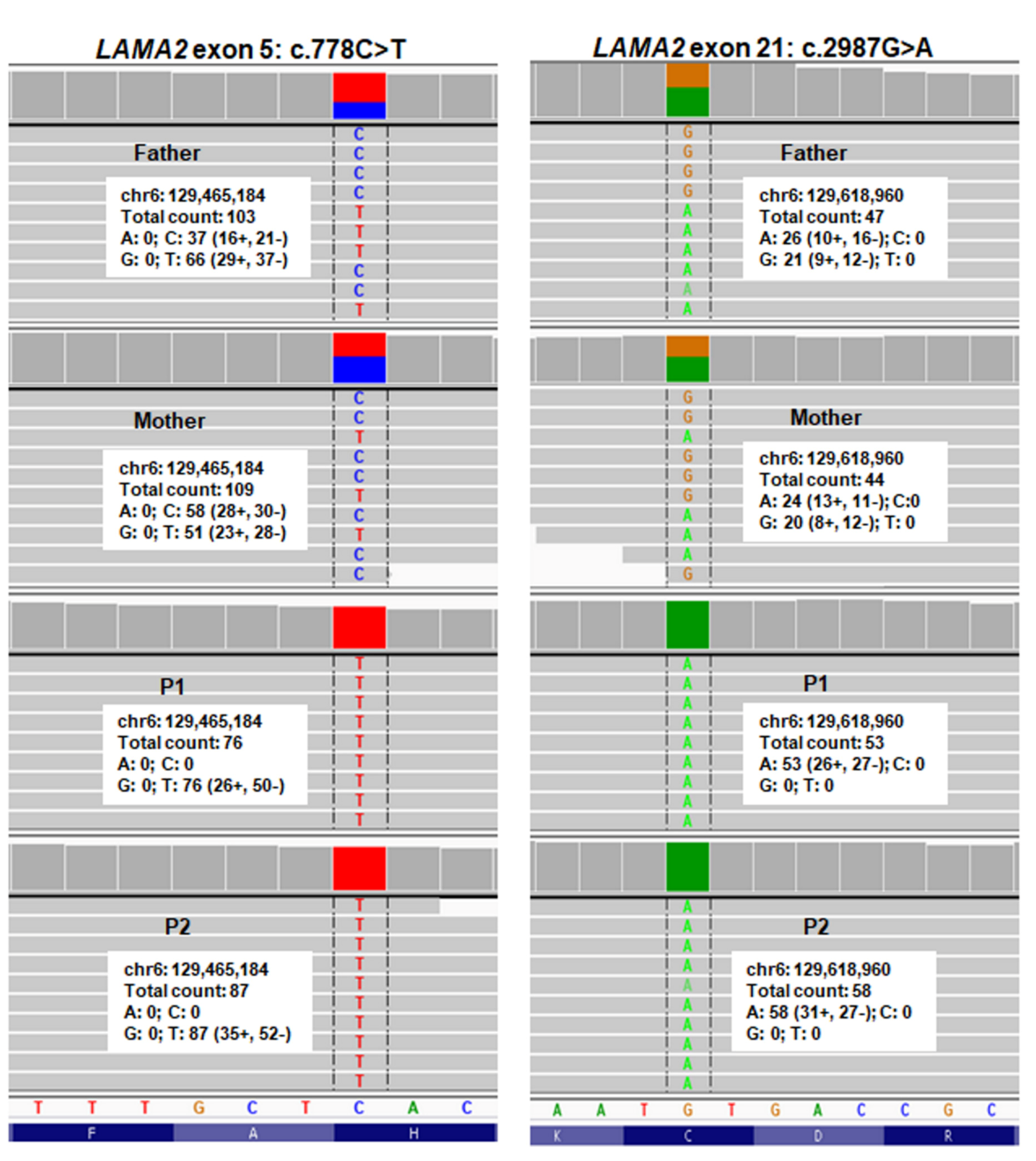

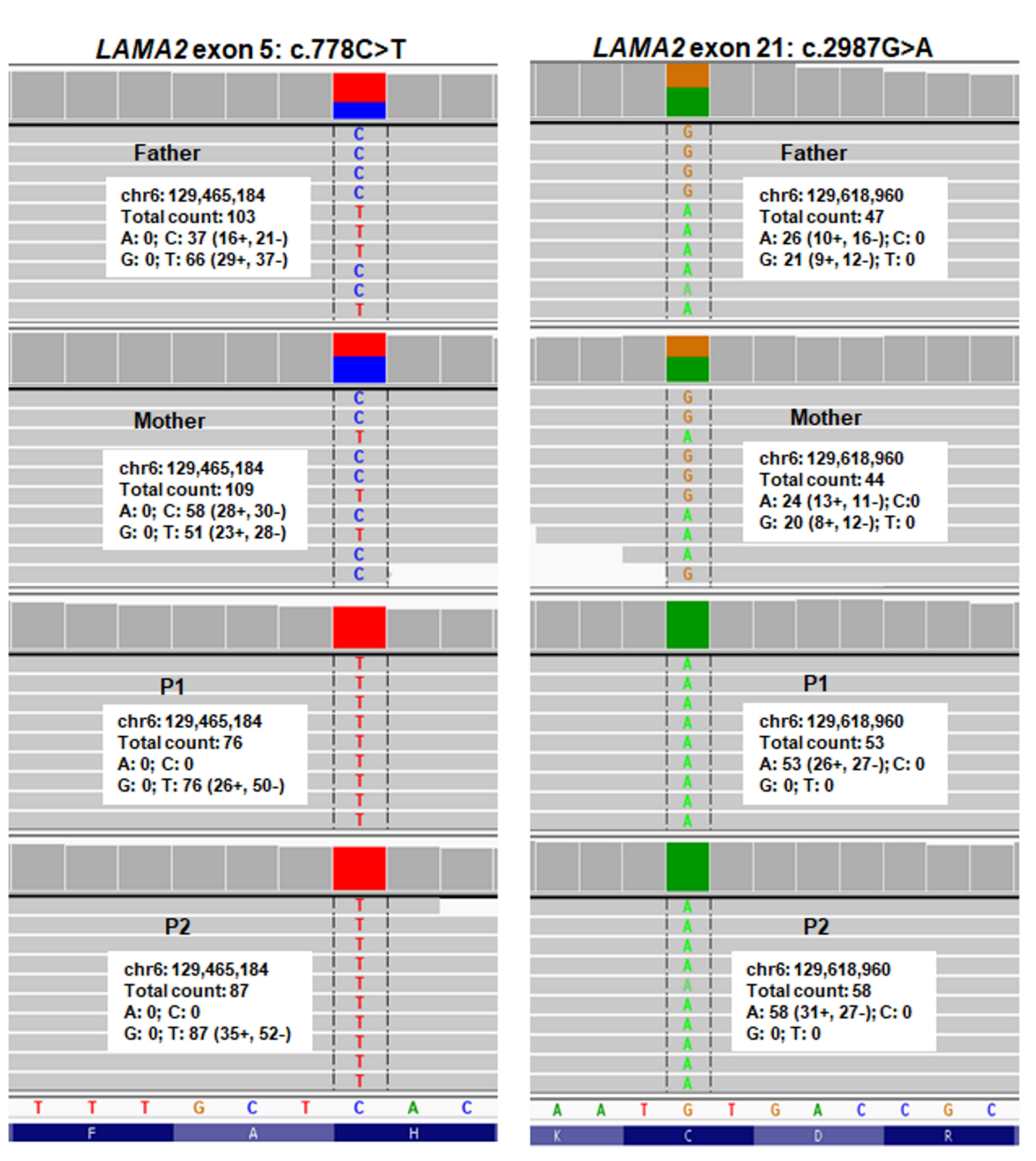{kind=link}
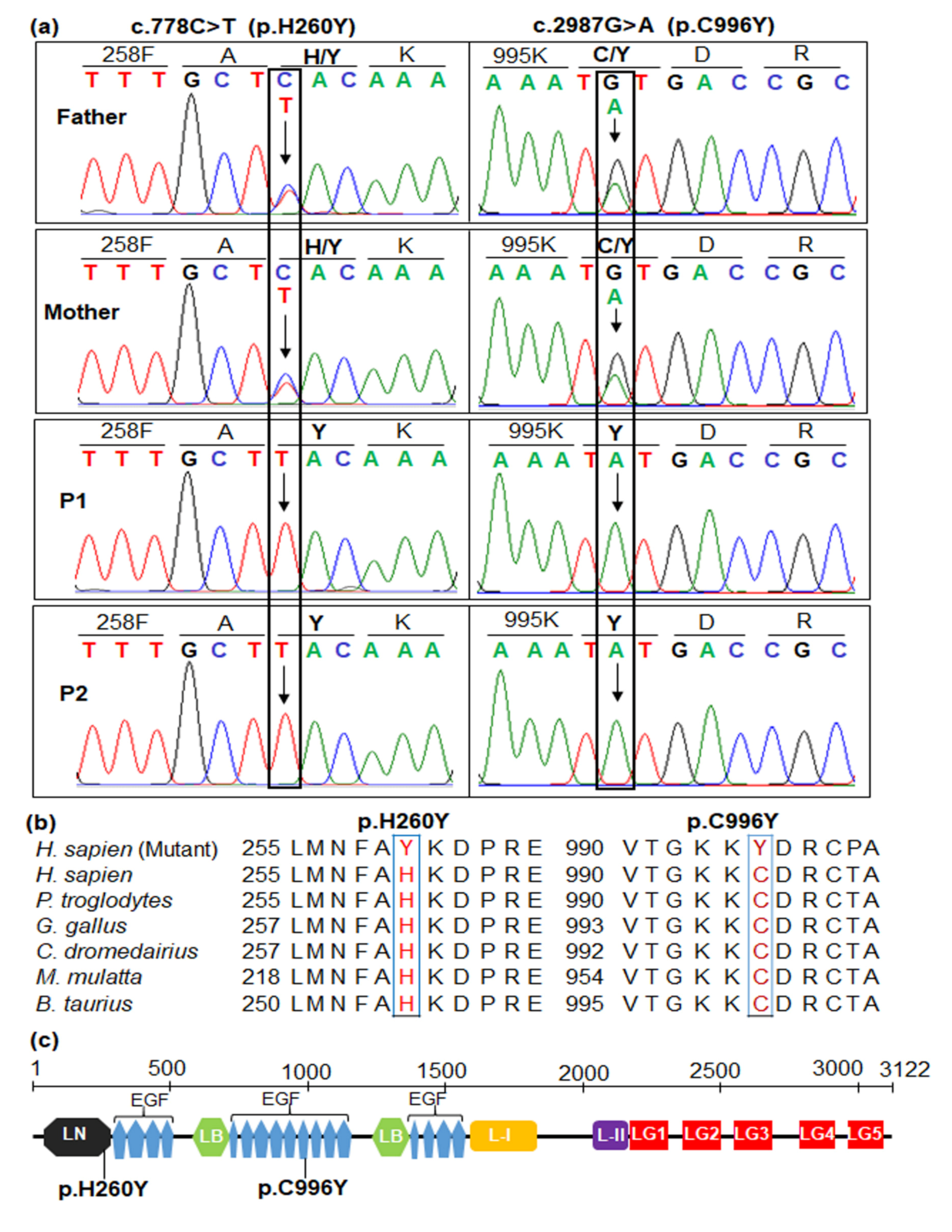{kind=link}
| Patients | P1 | P2 |
|---|---|---|
| Gender | Male | Female |
| Age of first admission | 6-year-old | 4-year-old |
| CK (UI/L) (Normal range: 24–229) | 942 | 523 |
| AST (UI/L) (Normal range: 15–55) | 33.9 | 40.3 |
| ALT (UI/L) (Normal range: 5–40) | 10.1 | 32.7 |
| Head and Neck | Myopathic face Open mouth Prominent jaw Bifid uvula | Myopathic face Open mouth Prominent jaw Broad uvula |
| Skeletal system | Hyperlordosis A wide-based stance Distal contractures of the fingers | Hyperlordosis A wide-based gait absent reflexes Distal contractures of the fingers |
| Musculature | Muscular atrophy Macroglossia Motor delay Positive Gowers sign No pseudohypertrophy of tongue and calf muscles Independent walking at 5 years of age Toe walking from 6 years of age Inability to walk from 9 years of age | Muscular atrophy Macroglossia Motor delay Positive Gowers sign No pseudohypertrophy of tongue and calf muscles Independent walking at 4 years of age Toe walking from 5 years of age |
| Growth development | Delay Normal verbal cognition | Delay Normal verbal cognition |
| Nervous system | Diffuse white matter changes: hyperintense on T2W and FLAIR sequences, hypointense on T1W sequence Not restricted on diffusion with symmetric appearance Normal corpus callosum, ventricular, pontocerebellar angle, cerebral nerves, medulla, pons and high cervical spinal cord. No fluid collection in meningeal space. No history of seizure | Diffuse white matter changes: hyperintense on T2W and FLAIR sequences, hypointense on T1W sequence Not restricted on diffusion with symmetric appearance Normal corpus callosum, ventricular, pontocerebellar angle, cerebral nerves, medulla, pons and high cervical spinal cord. No fluid collection in meningeal space. No history of seizure |
| Heart ultrasound | Normal | Normal |
| Respiratory system | Normal | Normal |
| Digestive system | Normal | Normal |
| Karyotype | 46, XY | 46, XX |
| Screening pathogenic variants in the SMN and GAA genes | Negative | Negative |
| Variant | c.778C>T (p.H260Y) | c.2987G>A (p.C996Y) |
|---|---|---|
| Prediction (Score) | Prediction (Score) | |
| Align-GVGD | Pathogenic (C65) | Pathogenic (C65) |
| CADD (Phred-scaled score) | Likely deleterious (24.1) | Likely deleterious (29.7) |
| Fathmm | Tolerated (1.41) | Damaging (−3.40) |
| Mutation Taster | Disease causing (1) | Disease causing (1) |
| Mutation Assessor | Low impact (0.9) | High impact (4.14) |
| MutPred | Pathogenicity (0.822) | Pathogenicity (0.985) |
| PANTHER | Possibly damaging (361) | Probably damaging (1036) |
| PhD-SNP | Neutral (RI 6) | Disease (RI 7) |
| PON-P2 | Unknown (0.272) | Pathogenic (0.973) |
| PolyPhen-2 | Damaging (0.988) | Damaging (0.998) |
| PROVEAN | Deleterious (−3.247) | Deleterious (−10.385) |
| SIFT | Tolerated (0.148) | Damaging (0) |
| SNP&GO | Neutral (RI 5) | Disease (RI 9) |
| SNAP | Neutral (−32) | Effect (53) |
| UMD-Predictor | Pathogenic (78) | Pathogenic (100) |
| ACMG classification | Likely pathogenic (PM2, PP1–PP4) | Likely pathogenic (PM2, PM5, PP1–PP4) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.-L.; Ngoc, C.T.B.; Vu, C.D.; Nguyen, T.T.H.; Nguyen, H.H. Whole Exome Sequencing as a Diagnostic Tool for Unidentified Muscular Dystrophy in a Vietnamese Family. Diagnostics 2020, 10, 741. https://doi.org/10.3390/diagnostics10100741
Nguyen N-L, Ngoc CTB, Vu CD, Nguyen TTH, Nguyen HH. Whole Exome Sequencing as a Diagnostic Tool for Unidentified Muscular Dystrophy in a Vietnamese Family. Diagnostics. 2020; 10(10):741. https://doi.org/10.3390/diagnostics10100741
Chicago/Turabian StyleNguyen, Ngoc-Lan, Can Thi Bich Ngoc, Chi Dung Vu, Thi Thu Huong Nguyen, and Huy Hoang Nguyen. 2020. "Whole Exome Sequencing as a Diagnostic Tool for Unidentified Muscular Dystrophy in a Vietnamese Family" Diagnostics 10, no. 10: 741. https://doi.org/10.3390/diagnostics10100741
APA StyleNguyen, N.-L., Ngoc, C. T. B., Vu, C. D., Nguyen, T. T. H., & Nguyen, H. H. (2020). Whole Exome Sequencing as a Diagnostic Tool for Unidentified Muscular Dystrophy in a Vietnamese Family. Diagnostics, 10(10), 741. https://doi.org/10.3390/diagnostics10100741