Long Term Follow-Up of Polish Patients with Isovaleric Aciduria. Clinical and Molecular Delineation of Isovaleric Aciduria

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
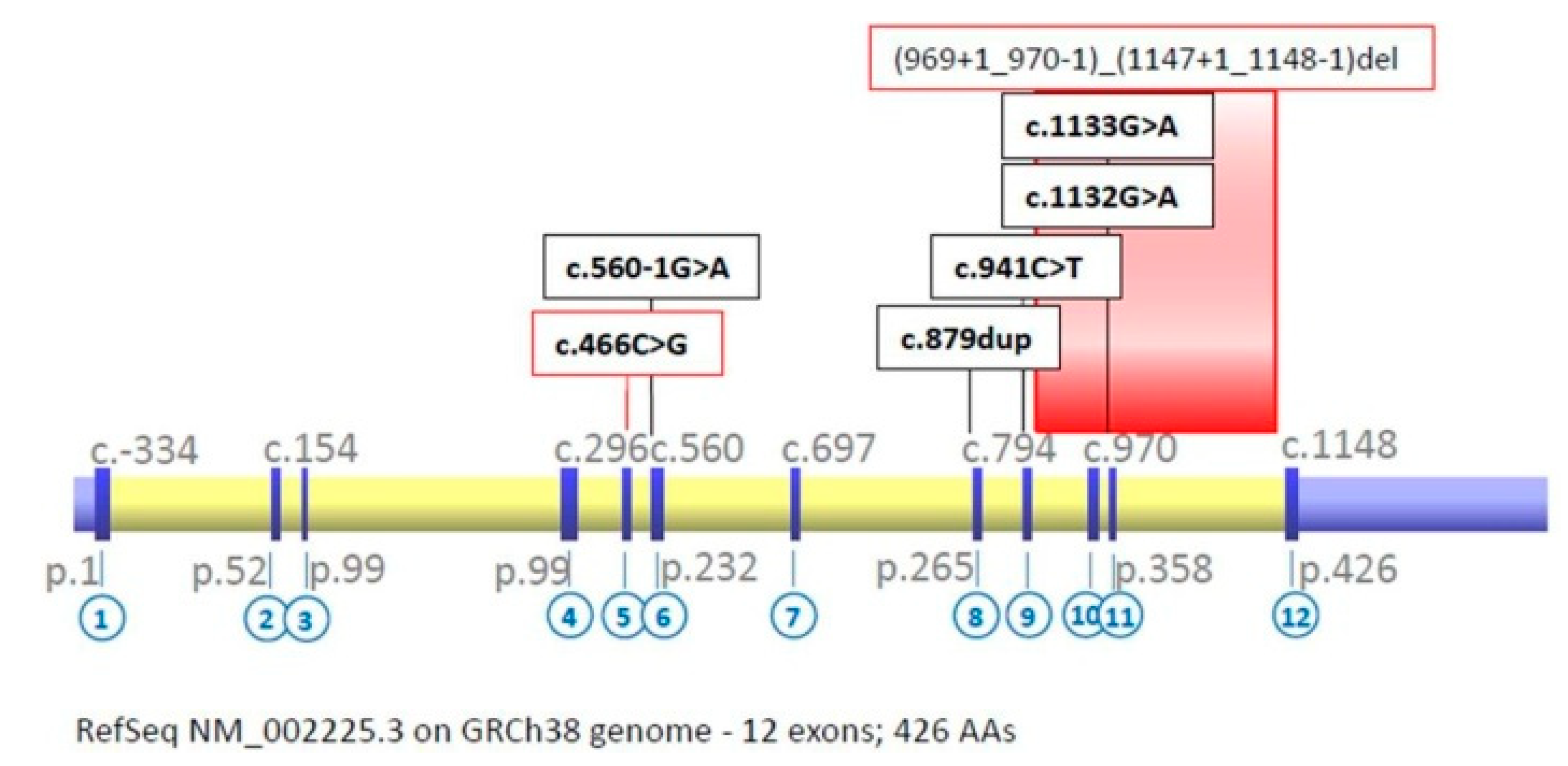
Molecular Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sweetman, L.; Williams, J.D. Branched chain organic acidurias. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Scriver, C., Beaudet, A.L., Sly, W., Valle, D., Eds.; McGraw Hill: New York, NY, USA, 2001; pp. 2125–2164. [Google Scholar]
- Parimoo, B.; Tanaka, K. Structural Organization of the Human Isovaleryl-CoA Dehydrogenase Gene. Genom. 1993, 15, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.R.; Gallo, G.; Scolamiero, E.; Salvatore, F.; Ruoppolo, M. “Classical organic acidurias”: Diagnosis and pathogenesis. Clin. Exp. Med. 2016, 17, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Dercksen, M.; Duran, M.; Ijlst, L.; Mienie, L.J.; Reinecke, C.J.; Ruiter, J.P.N.; Waterham, H.R.; Wanders, R.J.A. Clinical variability of isovaleric acidemia in a genetically homogeneous population. J. Inherit. Metab. Dis. 2012, 35, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Ensenauer, R. Isovaleric acidemia: New aspects of genetic and phenotypic heterogeneity. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2006, 15, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Ensenauer, R.; Vockley, J.; Willard, J.-M.; Huey, J.C.; Sass, J.O.; Edland, S.D.; Burton, B.K.; Berry, S.A.; Santer, R.; Grünert, S.; et al. A Common Mutation Is Associated with a Mild, Potentially Asymptomatic Phenotype in Patients with Isovaleric Acidemia Diagnosed by Newborn Screening. Am. J. Hum. Genet. 2004, 75, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Worthen, H.G.; Al Ashwal, A.; Ozand, P.T.; Garawi, S.; Rahbeeni, Z.; Al Odaib, A.; Subramanyam, S.B.; Rashed, M. Comparative frequency and severity of hypoglycemia in selected organic acidemias, branched chain amino acidemia, and disorders of fructose metabolism. Brain Dev. 1994, 16. [Google Scholar] [CrossRef]
- Kelleher, J.F.; Yudkoff, M.; Hutchinson, R.; August, C.S.; Cohn, R.M. The pancytopenia of isovaleric acidemia. Pediatr. 1980, 65, 1023–1027. [Google Scholar]
- Pinto, A.; Daly, A.; Evans, S.; Almeida, M.F.; Assoun, M.; Belanger-Quintana, A.; Bernabei, S.; Bollhalder, S.; Cassiman, D.; Champion, H.; et al. Dietary practices in isovaleric acidemia: A European survey. Mol. Genet. Metab. Rep. 2017, 12, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, E.; Lee, P.J.; Micciche, A.; Grunewald, S.; Lachmann, R.H. Long-term needs of adult patients with organic acidaemias: Outcome and prognostic factors. J. Inherit. Metab. Dis. 2009, 32, 523–533. [Google Scholar] [CrossRef] [PubMed]
- WHO. Protein and amino acid requirements in human nutrition. Available online: https://apps.who.int/iris/bitstream/handle/10665/43411/WHO_TRS_935_eng.pdf?ua=1 (accessed on 24 August 2020).
- Grünert, S.C.; Wendel, U.; Lindner, M.; Leichsenring, M.; Schwab, K.O.; Vockley, J.; Lehnert, W.; Ensenauer, R. Clinical and neurocognitive outcome in symptomatic isovaleric acidemia. Orphanet J. Rare Dis. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Valayannopoulos, V.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; Baric, I.; Karall, D.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: The evolving clinical phenotype. J. Inherit. Metab. Dis. 2015, 38, 1059–1074. [Google Scholar] [CrossRef]
- Nizon, M.; Ottolenghi, C.; Valayannopoulos, V.; Arnoux, J.-B.; Barbier, V.; Habarou, F.; Desguerre, I.; Boddaert, N.; Bonnefont, J.-P.; Acquaviva, C.; et al. Long-term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J Rare Dis 2013, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Heringer, J.; Valayannopoulos, V.; Lund, A.M.; Wijburg, F.A.; Freisinger, P.; Baric, I.; Baumgartner, M.R.; Burgard, P.; Burlina, A.B.; Chapman, K.A.; et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J Inherit Metab Dis 2016, 39, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Hertecant, J.L.; Ben-Rebeh, I.; Marah, M.A.; Abbas, T.; Ayadi, L.; Ben-Salem, S.; Al-Jasmi, F.A.; Al-Gazali, L.; Al-Yahyaee, S.A.; Ali, B.R. Clinical and molecular analysis of isovaleric acidemia patients in the United Arab Emirates reveals remarkable phenotypes and four novel mutations in the IVD gene. Eur. J. Med. Genet. 2012, 55, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Dionisi-Vici, C.; Deodato, F.; Röschinger, W.; Rhead, W.; Wilcken, B. ‘Classical’ organic acidurias, proionic aciduria, methylmalonic aciduria and isovaleric aciduria: Long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J. Inherit. Metab. Dis. 2006, 29, 383–389. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gender | Age at Diagnosis | Method of Screening (GCMS); -Neonatal -Newborn -Family | First Symptoms | Therapeutic Management | Current Age, Clinical Status and History | IVD Genotype * | |
|---|---|---|---|---|---|---|---|
| 1 | F | neonatal | newborn | adaptation period complicated with congenital infection | special nutritional formula for IVA, Supplementation of Gly and carnitine introduced since diagnosis; good adherence | Age: 5 years normal clinical condition with no complications-4 mild decompensations with no severe complications | c.[1133G>A]; [(1133G>A)] p.(Gly378Asp)/p.(Gly378Asp) |
| 2 | M | neonatal | symptomatic | intoxication syndrome: failure to thrive, dehydration, hypotonia ammonia level—761 µmol/l at diagnosis-decompensation | low-protein diet+ special nutrition formula for IVA, Supplementation of Gly and carnitine introduced since diagnosis; satisfactory adherence | Age: 7 years moderate developmental delay, gastrostomy at the age of 4.5 during one of metabolic decompensation, than removed | Na |
| 3 | M | neonatal | symptomatic | since birth: hypotonia, apathy, failure to thrive, vomits, specific odour | special nutrition formula for IVA, supplementation of Gly and carnitine introduced since diagnosis; good adherence | Age: 5 years normal clinical condition, average IQ—lower limit of the norm no metabolic decompensations | c.[466C>G(;)879dup] p.(Leu156Val)/p.(Pro294Alafs*38) |
| 4 | F | neonatal | Family history positive for IVA (first child died at the age of 8 days due to IVA) | besides light hypotonia no worrying symptoms | special nutrition formula for IVA, supplementation of Gly and carnitine introduced since diagnosis; good adherence | Age: 4 years normal clinical condition and development—2 metabolic decompensations triggered by infections | c.[466C>G(;)1133G> A] p.(Leu156Val)/p.(Gly378Asp) |
| 5 | M | neonatal | symptomatic | intoxication syndrome: vomiting, poor general condition—respiratory failure, sweaty foot odour | low-protein diet, supplementation of carnitine introduced since diagnosis; poor adherence | Age: 22 years depression treated with antidepressants, poor adherence to dietary restrictions; 2 metabolic decompensations: first one at diagnosis, second one triggered by infections | Na |
| 6 | f | 10y | symptomatic | mild mental delay; during 9 and 10 years of age recurrent vomiting and ketosis; inborn toxoplasmosis | low-protein diet, supplementation of carnitine introduced since diagnosis (10 y); poor adherence | Age: 32 years normal clinical condition, gave birth to 2 children -2 decompensations: first at diagnosis, second after 1st childbirth | c.[466C>G(;)560- 1G>A] p.Leu156Val/p.? |
| 7 | M | 6y | symptomatic | intoxication syndrome: vomiting, encephalopaty, food intolerance | low-Leu diet, supplementation of Gly introduced since diagnosis (6 y); good adherence | Age: 25 years normal psycho-motor development, but recurrent vomiting, multiple decompensatios till diagnosis—since diet introduction—no episodes | c.[(969+1_970-1)_(1147+1_1148-1)del]; [((969+1_970-1)_(1147+1_1148-1)del)] p.?/p.? |
| 8 | F | neonatal | symptomatic | good clinical condition | low-protein diet, supplementation of carnitine introduced since diagnosis; good adherence | Age: 11 years normal psycho-motor development—one metabolic decompensation —at diagnosis | c.[941C>T(;)1132G> A] p.(Ala314Val)/p.(Gly378Ser) |
| 9 | M | 5y | symptomatic | metabolic decompensation: vomiting, poor nutritional status; cerebral palsy | low-protein diet, supplementation of carnitine introduced since diagnosis (5 y); satisfactory adherence | Age: 25 years delayed psycho-motor development—one metabolic decompensation—at diagnosis | Na |
| 10 | M | neonatal | family (first child died) | good clinical status | low-protein diet + special formula (Prosobee), supplementation of Gly and carnitine; good adherence | Age: 29 years good clinical, and psychological condition—one decompensation due to bacterial tonsillitis (fever and vomiting) | c.[(969+1_970-1)_(1147+1_1148-1)del]; [((969+1_970-1)_(1147+1_1148-1)del)] p.?/p.? |
| IVD Variant | Reporting Data * | ||||||
|---|---|---|---|---|---|---|---|
| cDNA Level | Protein Level | No of Alleles | Type | Status ** | HGMD | ClinVar | PIMID |
| c.466C>G | p.(Leu156Val) | 3 | missense | novel 1 | na | Na | no citation |
| c.560-1G>A | p.? | 1 | splice-site | known | na | RCV000410323.1 (likely pathogenic-IVA) | no citation |
| c.879dup | p.(Pro294Alafs * 38) | 1 | frameshift | known | CI001574 (IVA) | RCV000555769.1 (pathogenic–IV) | 10677295 |
| c.941C>T | p.(Ala314Val) | 1 | missense | known | CM983435 (IVA) | RCV000080003.11(pathogenic–not provided) RCV000003749.9 (pathogenic-IVA) | 9665741, 23757202 26018748, 26937393 |
| 969+1_970-1)_(1147+1_1148-1)del | p.? | 4 | deletion (ex.10-11) | known | CP124273 (IVA) | Na | 22090376 |
| c.1132G>A | p.(Gly378Ser) | 1 | missense | Novel 1 | na | Na | no citation |
| c.1133G>A | p.(Gly378Asp) | 3 | missense | known | BM0867669 (IVA?) | Na | 19089597 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymańska, E.; Jezela-Stanek, A.; Bogdańska, A.; Rokicki, D.; Ehmke vel Emczyńska-Seliga, E.; Pajdowska, M.; Ciara, E.; Tylki-Szymańska, A. Long Term Follow-Up of Polish Patients with Isovaleric Aciduria. Clinical and Molecular Delineation of Isovaleric Aciduria. Diagnostics 2020, 10, 738. https://doi.org/10.3390/diagnostics10100738
Szymańska E, Jezela-Stanek A, Bogdańska A, Rokicki D, Ehmke vel Emczyńska-Seliga E, Pajdowska M, Ciara E, Tylki-Szymańska A. Long Term Follow-Up of Polish Patients with Isovaleric Aciduria. Clinical and Molecular Delineation of Isovaleric Aciduria. Diagnostics. 2020; 10(10):738. https://doi.org/10.3390/diagnostics10100738
Chicago/Turabian StyleSzymańska, Edyta, Aleksandra Jezela-Stanek, Anna Bogdańska, Dariusz Rokicki, Ewa Ehmke vel Emczyńska-Seliga, Magdalena Pajdowska, Elżbieta Ciara, and Anna Tylki-Szymańska. 2020. "Long Term Follow-Up of Polish Patients with Isovaleric Aciduria. Clinical and Molecular Delineation of Isovaleric Aciduria" Diagnostics 10, no. 10: 738. https://doi.org/10.3390/diagnostics10100738
APA StyleSzymańska, E., Jezela-Stanek, A., Bogdańska, A., Rokicki, D., Ehmke vel Emczyńska-Seliga, E., Pajdowska, M., Ciara, E., & Tylki-Szymańska, A. (2020). Long Term Follow-Up of Polish Patients with Isovaleric Aciduria. Clinical and Molecular Delineation of Isovaleric Aciduria. Diagnostics, 10(10), 738. https://doi.org/10.3390/diagnostics10100738