The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean




Abstract
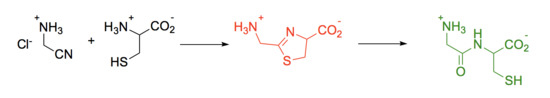
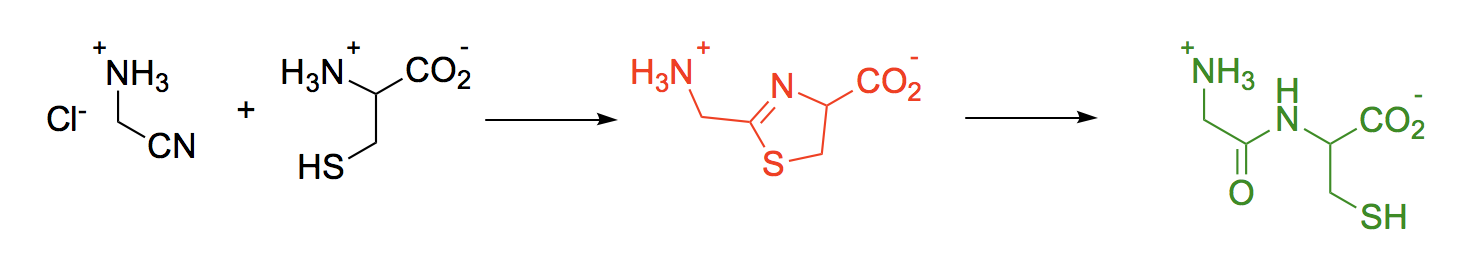
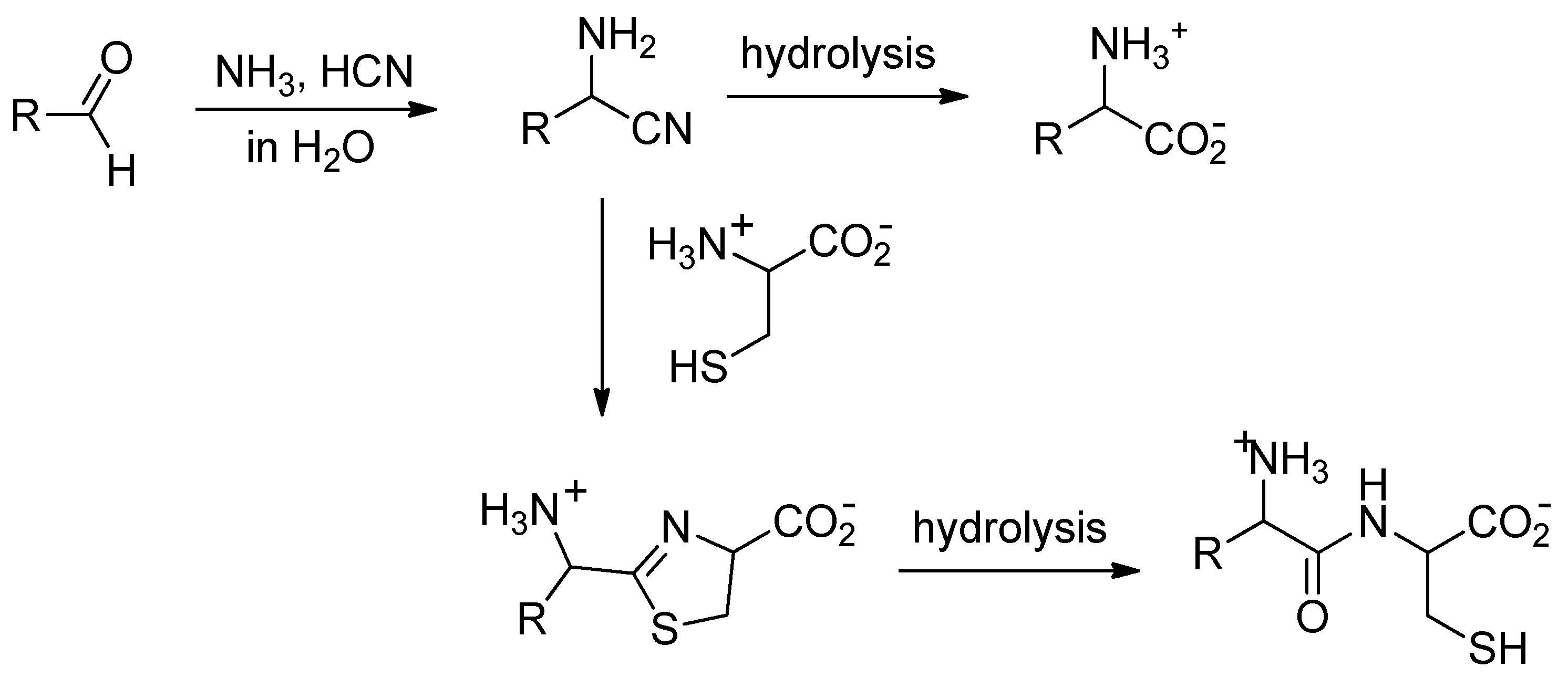
1. Introduction
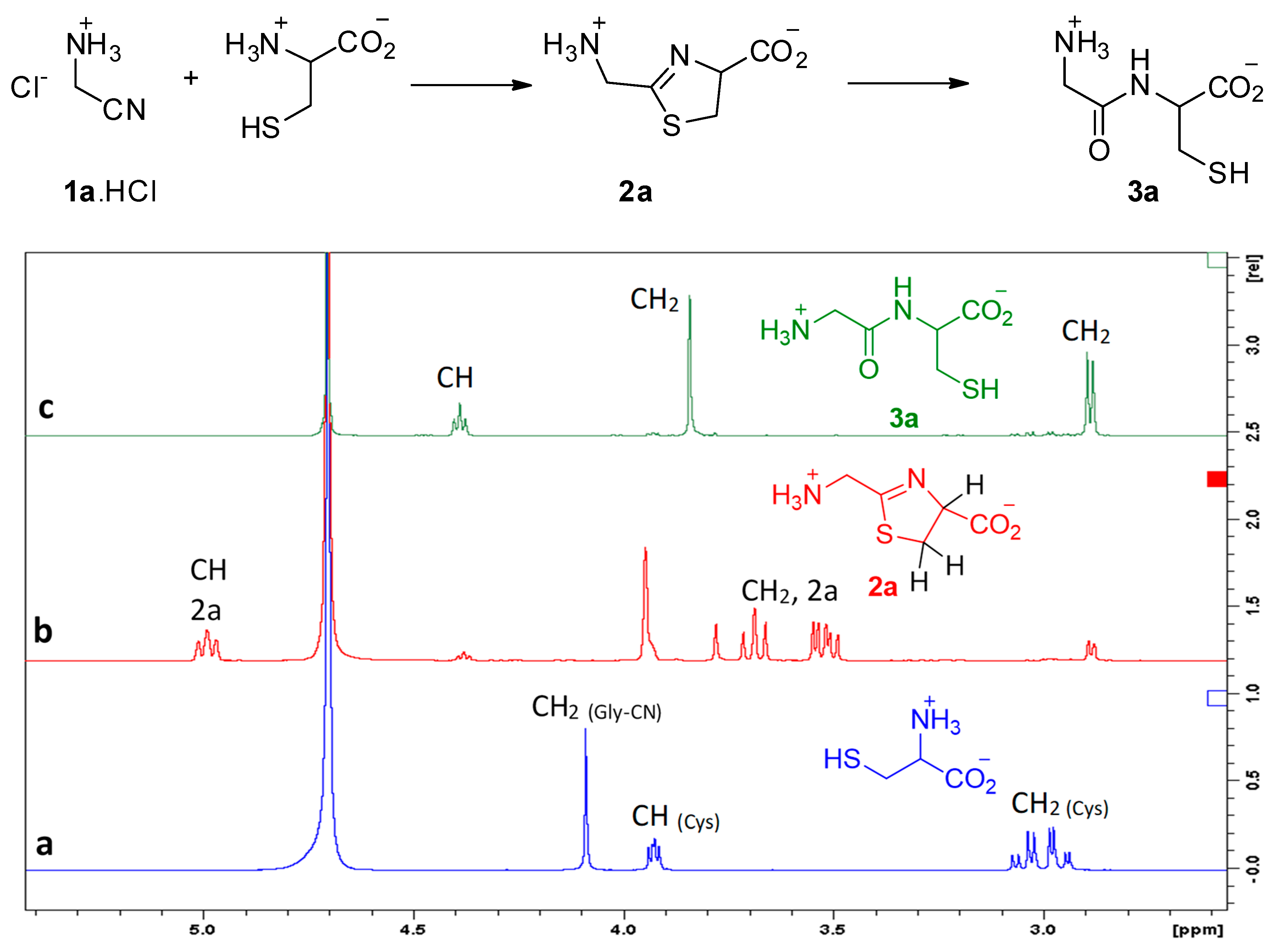
2. Experimental Section


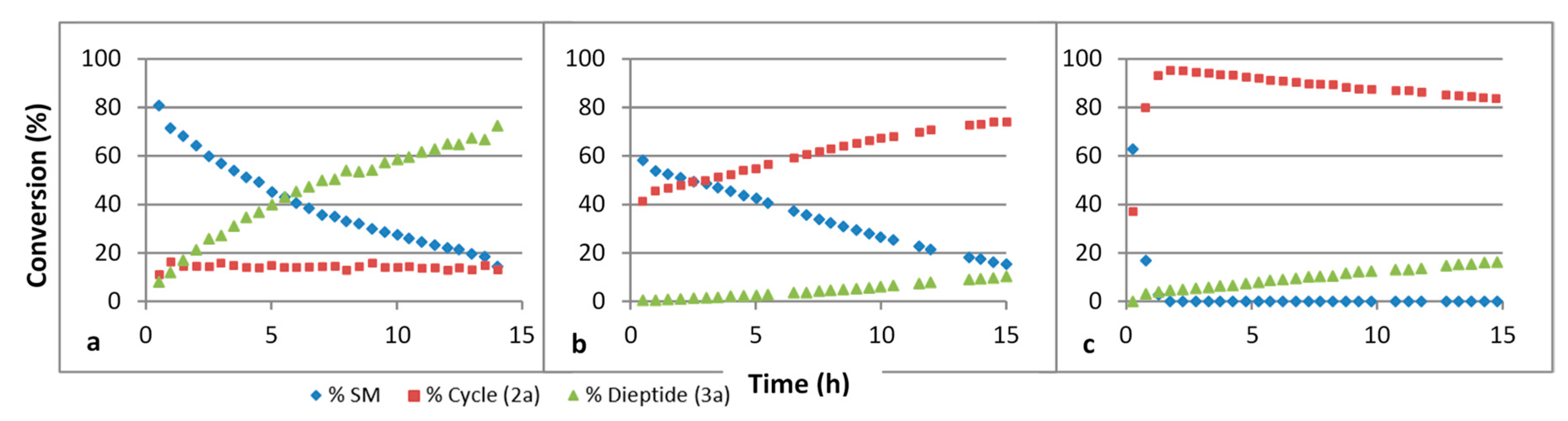
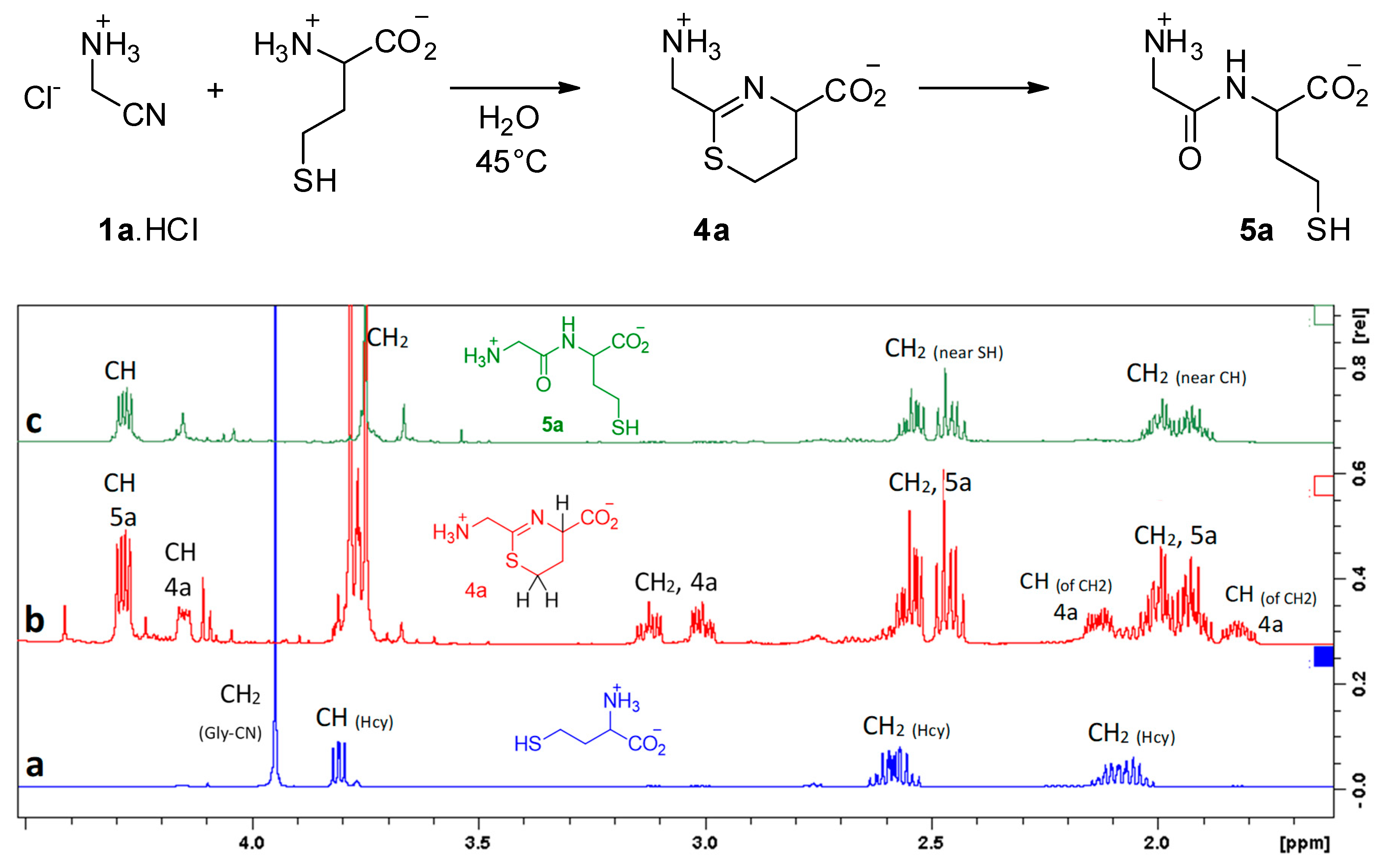
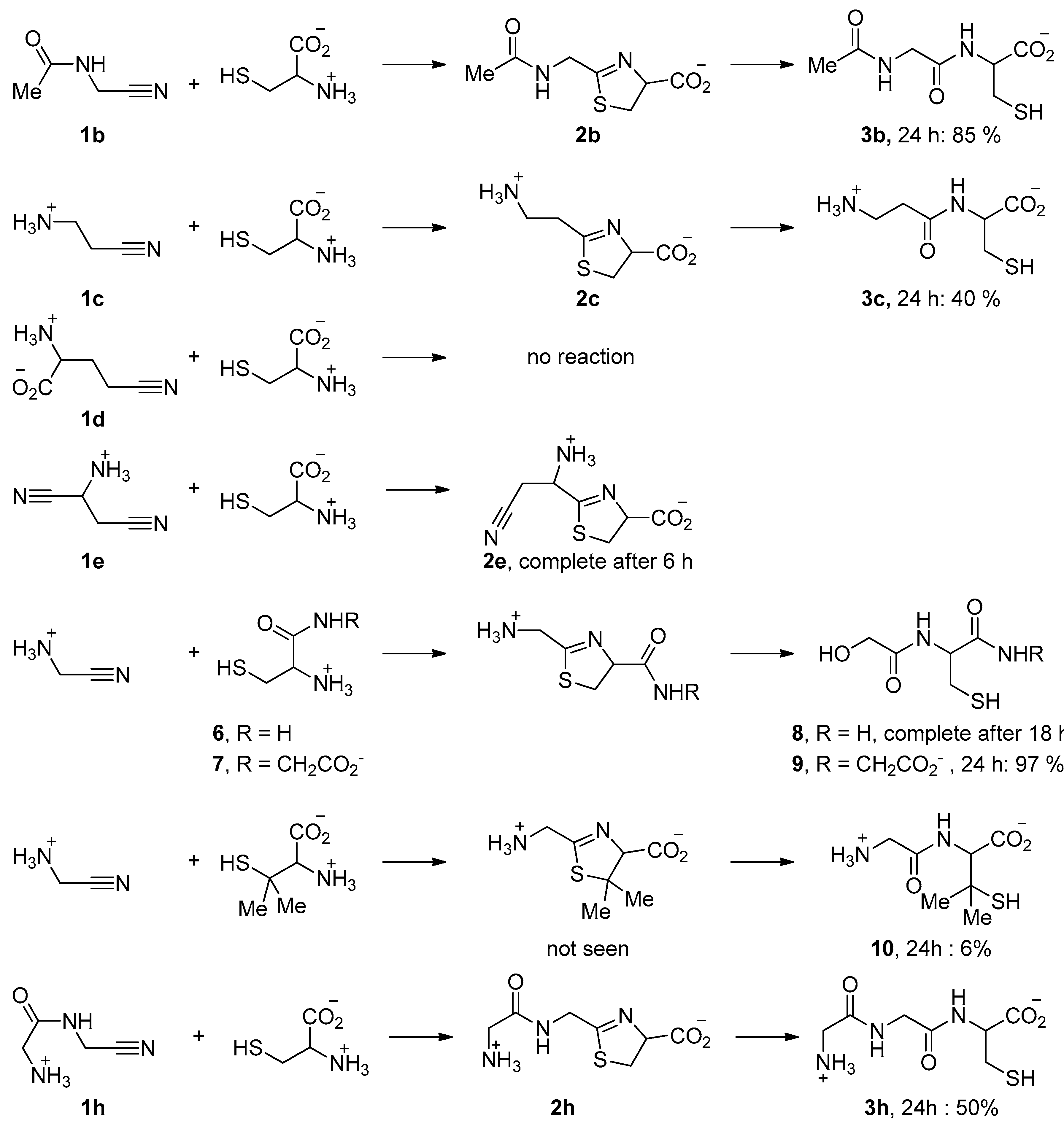
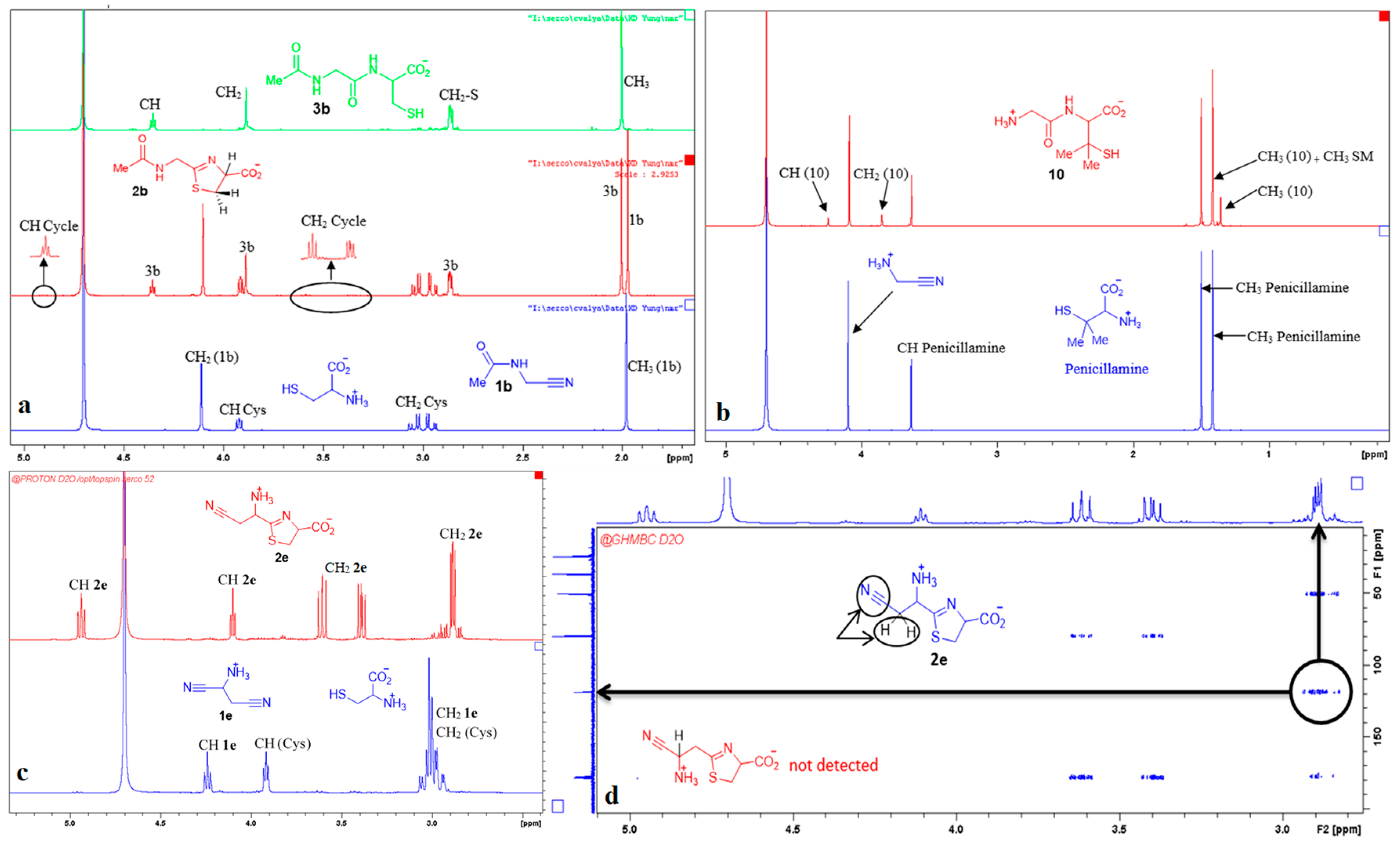
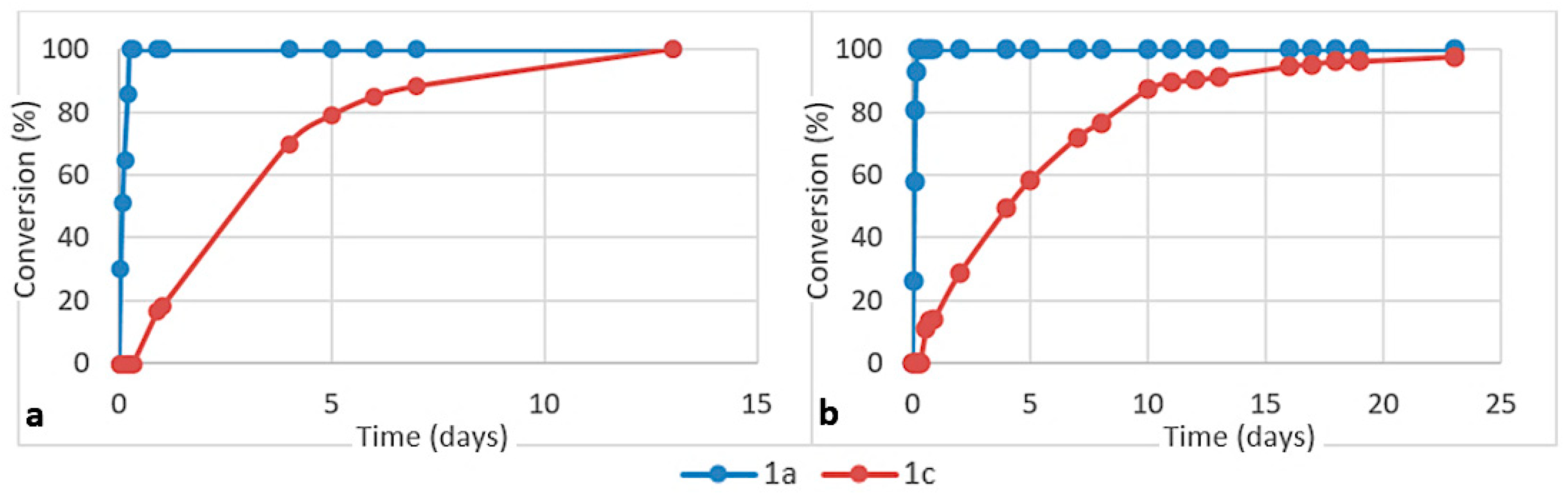
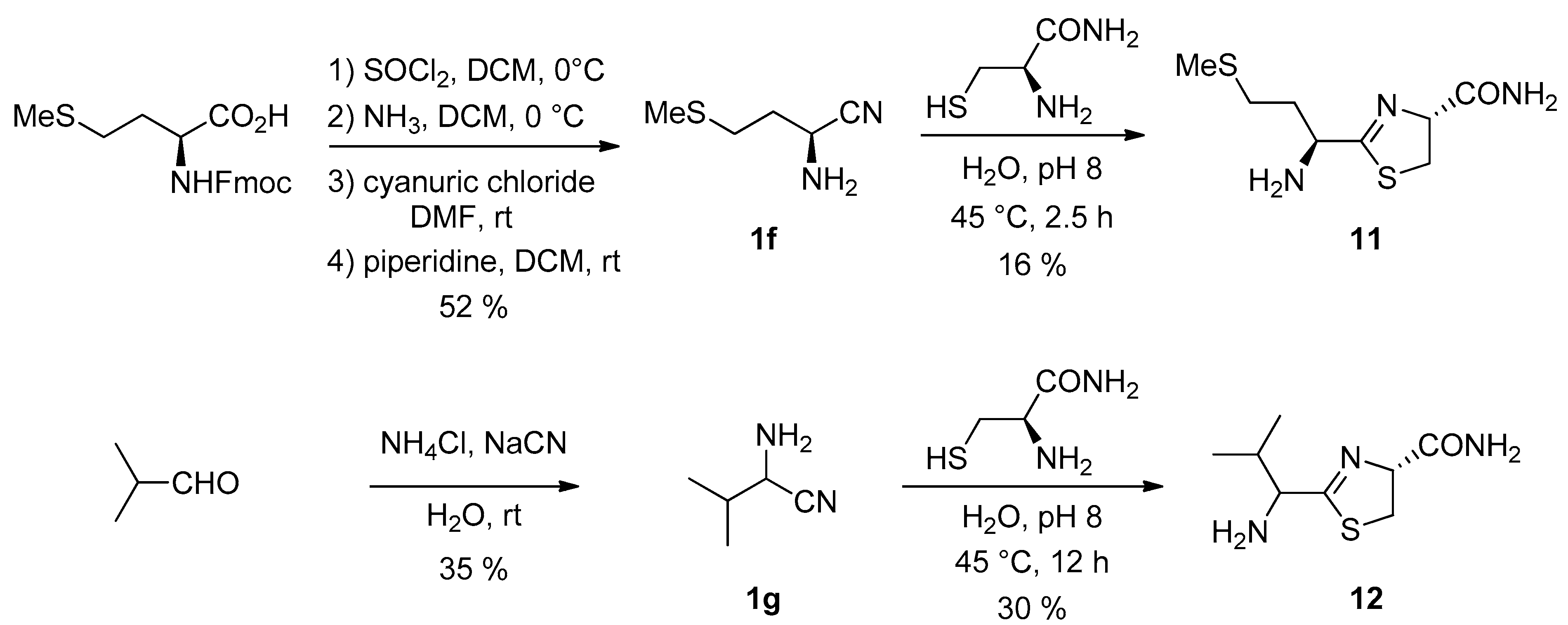
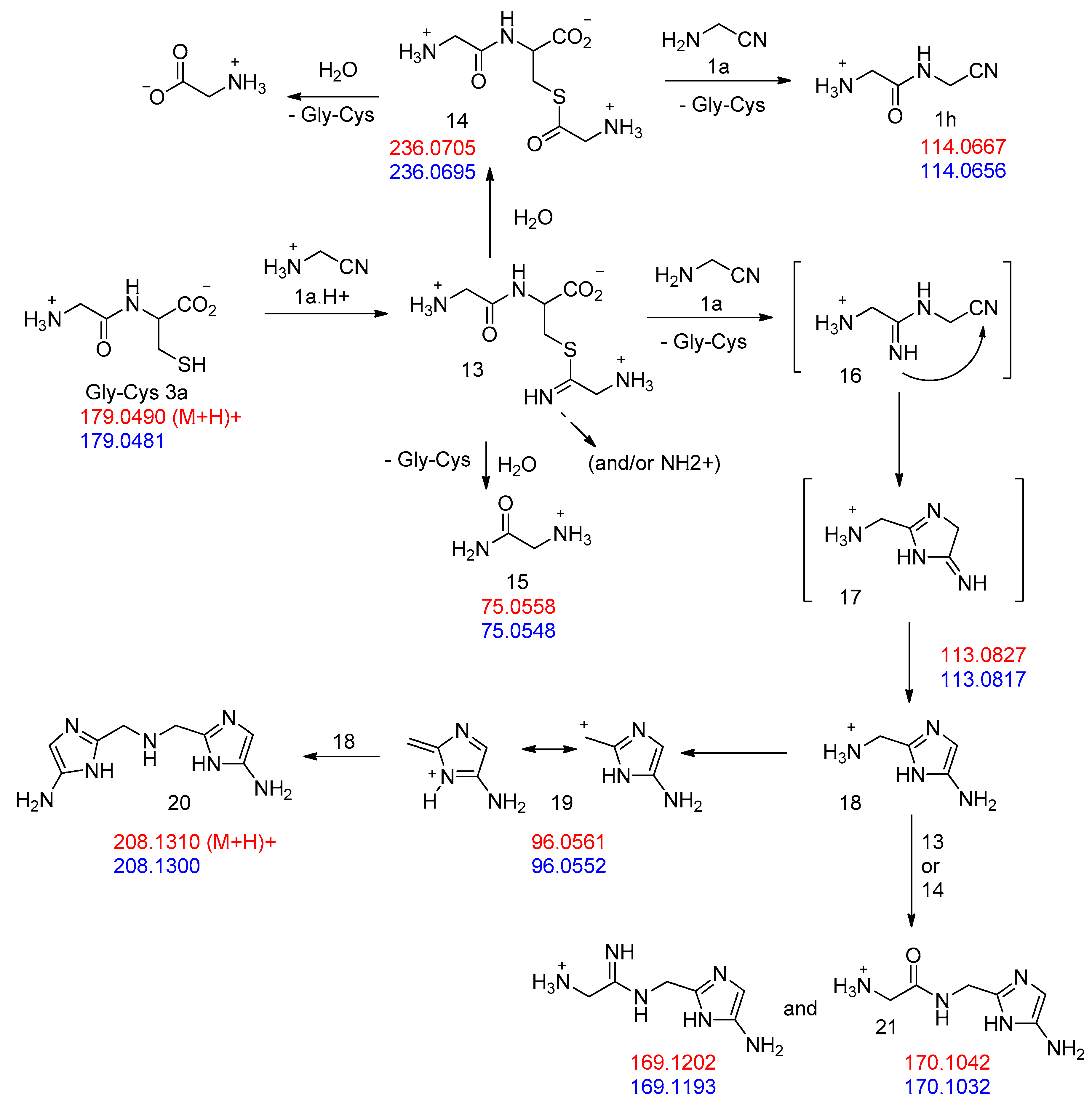
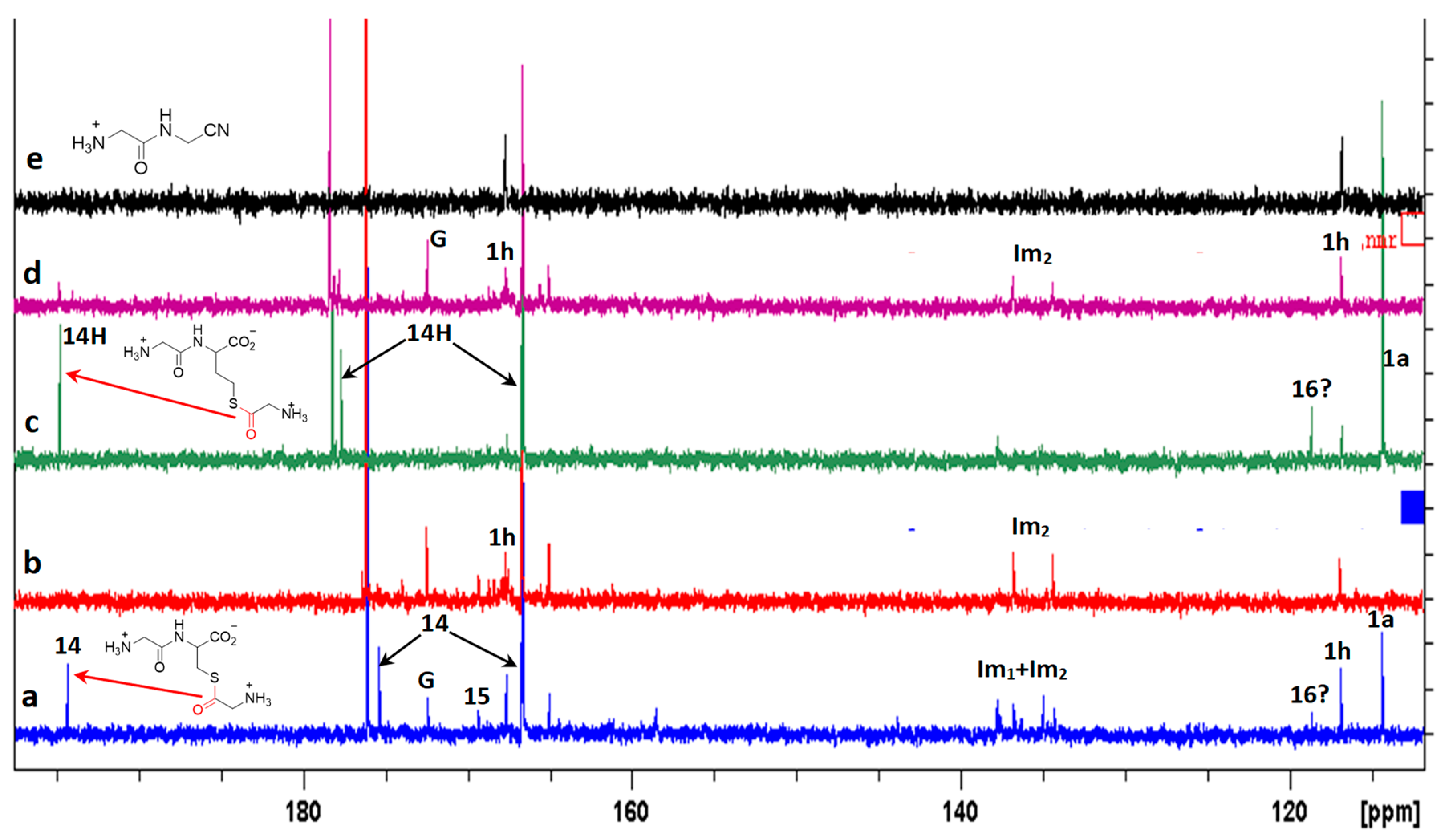
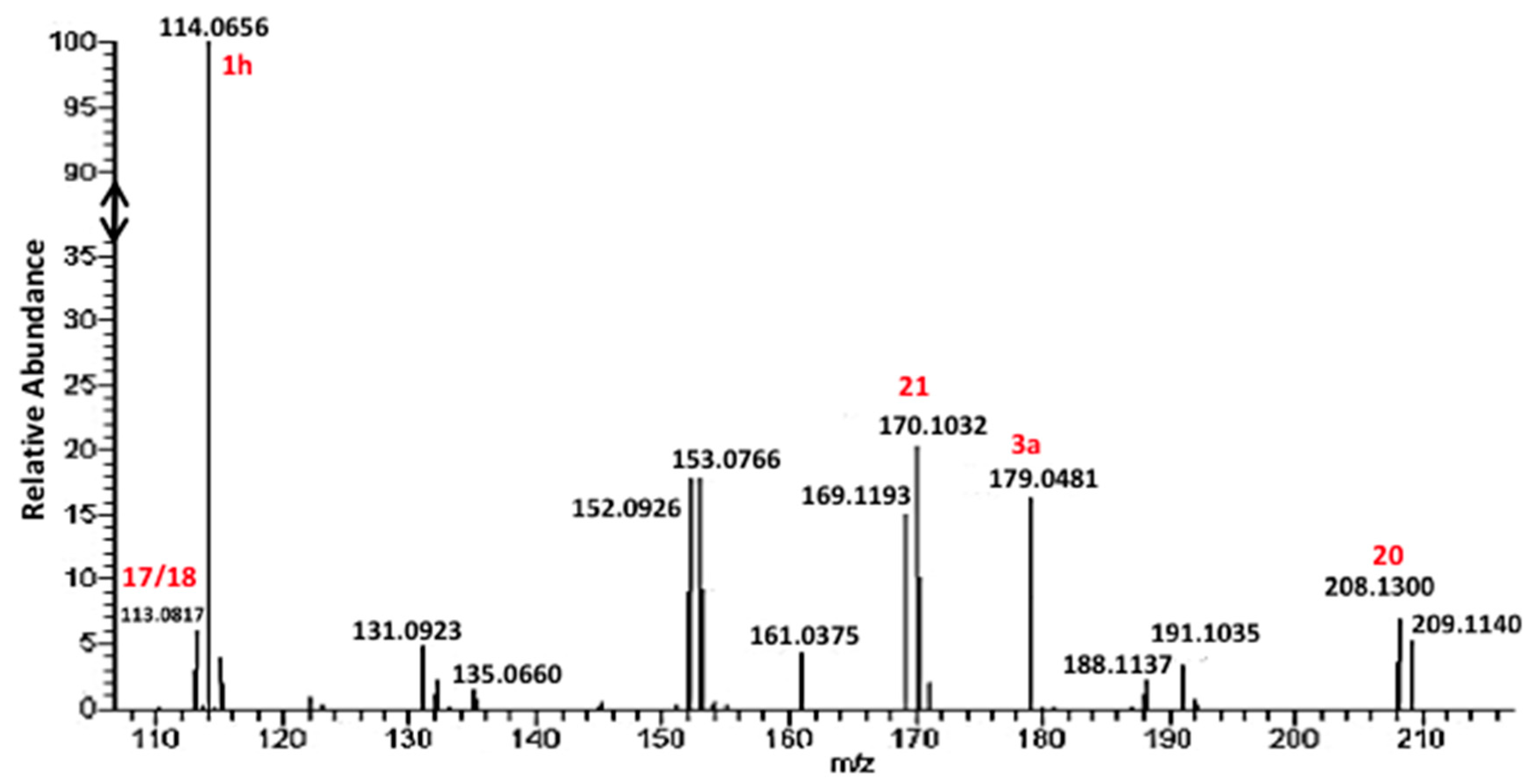
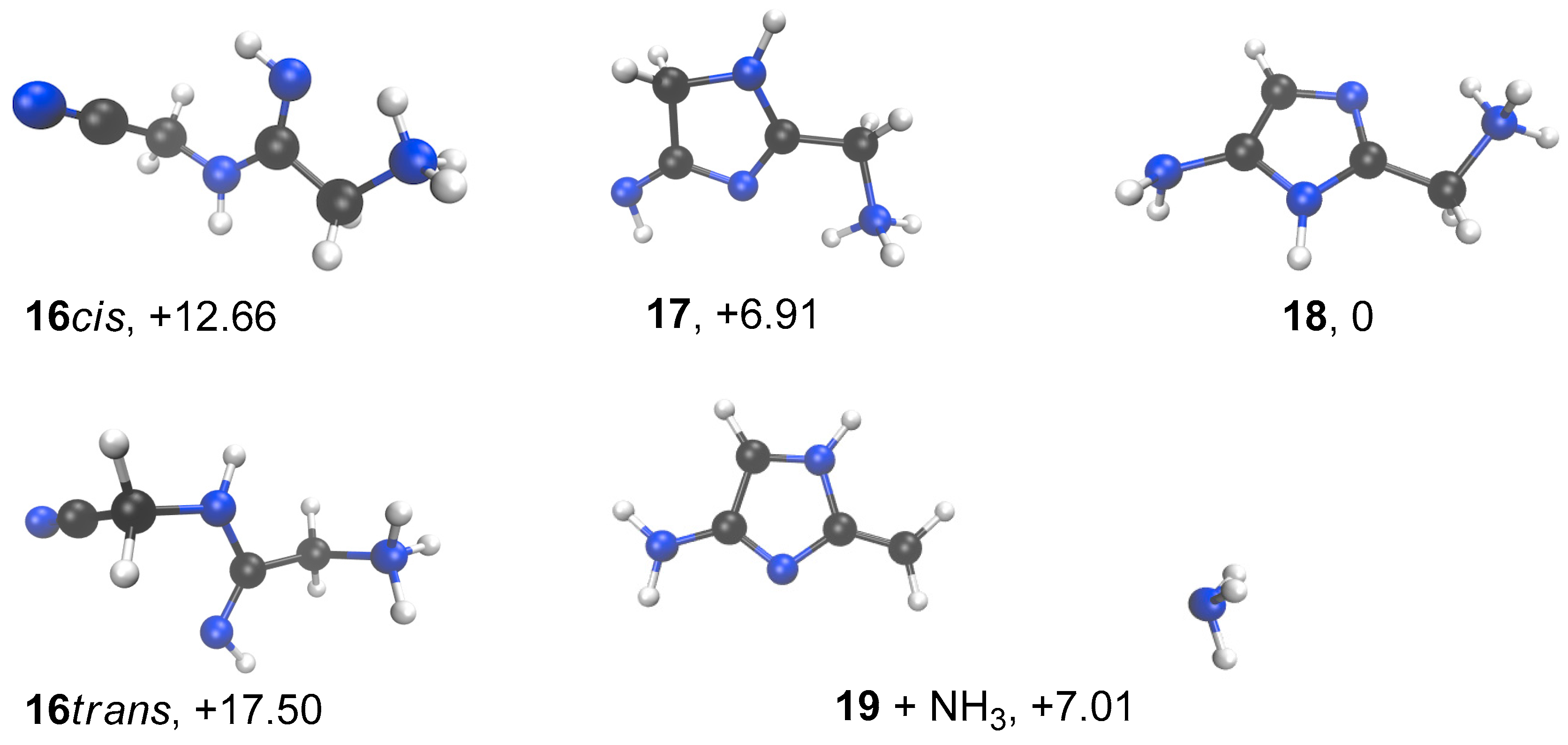
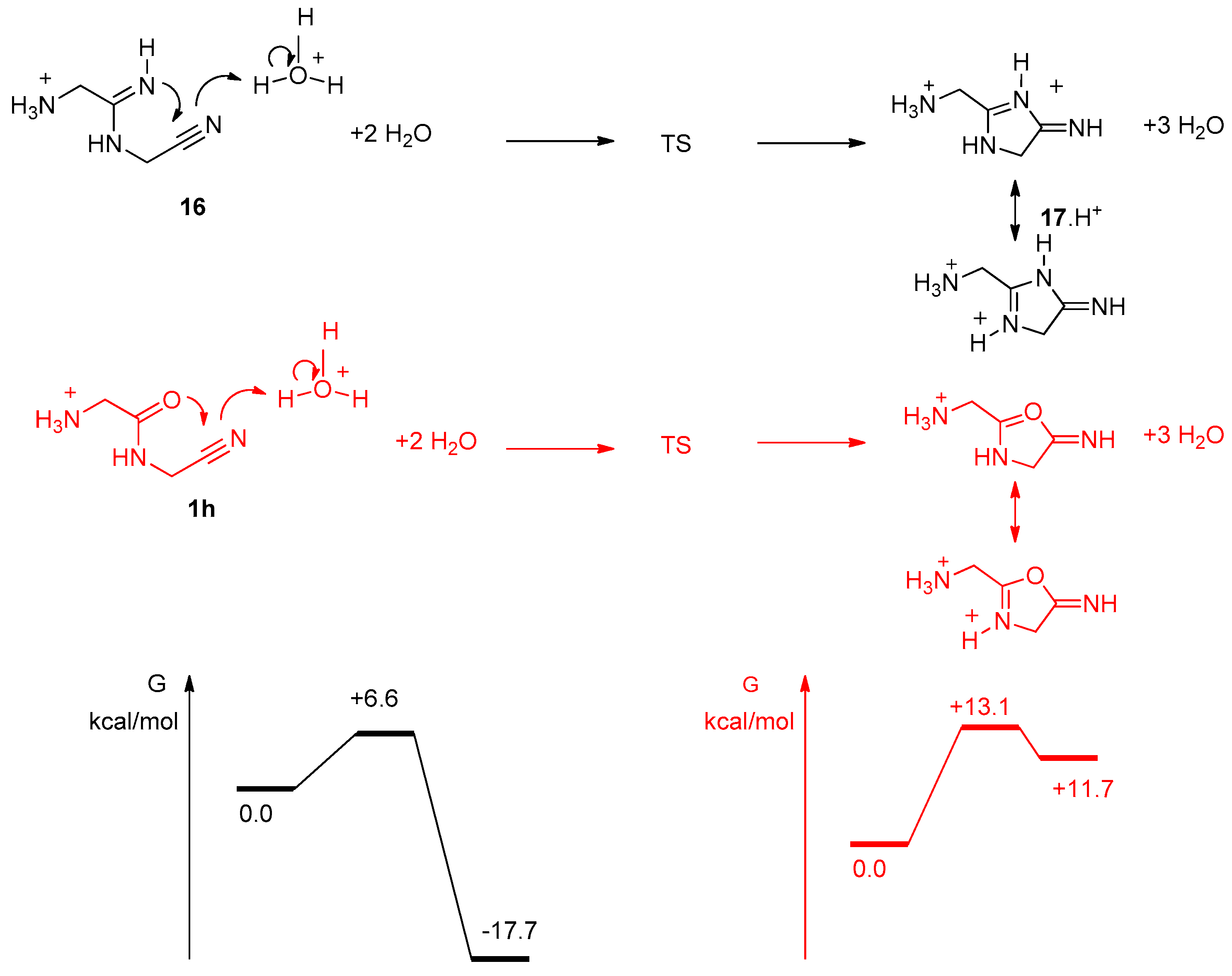
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hashimoto, S.I. Ribosome-independent peptide synthesis and their application to dipeptide production. J. Biol. Macromol. 2008, 8, 28–37. [Google Scholar]
- Chalmet, S.; Harb, W.; Ruiz-Lòpez, M.F. Computer simulation of amide bond formation in aqueous solution. J. Phys. Chem. A 2001, 105, 11574–11581. [Google Scholar] [CrossRef]
- Gajewski, E.; Steckler, D.K.; Goldberg, R.N. Thermodynamics of the hydrolysis of adenosine 5'-triphosphate to adenosine 5′-diphosphate. J. Biol. Chem. 1988, 261, 12733–12737. [Google Scholar]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic systems chemistry: New perspectives for the origins of life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef] [PubMed]
- Strecker, A. Ueber die künstliche bildung der milchsäure und einen neuen, dem glycocoll homologen körper. Liebigs Ann. Chem. 1850, 75, 27–45. [Google Scholar] [CrossRef]
- Guthrie, J.P.; Yim, J.C.H.; Wang, Q. Hydration of nitriles: An examination in terms of no barrier theory. J. Phys. Org. Chem. 2014, 27, 27–37. [Google Scholar] [CrossRef]
- Krimmer, H.P.; Drauz, K.; Kleemann, A. Umsetzung von β-mercapto-α-aminosäuren mit nitrilen. Chemiker-Zeitung 1987, 111, 357–361. [Google Scholar]
- Vallée, Y.; Shalayel, I.; Dung, L.K.; Raghavendra Rao, K.V.; de Paëpe, G.; Märker, K.; Milet, A. At the very beginning of life on Earth: The thiol rich peptide (TRP) world hypothesis. Int. J. Dev. Biol. 2017, 61, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Hagan, W.J., Jr. HCN and chemical evolution: The possible role of cyano compounds in prebiotic synthesis. Tetrahedron 1984, 1093–1120. [Google Scholar] [CrossRef]
- Schilke, P.; Menten, K.M. Detection of a second, strong submillimeter HCN laser line toward carbon stars. Astrophys. J. 2003, 583, 446–450. [Google Scholar] [CrossRef]
- Thorwirth, S.; Wyrowski, F.; Schilke, P.; Menten, K.M.; Brünken, S.; Müller, H.S.P.; Winnewisser, G. Detection of HCN direct l-type transitions probing hot molecular gas in the proto-planetary nebula CRL 618. Astrophys. J. 2003, 586, 338–343. [Google Scholar] [CrossRef]
- Irvine, W.M.; Dickens, J.E.; Lovell, A.J.; Schoerb, F.P.; Senay, M.; Bergin, E.A.; Jewitt, D.; Matthews, H.E. The HNC/HCN ratio in comets. Earth Moon Planets 1997, 78, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Schloerb, F.P.; Kinzel, W.M.; Swade, D.A.; Irvine, W.M. Observations of HCN in comet P/Halley. Astron. Astrophys. 1987, 187, 475–480. [Google Scholar] [PubMed]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nature Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Zahnle, K.J. Photochemistry of methane and the formation of hydrocyanic acid (HCN) in the Earth’s early atmosphere. J. Geophys. Res. 1986, 91, 2819–2834. [Google Scholar] [CrossRef]
- Martin, R.S.; Mather, T.A.; Pyle, D.M. Volcanic emissions and the early Earth atmosphere. Geochim. Cosmochim. Acta 2007, 71, 3673–3685. [Google Scholar] [CrossRef]
- Mukhin, L.M. Volcanic processes and synthesis of simple organic compounds on primitive Earth. Orig. Life Evol. Biosph. 1976, 7, 355–368. [Google Scholar] [CrossRef]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Thaddeus, P.; Kutner, M.L.; Penzias, A.A.; Wilson, R.W.; Jefferts, K.B. Interstellar hydrogen sulfide. Astrophys. J. 1972, 176, L73–L76. [Google Scholar] [CrossRef]
- Minh, Y.C.; Ziurys, L.M.; Irvine, W.M.; McGonagle, D. Abundances of hydrogen sulfide in star-forming regions. Astrophys. J. 1991, 366, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Minh, Y.C.; Irvine, W.M.; Ziurys, L.M. Detection of interstellar hydrogen sulfide in cold, dark clouds. Astrophys. J. 1989, 345, L63–L66. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, P.; Meier, R.; Krankowsky, D.; Hodges, R.R. Methanol and hydrogen sulfide in comet P/Halley. Astron. Astrophys. 1994, 288, 315–329. [Google Scholar]
- Carapezza, M.L.; Badalamenti, B.; Cavarra, L.; Scalzo, A. Gas hazard assessment in a densely inhabited area of Colli Albany Volcano (Cava dei Selci, Roma). J. Volcanol. Geotherm. Res. 2003, 123, 81–94. [Google Scholar] [CrossRef]
- Oppenheimer, C.; Scaillet, B.; Martin, R.S. Sulfur degassing from volcanoes: Source conditions, surveillance, plume chemistry and Earth system impacts. Rev. Min. Geochem. 2011, 73, 363–421. [Google Scholar] [CrossRef]
- Parker, E.T.; Cleaves, H.J.; Dworkin, J.P.; Glavin, D.P.; Callahan, M.; Aubrey, A.; Lazcano, A.; Bada, J.L. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc. Natl. Acad. Sci. USA 2011, 108, 5526–5531. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of the solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: Theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Bounama, C.; Franck, S.; von Bloh, W. The fate of Earth’s ocean. Hydrol. Earth Syst. Sci. 2001, 5, 569–575. [Google Scholar] [CrossRef]
- Pinti, D.L. The origin and evolution of the oceans. In Lectures in Astrobiology, Vol. 1; Gargaud, M., Barbier, B., Martin, H., Reisse, J., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2005; pp. 83–112. [Google Scholar]
- Boger, D.L.; Keim, H.; Oberhauser, B.; Schreiner, E.P.; Foster, C.A. Total synthesis of HUN-7293. J. Am. Chem. Soc. 1999, 121, 6197–6205. [Google Scholar] [CrossRef]
- Xiang, Y.B.; Drenkard, S.; Baumann, K.; Hickey, D.; Eschenmoser, A. Chemie von α-aminonitrilen. Sondierungen über thermische umwandlungen von α-aminonitrilen. Helv. Chem. Acta 1994, 77, 2209–2250. [Google Scholar] [CrossRef]
- Léger, S.; Bayly, C.I.; Black, W.C.; Desmarais, S.; Falgueyret, J.P.; Massé, F.; Percival, M.D.; Truchon, J.F. Primary amides as selective inhibitors of cathepsin K. Bioorg. Med. Chem. Lett. 2007, 17, 4328–4332. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, J.C.; Denis, J.M. Synthèse d’imines linéaires non-stabilisées par réactions gaz-solide sous vide. Tetrahedron 1988, 44, 4431–4446. [Google Scholar] [CrossRef]
- Martin, T.A.; Causey, D.H.; Sheffner, A.L.; Wheeler, A.G.; Corrigan, J.R. Amides of N-acylcysteines as mucolytic agents. J. Med. Chem. 1967, 10, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. A research proposal on the origin of life. Orig. Life Evol. Biosph. 2003, 33, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Song, B.D.; Jencks, W.P. Aminolysis of benzoyl fluorides in water. J. Am. Chem. Soc. 1989, 111, 8479–8484. [Google Scholar] [CrossRef]
- Lam, A.K.Y.; Ryzhov, V.; O’Hair, R.A.J. Mobile protons versus mobile radicals: Gas-phase unimolecular chemistry of radical cations of cysteine-containing peptides. J. Am. Soc. Mass Spectrom. 2010, 21, 1296–1312. [Google Scholar] [CrossRef] [PubMed]
- Elkholy, Y.M.; Erian, A.W. An aminoimidazole and its utility in heterocyclic synthesis. Heteroatom Chem. 2003, 14, 503–508. [Google Scholar] [CrossRef]
- Buller, A.R.; Townsend, C.A. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, E653–E661. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.; Groziak, M.P.; Leonard, N.J. Nonenzymatic synthesis and properties of 5-aminoimidazole ribonucleotide (AIR). Synthesis of specifically 15N-labeled 5-aminoimidazole ribonucleoside (AIRs) derivatives. J. Am. Chem. Soc. 1990, 112, 4891–4897. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nitrile | π* Value | Reaction Rate | |
|---|---|---|---|
| H3N(+)CH2CN 1a protonated | −0.03632 | quick | |
| H2NCH2CN 1a | 0.01698 | No reaction? | |
| H3N(+)CH(CH3)CN | −0.03010 | quick | |
| H3CCONHCH2CN 1b | 0.00504 | slower | |
| H3N(+)CH2CONHCH2CN 1h | 0.00353 | quick | |
| H3N(+)CH2CONHCH2CONHCH2CN | −0.00547 | quick | |
| H3N(+)CH2CH2CN 1c | 0.00616 | slower | |
| H3N(+)CH2CH2CH2CN | 0.01556 | No reaction | |
| H3CCN | 0.03491 | No reaction | |
| Aspartic acid bis-nitrile 1e | αCN | −0.03566 | Reacts at αCN |
| βCN | −0.00500 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalayel, I.; Coulibaly, S.; Ly, K.D.; Milet, A.; Vallée, Y. The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean. Life 2018, 8, 47. https://doi.org/10.3390/life8040047
Shalayel I, Coulibaly S, Ly KD, Milet A, Vallée Y. The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean. Life. 2018; 8(4):47. https://doi.org/10.3390/life8040047
Chicago/Turabian StyleShalayel, Ibrahim, Seydou Coulibaly, Kieu Dung Ly, Anne Milet, and Yannick Vallée. 2018. "The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean" Life 8, no. 4: 47. https://doi.org/10.3390/life8040047
APA StyleShalayel, I., Coulibaly, S., Ly, K. D., Milet, A., & Vallée, Y. (2018). The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean. Life, 8(4), 47. https://doi.org/10.3390/life8040047