Abstract
The Strecker reaction of aldehydes with ammonia and hydrogen cyanide first leads to α-aminonitriles, which are then hydrolyzed to α-amino acids. However, before reacting with water, these aminonitriles can be trapped by aminothiols, such as cysteine or homocysteine, to give 5- or 6-membered ring heterocycles, which in turn are hydrolyzed to dipeptides. We propose that this two-step process enabled the formation of thiol-containing dipeptides in the primitive ocean. These small peptides are able to promote the formation of other peptide bonds and of heterocyclic molecules. Theoretical calculations support our experimental results. They predict that α-aminonitriles should be more reactive than other nitriles, and that imidazoles should be formed from transiently formed amidinonitriles. Overall, this set of reactions delineates a possible early stage of the development of organic chemistry, hence of life, on Earth dominated by nitriles and thiol-rich peptides (TRP).
1. Introduction
In ribosomes, peptide bonds are formed by the reaction of the amine group of an amino acid with an ester function. For non-ribosomal peptides, the amide formation involves the reaction of an amine on a thioester [1]. In both cases, mixed phosphoric carboxylic anhydrides are transiently formed. Esters, thioesters, and anhydrides are activated forms of the carboxylic acid function. Their intermediacy is mandatory and no significant C-N bond formation would occur directly from the reaction of an acid function with an amine [2]. What is true in today’s biology, was also true four billion years ago, when life was beginning its development in the terrestrial ocean. Activated derivatives had to be involved in the formation of prebiotic polymers. As a consequence, if acids were involved at some stage, a strong energy source was necessary. Nowadays, it is furnished by the cleavage of the triphosphate group of adenosine triphosphate [3].
Many simple aldehydes were probably present in the primitive ocean [4] and are plausible precursors for α-amino acids. Reacting with ammonia and hydrogen cyanide, they would have first given α-aminonitriles, which, upon hydrolysis, would have delivered amino acids (Figure 1) [5]. However, even though it is exothermic, the reaction of nitriles with water is a slow process [6]; so slow that, once formed in the ocean, aminonitriles would have had ample time to react with species more nucleophilic than water.
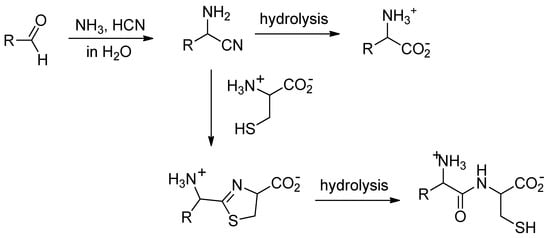
Figure 1.
Strecker reaction followed by condensation of the obtained aminonitrile with cysteine.
Nitriles are known to react with aminothiols to give thiazolines, which in turn can be hydrolyzed to mercaptoamides [7]. Starting from α-aminonitriles and cysteine, the expected products of this two-step process are dipeptides (Figure 1). In the early ocean, this could have been an efficient and selective process to thiol-containing dipeptides [8].
Compared to any activation process starting from acids, this nitrile scenario has the advantage of not necessitating any strong energy source. The acid does not need to be activated as it is delivered directly in an activated form by the Strecker reaction.
HCN has long been given an important role in prebiotic molecular evolution [9]. As it is largely distributed in space, having been observed in various regions, for instance, near carbon stars [10] and in a proto-planetary nebula [11], as well as in comets [12,13], it is highly possible that HCN was present on the early Earth. Furthermore, it has been postulated that it could have been formed when numerous asteroids struck our planet during the Late Heavy Bombardment [14]. It might have been produced photochemically in the atmosphere [15,16]. It was ejected from volcanoes [17] and submarine hydrothermal vents [18].
Hydrogen sulfide is another important small molecule in our hypothesis. It would have been necessary for the formation of cysteine. It has often been detected in space [19], inter alia in star forming regions [20], and in cold clouds [21], as well as in comets [22]. Furthermore, it is abundantly ejected from volcanoes [23,24], so there is no doubt that it was effectively present on the primitive Earth. Its presence permitted the synthesis of cysteine and homocysteine [25]. Homocysteine would have been obtained by a Strecker reaction starting from the addition of the product H2S onto acrolein (HSCH2CH2CHO). In a similar way, cysteine would have been synthesized from HSCH2CHO, itself possibly obtained from glycolaldehyde.
2. Experimental Section
Products (thiazolines, dipeptides…) were identified in reaction mixtures by NMR spectroscopy (1H and 13C) and mass spectrometry. No attempt at purifying them was made (except for 11 and 12).
NMR monitored reactions were run in D2O solutions, in NMR tubes. NMR apparatus: Bruker Avance III 400 or 500. Classically, NMR experiments were run at concentrations of 5 × 10−3 to 5 × 10−2 mol/L.
For the mass experiment, H2O was used as the solvent. High-resolution mass spectra were recorded on a Waters G2-S Q-TOF mass spectrometer or on a LTQ Orbitrap XL (Thermo Scientific) spectrometer. Low resolution ESI analysis was performed on an Amazon speed (Brucker Daltonics) IonTrap spectrometer.
(R)-2-((S)-1-amino-3-(methylthio)propyl)-4,5-dihydrothiazole-4-carboxamide (11)

Met-CN (168 mg, 1.29 mmol) was dissolved in 15 mL H2O. Cys-NH2.TFA (280 mg; 1.29 mmol) was added. The pH of the solution was adjusted to 8 by adding Na2CO3. The solution was stirred at 45 °C for 2.5 h. The aqueous phase was extracted three times with ethyl acetate. The organic layer was dried over Na2SO4, filtered, and concentrated under vacuum. After purification by silica gel chromatography (1–10% MeOH/DCM), the thiazoline 11 was obtained as an orange oil (16% yield).
HRMS (ESI) for C8H16ON3S2: calc. m/z = 234.0735, Found m/z = 234.0740 [M + H]+. 1H-NMR (D2O, 400 MHz) (δ, ppm): 5.08 (1H, t, J = 8.98 Hz, CH), 3.95 (1H, t, J = 6.57 Hz, CH), 3.65 (1H, t, J = 10.82 Hz, CH2), 3.46 (1H, dd, J = 11.30; 8.20, CH2), 2.56 (2H, t, J = 7.08 Hz, CH2), 2.07 (3H, s, CH3), 1.97 (2H, sep, J = 7.0, CH2). 13C-NMR (D2O, 100 MHz) (δ, ppm): 182.99, 176.14, 77.00, 52.93, 34.89, 34.43, 29.18, 14.12.
(4R)-2-(1-amino-2-methylpropyl)-4,5-dihydrothiazole-4-carboxamide (12)

Val-CN.HCl (35 mg, 0.26 mmol) was dissolved in 5 mL H2O. Cys-NH2.TFA (57 mg; 0.26 mmol) was added. The solution was adjusted to pH = 7 by adding Na2CO3. The solution was stirred at 45 °C for 24 h. The aqueous phase was extracted three times with ethyl acetate. The organic layer was dried over Na2SO4, filtered, and concentrated under vacuum. After purification by silica gel chromatography (1–10% MeOH/DCM), the thiazoline 12 was obtained as a yellow oil (30% yield).
HRMS (ESI) for C8H16ON3S: calc. m/z = 202.1014, Found m/z = 202.1016 [M + H] +. 1H-NMR (D2O, 500 MHz) (δ, ppm): 4.40 (1H, m, CH), 3.75 (1H, dd, 5.68; 2.59 Hz, CH), 2.70–2.99 (2H, m, CH2), 2.12 (1H, sep, J = 6.60, CH), 0.85–0.94 (6H, m, CH3). 13C-NMR (D2O, 125 MHz, both isomers were observed) (δ, ppm): 174.02–173.74, 169.70–169.43, 58.66–58.38, 55.65–55.63, 30.01–29.94, 25.30–25.02, 17.79–17.62, 16.85–16.65.
Theoretical calculations were carried out using the Gaussian09, Revision D.01 software. All the geometries were optimized using the B3LYP functional in conjunction with the 6-31g(d,p) basis set and the water solvent effects were described by using the polarizable continuum model (PCM), namely IEFPCM (integral equation formalism PCM) [26]. These optimizations were followed by a frequency calculation at the same level to ensure that the geometry was indeed a real minimum, i.e., all the second derivatives were positive.
3. Results
We first studied the reaction of aminoacetonitrile (GlyCN 1a, the nitrile derivative of glycine) with cysteine. Reactions were conducted in D2O solutions and followed by NMR spectroscopy. Representative 1H NMR spectra are shown in Figure 2. Formation of the expected thiazoline ring 2a was evidenced by the apparition of signals at ca. 5 ppm (a triplet-like dd), and from 3.4 to 3.7 ppm (2 dd). After some time, new signals grew, including a triplet at 4.4 ppm and a thin doublet-like signal at ca. 2.9 ppm, both characteristic of Gly-Cys 3a. We repeated this experiment many times, generally at a concentration of 5 × 10−3 to 5 × 10−2 mol/L, for practical NMR measurements. However, we also tested it at 3 10−4 mol/L, a concentration at which 2a and 3a were also obtained.
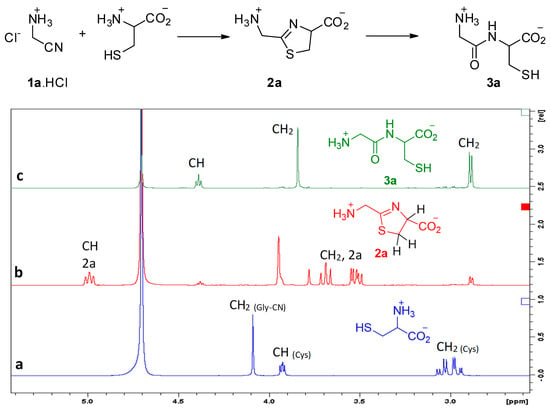
Figure 2.
Reaction of aminoacetonitrile with cysteine, (a) mixture of starting materials, (b) mostly 2a, (c) GlyCys 3a. Conditions: room temperature, pH 6.5, concentration 10−2 mol/L.
We have studied the influence of the pH on these reactions. The results are summarized in Figure 3. The ring formation is quicker under basic conditions. We believe that under such conditions, the thiol function is deprotonated, giving the more nucleophilic thiolate species. Under an acidic condition, the nucleophilic species is probably the thiol itself. The hydrolysis step is quicker under acidic conditions. This probably implies that the thiazoline ring is activated through protonation of the double bonded nitrogen atom before H2O addition.
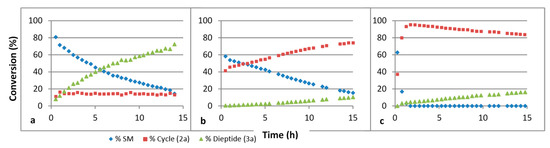
Figure 3.
Evolution of a mixture of GlyCN and cysteine in D2O at 45 °C followed by 1H NMR, at various pH’s. (a) pH 4, (b) pH 6, (c) pH 8. SM: starting materials. Concentration 4 × 10−2 mol/L.
We have also tested these reactions at 24 °C and 70 °C. Not surprisingly the process is quicker at a higher temperature, but also goes well at room temperature.
The conditions in the ocean four billion years ago are not precisely known. However, water was probably still hotter than now [27] and the presence of large amounts of CO2 in the atmosphere might imply that it was slightly acidic (nowadays, ocean’s pH is 8.1) [28]. Taking these considerations into account, we chose a temperature of 45 °C and a pH of ca. 5.5–6.5 as standard conditions.
Under such conditions, we observed no reaction between aminoacetonitrile and any other proteinogenic amino acid that we tested (glycine, alanine, serine, methionine, aspartic acid, histidine, and lysine). It is worth noting that serine did not react. It appears that its alcohol function is not nucleophilic enough to attack the CN triple bond. Hence, the presence of a thiol function is mandatory. Indeed, homocysteine (Hcy) did react with a reaction rate similar to that observed with cysteine. In this case, the intermediate is the six-membered ring 4a, and the final product is Gly-Hcy 5a (Figure 4).
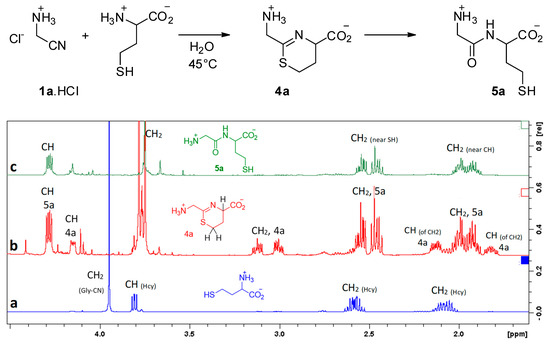
Figure 4.
Reaction of homocysteine with GlyCN at 45 °C, pH = 6.5, 10−2 mol/L. 1H NMR’s show: (a) starting mixture, (b) reaction mixture after 6 h (4a/5a = 3/7), (c) after 24 h.
Some other representative results are presented in Figure 5. They show that the acid function of cysteine can be replaced by a primary or secondary amide. When Cys-Gly was used, the tripeptide Gly-Cys-Gly 9 was obtained with a very good conversion. N-Acetyl aminoacetonitrile 1b, which can be considered as a model for any other N-acyl acetonitrile, including cyano-terminated peptides, also reacted with a good rate (Figure 6a). In contrast, the reaction was slower when aminoacetonitrile was replaced by β-aminopropionitrile 1c (Figure 7). In these two last examples, the hydrolysis step was quick. No reaction was observed with the γ-nitrile of glutamic acid 1d [29]. The selectivity in favor of α-aminonitriles was also exemplified when the bis-nitrile derived from aspartic acid 1e [30] was used. In this case, only the α-aminonitrile reacted, giving the corresponding thiazoline 2e, which was stable under these conditions (Figure 6c,d).
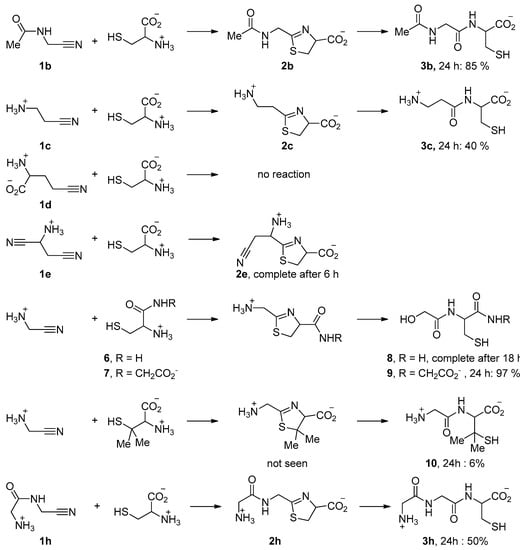
Figure 5.
Some reactions of aminothiols with nitriles (45 °C, pH 5–6). The solvent was D2O. Reactions were monitored by 1H NMR.
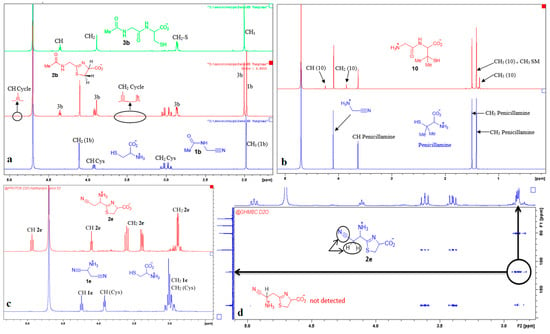
Figure 6.
NMR spectra recorded during representative aminothiol + aminonitrile reactions. (a) Reaction of cysteine with N-acetyl aminoacetonitrile; (b) reaction of penicillamine with GlyCN; (c) reaction of aspartic acid bis-nitrile with cysteine; (d) 2D experiment demonstrating the regioselectivity of this last reaction towards α-nitrile.
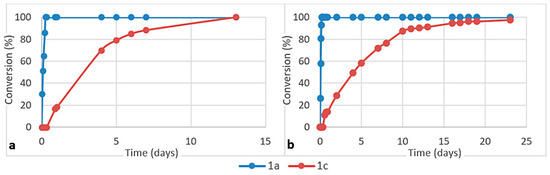
Figure 7.
Consumption of aminoacetonitrile 1a and β-aminopropionitrile 1c in competition reactions with (a) cysteine and (b) homocysteine (ratio 1a/1c/amino acid 1/1/2, pH ca. 6, 45 °C).
Finally, we also tested the reactivity of penicilamine, a sterically hindered aminothiol. In this case, the reaction was very slow, probably because of the bulkiness of the gem-dimethyl substituents. Furthermore, the only detected product was the final dipeptide 10 (Figure 6b). This might be due to the electron donating property of the methyl groups, making the nitrogen atom of the intermediate thiazoline ring more basic. Protonation of this nitrogen atom would thus be easier, hence the hydrolysis step quicker.
In order to explain the observed selectivity, we calculated the level of the π* orbital of a series of nitriles (Table 1).

Table 1.
Calculated level of the π* orbital of various nitriles.
The lowest calculated orbital was that of the protonated form of aminoacetonitrile. Such a level would explain its greater reactivity compared to other nitriles. Noticeably, the non-protonated form of aminoacetonitrile is predicted to be much less reactive and so is probably not involved in the reaction mechanism. Also, the π* orbital of β-propionitrile is higher (it is less reactive) and the simplest γ-aminonitrile is predicted to be even less reactive (no reaction from glutamic nitrile). In contrast, α-substitution of aminoacetonitrile, as in α-propionitrile, should not significantly alter its reactivity.
We studied this substitution effect using the nitriles derived from two other amino acids (Figure 8). l-Methionine nitrile 1f [31] was prepared from N-protected L-methionine in a three-step process. Valine nitrile 1g, was prepared as a racemic mixture using a Strecker reaction from the corresponding aldehyde [32].
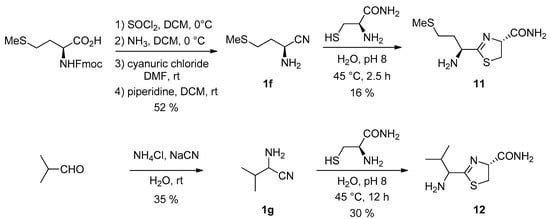
Figure 8.
Synthesis of MeCN 1f and ValCN 1g and their reaction with cysteine amide.
Their reaction with cysteine amide [33] was studied (Figure 8). In these cases, we were able to isolate the intermediate cycles in pure form (as a 1/1 mixture of diastereoisomers from racemic ValCN). The deceptive isolated yields, despite the slightly basic conditions we used, which should have slowed the hydrolysis step, were probably due to important hydrolysis during column chromatography on silica. In addition, we found that the rate of hydrolysis in water of the valine-derived thiazoline 12 was much slower than the one of the methionine derivative 11 (and of the simplest Glycine derivative 2a). This is probably due to the presence of the bulky isopropyl group in 12.
On the basis of our experiments, we propose that AA-Cys and AA-Hcy dipeptides were over-represented in the primitive ocean (compared to non-thiol-containing dipeptides).
These dipeptides are thiols and as such, could be major players in a “thioester world” [34]. Indeed, when we mixed Gly-Cys 3a (obtained from a 1 to 1 mixture of GlyCN and cysteine) with an excess of GlyCN in D2O solution at 45 °C (Figure 9), a peak was observed at 194.16 ppm in the 13C NMR of the reaction mixture (Figure 10). Such a chemical shift is characteristic of the thioester function. We believe that it belongs to compound 14. We also noticed the formation of glycine amide 15. These products would both derive from the first formed C=N double-bonded addition product 13. The thioester was partly hydrolyzed to give glycine, but we were also able to characterize, among the reaction products, the amidonitrile Gly-GlyCN 1h (13C NMR: 27.58, 40.34, 116.74, 167.58 ppm), meaning that the thioester reacted with the non-protonated amino group of GlyCN (which is possible because of the low pKa of GlyCN: 5.55 [35]). For instance, in an experiment in which we used globally 4 eq. of GlyCN (relative to cysteine), after two days at 45 °C, the observed GlyOH/GlyNH2/GlyGlyCN ratio was found to be 21/37/42. This demonstrates that Gly-Cys is able to promote the formation of a peptide bond from a nitrile. Similar results were obtained for Gly-Hcy. In addition, as our theoretical calculations predicted (Table 1), when Gly-GlyCN 1h was mixed with cysteine, Gly-Gly-Cys 3h [36] was readily formed (Figure 5), demonstrating that not only dipeptides, but also tripeptides, could have been formed by this process in the primitive ocean.
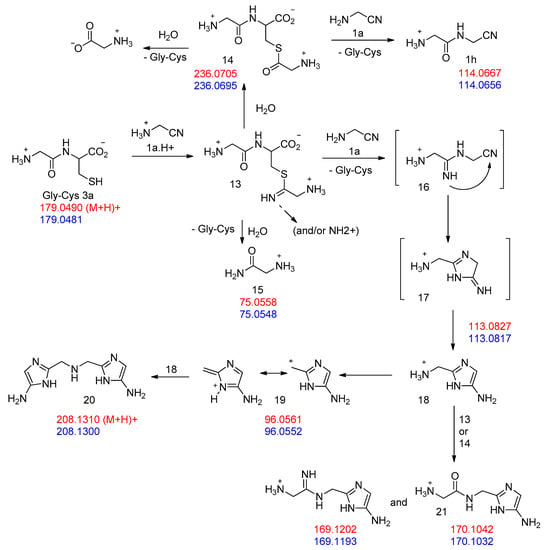
Figure 9.
Proposed pathways in the reaction of excess GlyCN 1a with GlyCys 3a. Mass attribution (red, calculated; blue, found).
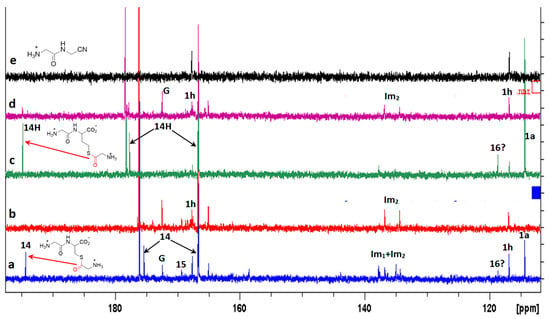
Figure 10.
13C NMR spectra recorded during the reaction of an excess GlyCN 1a with GlyCys 3a (from 1a. HCl + cysteine) or GlyHcy 5a (from 1a. HCl + homocysteine) at 45 °C, pH 6.5. (a) with 3a 20 h after mixing 1a. HCl and cysteine; (b) after 70h; (c) with 5a 20 h after mixing 1a. HCl and homocysteine; (d) after 70h; (e) reference spectrum of 1h. G: glycine. Peaks corresponding to at least two products were detected near 135–140 ppm. They might correspond to two different imidazoles (named Im1 and Im2). 14H: the homocysteine thioester analogue of 14. Big peaks at 166.65 (166.66) and 176.07 (178.33) belong to 3a (and 5a). One peak of both 14 and 14H sticks to the foot of the 166.6 ppm peak of 3a and 5a.
However, the formation of other products was also evidenced in the reaction of cysteine with excess GlyCN. Thus, in the 13C NMR spectra, peaks at 130–140 ppm were observed (Figure 10). The mass spectrum of a reaction mixture in water also showed the formation of various products (Figure 11, see Supplementary Materials for complete spectrum and further mass attributions). This mass spectrum first confirmed the presence of Gly-GlyCN 1h (protonated, found 114.0656, calcd 114.0667). Another mass was detected at 113.0817. It could be attributed to the amidine 16 (calcd 113.0827). However, in accordance with the observed 13C NMR of the mixture, we propose that this amidine cyclized and that this mass peak should be attributed to the imidazole 18. Indeed, the cyclization of an amidinonitrile similar to 16 into an aminoimidazole (5-amino-2-methyl-1H-imidazole) has already been reported [37]. At least some of the other products observed in the mass spectrum would be evolution products of 18. For instance, this imidazole could lose ammonia to give the stabilized cation 19 that would in turn react with 18 (in its free amine form) to yield the bis-imidazole 20 (M+H, found 208.1300, calcd 208.1310). 18 itself could react with thiolester 14 to give the amide 21. Other structures are possible (see Supplementary Materials).
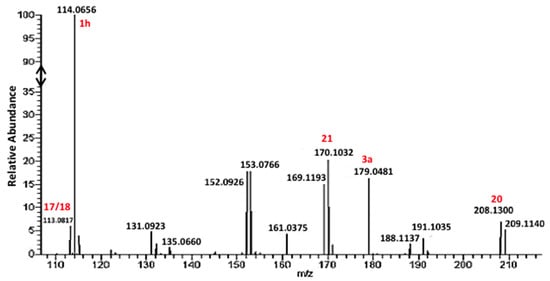
Figure 11.
Mass spectrum of a reaction of an excess GlyCN 1a with GlyCys.
In order to further ascertain the cyclization of the imino-compound 16, we calculated its stability in comparison to cyclized forms. We used the strategy described previously for Table 1 results, with the 6-31+G(d,p) basis set (Figure 12). Not surprisingly, 18 was calculated to have free enthalpy 12.7 kcal/mol lower than 16 and 6.9 kcal/mol more stable than 17.
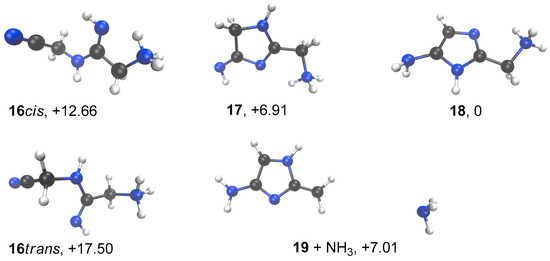
Figure 12.
Minimized conformation for compounds 16 to 19. B3LYP/6-31+G** with a continuum to mimic the solvent effect of water. ∆G’s relative to 18 in kcal/mol.
Interestingly, it was found that the dissociation of 18 into 19 + NH3 only costs around 7 kcal/mol. The ΔH of dissociation is nearly 19 kcal/mol (18.67 kcal/mol), but due to the dissociative character of the process, the ∆G value drops to 7.01 kcal/mol. This process does not show a well-defined TS. Thus, we think that the formation of cation 19 proposed in Figure 9 is a plausible event.
We studied more precisely the cyclization step from 16 to 17 (Figure 13). H3O+ was used to promote the reaction and to give a proton to the nitrogen atom of the nitrile group, which becomes part of the exocyclic imine of 17. Two explicit molecules of water were introduced, in addition to the water continuum. It appeared that the cyclization step should be exocyclic and quick, with a low level TS (activation energy of 6.6 kcal/mol). This is another confirmation that the compound of mass 113.0817 we observed was not 16, but indeed the imidazole 18 (resulting from a simple proton migration from 17). It is noticeable that similar calculations for the potential cyclization of the amide 1h into an oxazole indicated that this reaction should not happen. Indeed, we never observed it experimentally. In sharp contrast with 16, 1h (GlyGlyCN) is stable.
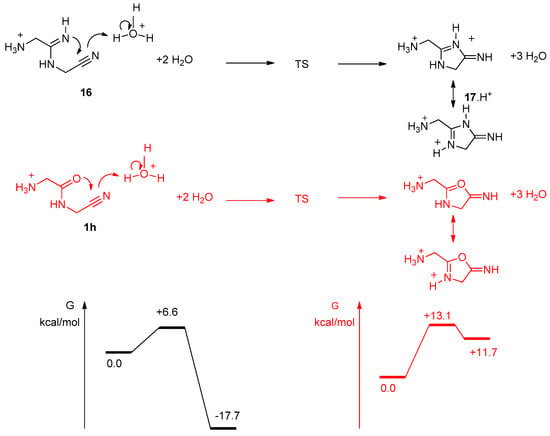
Figure 13.
Reaction pathways for the cyclisation step from 16 (black), and 1h (red). Level of calculation B3LYP/6-31+G**/SCRF(water).
4. Conclusions
A world containing small peptides and heterocycles, based on the chemistry of thiols and nitriles, can be delineated. It could have persisted as long as a significant amount of HCN was present in the ocean and permitted the synthesis of aminonitriles from aldehydes. In this “cyano-sulfidic” world [14], thiol-containing peptides would have been the most important molecules. We propose to name it the “Thiol Rich Peptide (TRP) world” [8]. In such a world, not only dipeptides, but also tripeptides, would have been formed. For instance, any dipeptide nitrile AA1-AA2CN (the simplest example being Gly-GlyCN) produced from the reaction of H2NAA2CN with a thioester of AA1, would react with cysteine to give the tripeptide AA1-AA2-Cys, and with homocysteine to give AA1-AA2-Hcy. Could some of these tripeptides have been the very first catalytic triades [38]? Indeed, we have demonstrated that even dipeptides like GlyCys (but not monomeric cysteine) are able to promote the formation of peptide bonds from nitriles. They are also able to promote the formation of imidazoles. Such heterocycles play an important role in today’s biochemistry. Of special interest is the simplest aminoimidazole, which, as its ribonucleotide derivative (AIR) [39], is an intermediate in the de-novo synthesis of inosine monophosphate (IMP), hence of purine nucleotides. Thus, imidazoles could have established a bridge from peptides to nucleic acids.
Supplementary Materials
The following are available online at http://www.mdpi.com/2075-1729/8/4/47/s1. HRMS of 1a + cysteine and + homocysteine reaction mixtures. NMR spectra of 11 and 12. ESI mass spectrum and 13C NMR of a cysteine + excess GlyCN reaction mixture. Additional data for theoretical calculations.
Author Contributions
Conceptualization, Y.V.; methodology, I.S., A.M. and Y.V.; validation, Y.V. and A.M.; investigation, I.S., S.C. and K.D.L.; writing—original draft preparation, Y.V.; writing—review and editing, Y.V.; visualization, Y.V. and A.M.; supervision, Y.V.; project administration, Y.V.; funding acquisition, Y.V. and A.M.
Funding
This research was funded by the Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003), and the French National Research Agency (Investissements d’Avenir ANR-15-IDEX-02).
Acknowledgments
The authors thank le Ministère des Affaires Etrangères (Consulat Général de France à Jérusalem and Ambassade de France au Mali) for I.S. and S.C. grants, and l’Université de Bamako, Mali.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hashimoto, S.I. Ribosome-independent peptide synthesis and their application to dipeptide production. J. Biol. Macromol. 2008, 8, 28–37. [Google Scholar]
- Chalmet, S.; Harb, W.; Ruiz-Lòpez, M.F. Computer simulation of amide bond formation in aqueous solution. J. Phys. Chem. A 2001, 105, 11574–11581. [Google Scholar] [CrossRef]
- Gajewski, E.; Steckler, D.K.; Goldberg, R.N. Thermodynamics of the hydrolysis of adenosine 5'-triphosphate to adenosine 5′-diphosphate. J. Biol. Chem. 1988, 261, 12733–12737. [Google Scholar]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic systems chemistry: New perspectives for the origins of life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef] [PubMed]
- Strecker, A. Ueber die künstliche bildung der milchsäure und einen neuen, dem glycocoll homologen körper. Liebigs Ann. Chem. 1850, 75, 27–45. [Google Scholar] [CrossRef]
- Guthrie, J.P.; Yim, J.C.H.; Wang, Q. Hydration of nitriles: An examination in terms of no barrier theory. J. Phys. Org. Chem. 2014, 27, 27–37. [Google Scholar] [CrossRef]
- Krimmer, H.P.; Drauz, K.; Kleemann, A. Umsetzung von β-mercapto-α-aminosäuren mit nitrilen. Chemiker-Zeitung 1987, 111, 357–361. [Google Scholar]
- Vallée, Y.; Shalayel, I.; Dung, L.K.; Raghavendra Rao, K.V.; de Paëpe, G.; Märker, K.; Milet, A. At the very beginning of life on Earth: The thiol rich peptide (TRP) world hypothesis. Int. J. Dev. Biol. 2017, 61, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Hagan, W.J., Jr. HCN and chemical evolution: The possible role of cyano compounds in prebiotic synthesis. Tetrahedron 1984, 1093–1120. [Google Scholar] [CrossRef]
- Schilke, P.; Menten, K.M. Detection of a second, strong submillimeter HCN laser line toward carbon stars. Astrophys. J. 2003, 583, 446–450. [Google Scholar] [CrossRef]
- Thorwirth, S.; Wyrowski, F.; Schilke, P.; Menten, K.M.; Brünken, S.; Müller, H.S.P.; Winnewisser, G. Detection of HCN direct l-type transitions probing hot molecular gas in the proto-planetary nebula CRL 618. Astrophys. J. 2003, 586, 338–343. [Google Scholar] [CrossRef]
- Irvine, W.M.; Dickens, J.E.; Lovell, A.J.; Schoerb, F.P.; Senay, M.; Bergin, E.A.; Jewitt, D.; Matthews, H.E. The HNC/HCN ratio in comets. Earth Moon Planets 1997, 78, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Schloerb, F.P.; Kinzel, W.M.; Swade, D.A.; Irvine, W.M. Observations of HCN in comet P/Halley. Astron. Astrophys. 1987, 187, 475–480. [Google Scholar] [PubMed]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nature Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Zahnle, K.J. Photochemistry of methane and the formation of hydrocyanic acid (HCN) in the Earth’s early atmosphere. J. Geophys. Res. 1986, 91, 2819–2834. [Google Scholar] [CrossRef]
- Martin, R.S.; Mather, T.A.; Pyle, D.M. Volcanic emissions and the early Earth atmosphere. Geochim. Cosmochim. Acta 2007, 71, 3673–3685. [Google Scholar] [CrossRef]
- Mukhin, L.M. Volcanic processes and synthesis of simple organic compounds on primitive Earth. Orig. Life Evol. Biosph. 1976, 7, 355–368. [Google Scholar] [CrossRef]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Thaddeus, P.; Kutner, M.L.; Penzias, A.A.; Wilson, R.W.; Jefferts, K.B. Interstellar hydrogen sulfide. Astrophys. J. 1972, 176, L73–L76. [Google Scholar] [CrossRef]
- Minh, Y.C.; Ziurys, L.M.; Irvine, W.M.; McGonagle, D. Abundances of hydrogen sulfide in star-forming regions. Astrophys. J. 1991, 366, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Minh, Y.C.; Irvine, W.M.; Ziurys, L.M. Detection of interstellar hydrogen sulfide in cold, dark clouds. Astrophys. J. 1989, 345, L63–L66. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, P.; Meier, R.; Krankowsky, D.; Hodges, R.R. Methanol and hydrogen sulfide in comet P/Halley. Astron. Astrophys. 1994, 288, 315–329. [Google Scholar]
- Carapezza, M.L.; Badalamenti, B.; Cavarra, L.; Scalzo, A. Gas hazard assessment in a densely inhabited area of Colli Albany Volcano (Cava dei Selci, Roma). J. Volcanol. Geotherm. Res. 2003, 123, 81–94. [Google Scholar] [CrossRef]
- Oppenheimer, C.; Scaillet, B.; Martin, R.S. Sulfur degassing from volcanoes: Source conditions, surveillance, plume chemistry and Earth system impacts. Rev. Min. Geochem. 2011, 73, 363–421. [Google Scholar] [CrossRef]
- Parker, E.T.; Cleaves, H.J.; Dworkin, J.P.; Glavin, D.P.; Callahan, M.; Aubrey, A.; Lazcano, A.; Bada, J.L. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc. Natl. Acad. Sci. USA 2011, 108, 5526–5531. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of the solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: Theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Bounama, C.; Franck, S.; von Bloh, W. The fate of Earth’s ocean. Hydrol. Earth Syst. Sci. 2001, 5, 569–575. [Google Scholar] [CrossRef]
- Pinti, D.L. The origin and evolution of the oceans. In Lectures in Astrobiology, Vol. 1; Gargaud, M., Barbier, B., Martin, H., Reisse, J., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2005; pp. 83–112. [Google Scholar]
- Boger, D.L.; Keim, H.; Oberhauser, B.; Schreiner, E.P.; Foster, C.A. Total synthesis of HUN-7293. J. Am. Chem. Soc. 1999, 121, 6197–6205. [Google Scholar] [CrossRef]
- Xiang, Y.B.; Drenkard, S.; Baumann, K.; Hickey, D.; Eschenmoser, A. Chemie von α-aminonitrilen. Sondierungen über thermische umwandlungen von α-aminonitrilen. Helv. Chem. Acta 1994, 77, 2209–2250. [Google Scholar] [CrossRef]
- Léger, S.; Bayly, C.I.; Black, W.C.; Desmarais, S.; Falgueyret, J.P.; Massé, F.; Percival, M.D.; Truchon, J.F. Primary amides as selective inhibitors of cathepsin K. Bioorg. Med. Chem. Lett. 2007, 17, 4328–4332. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, J.C.; Denis, J.M. Synthèse d’imines linéaires non-stabilisées par réactions gaz-solide sous vide. Tetrahedron 1988, 44, 4431–4446. [Google Scholar] [CrossRef]
- Martin, T.A.; Causey, D.H.; Sheffner, A.L.; Wheeler, A.G.; Corrigan, J.R. Amides of N-acylcysteines as mucolytic agents. J. Med. Chem. 1967, 10, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. A research proposal on the origin of life. Orig. Life Evol. Biosph. 2003, 33, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Song, B.D.; Jencks, W.P. Aminolysis of benzoyl fluorides in water. J. Am. Chem. Soc. 1989, 111, 8479–8484. [Google Scholar] [CrossRef]
- Lam, A.K.Y.; Ryzhov, V.; O’Hair, R.A.J. Mobile protons versus mobile radicals: Gas-phase unimolecular chemistry of radical cations of cysteine-containing peptides. J. Am. Soc. Mass Spectrom. 2010, 21, 1296–1312. [Google Scholar] [CrossRef] [PubMed]
- Elkholy, Y.M.; Erian, A.W. An aminoimidazole and its utility in heterocyclic synthesis. Heteroatom Chem. 2003, 14, 503–508. [Google Scholar] [CrossRef]
- Buller, A.R.; Townsend, C.A. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, E653–E661. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.; Groziak, M.P.; Leonard, N.J. Nonenzymatic synthesis and properties of 5-aminoimidazole ribonucleotide (AIR). Synthesis of specifically 15N-labeled 5-aminoimidazole ribonucleoside (AIRs) derivatives. J. Am. Chem. Soc. 1990, 112, 4891–4897. [Google Scholar] [CrossRef]













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).