Chemical Diversity of Metal Sulfide Minerals and Its Implications for the Origin of Life


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
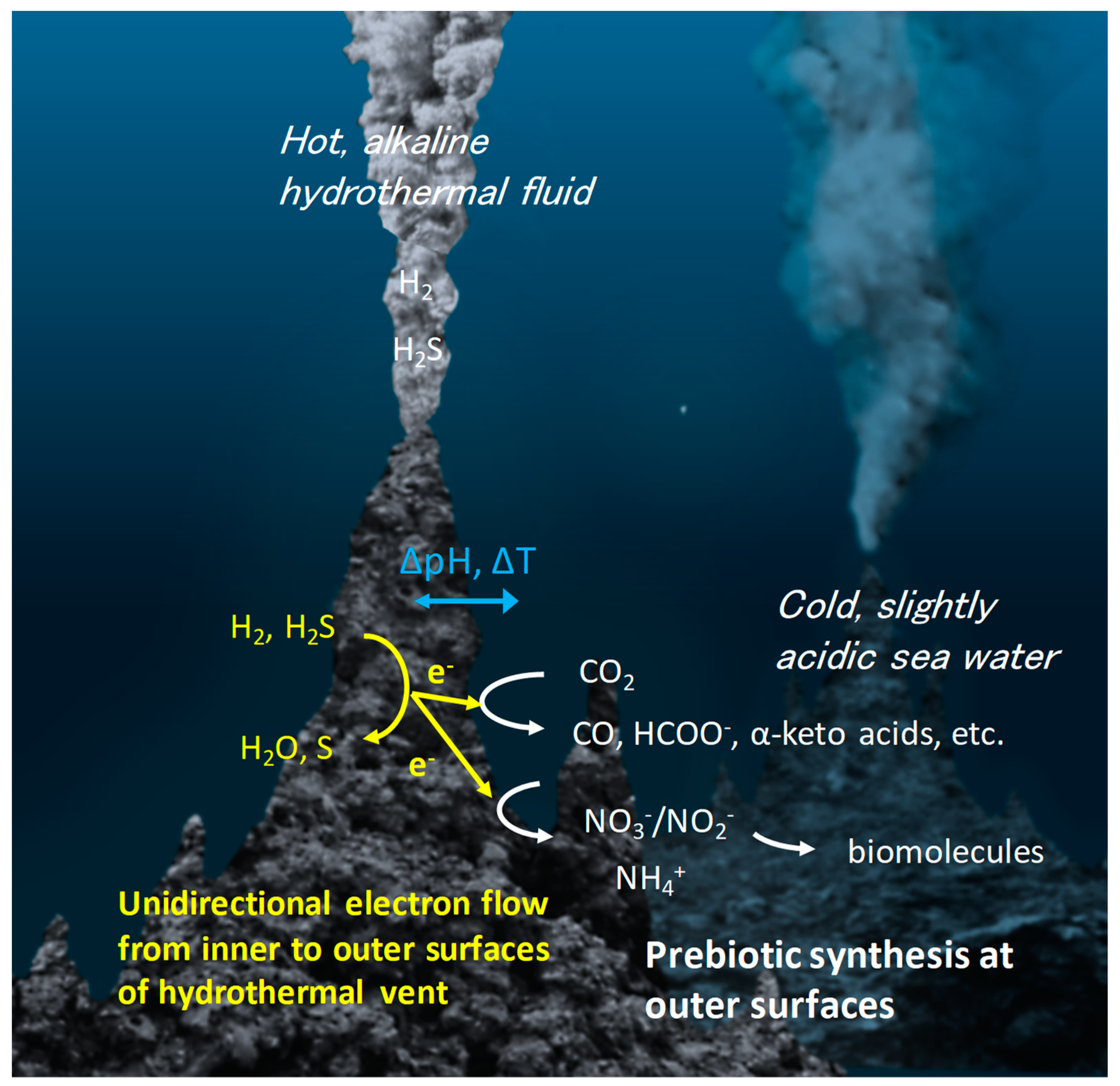
1. Introduction
2. Catalysis on Metal Sulfides Relevant to Prebiotic Chemistry
2.1. Thermal Catalysis on Metal Sulfides
2.2. Electrocatalysis on Metal Sulfides
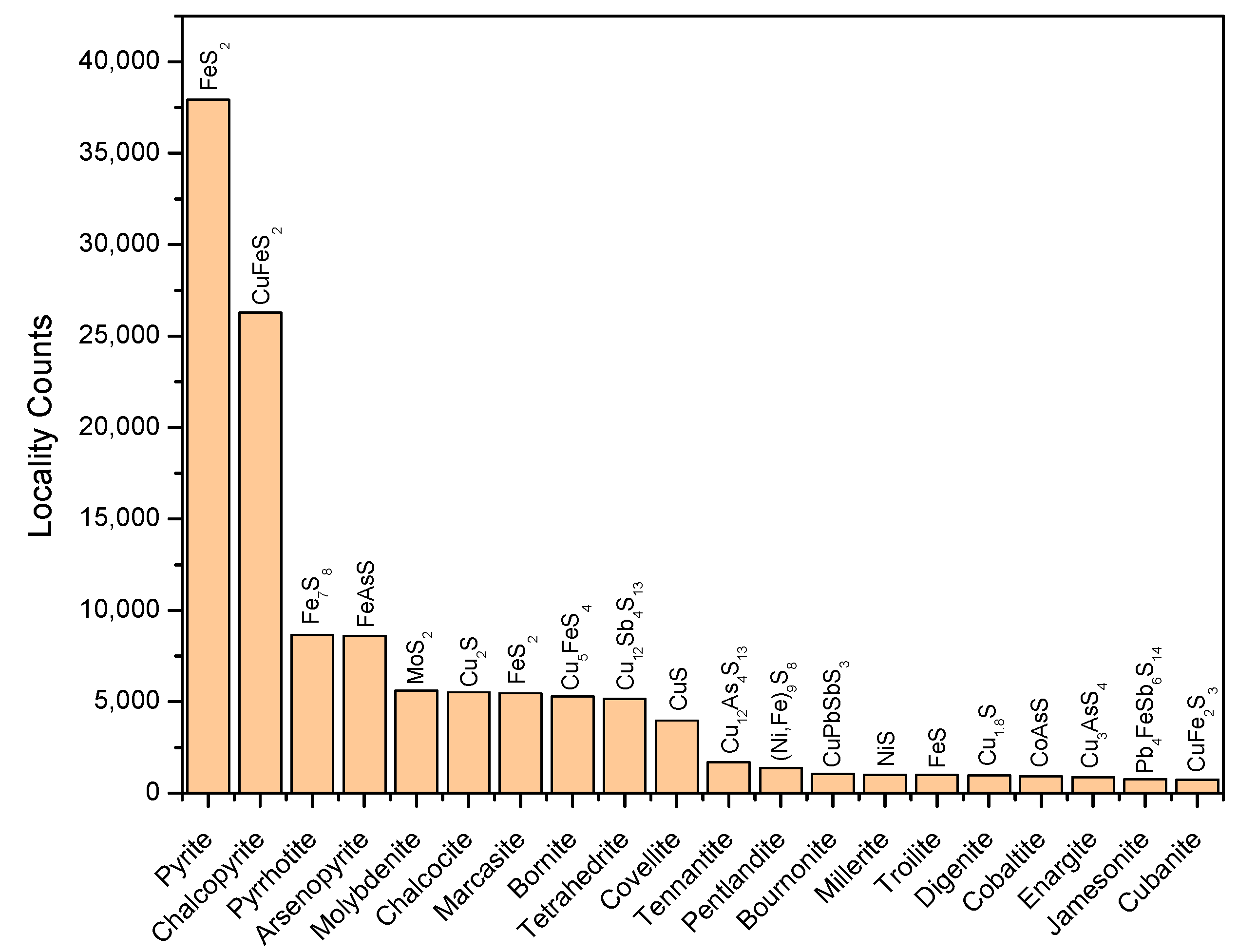
3. Chemical Diversity of Metal Sulfides in Mineralogy Libraries
- (1)
- Locality frequency. This parameter reflects the availability of minerals in natural environments. Although detection limits and metal-specific biases reflecting the metal’s economic value may exist, an estimate of the probability of natural occurrence is useful for evaluating the feasibility of mineral-promoted prebiotic reactions.
- (2)
- Multiple metal composition. Naturally observed metal sulfides are commonly coupled to more than one transition metal and occasionally exhibit ternary compositions. Because cation substitutions and the presence of dual metal surface sites influence catalytic activities of minerals, as described in Section 2.2, Section 4 and Section 5, metal-specific behaviors and tendency to form multiple compositions are important factors for screening potential mineral catalysts.
- (3)
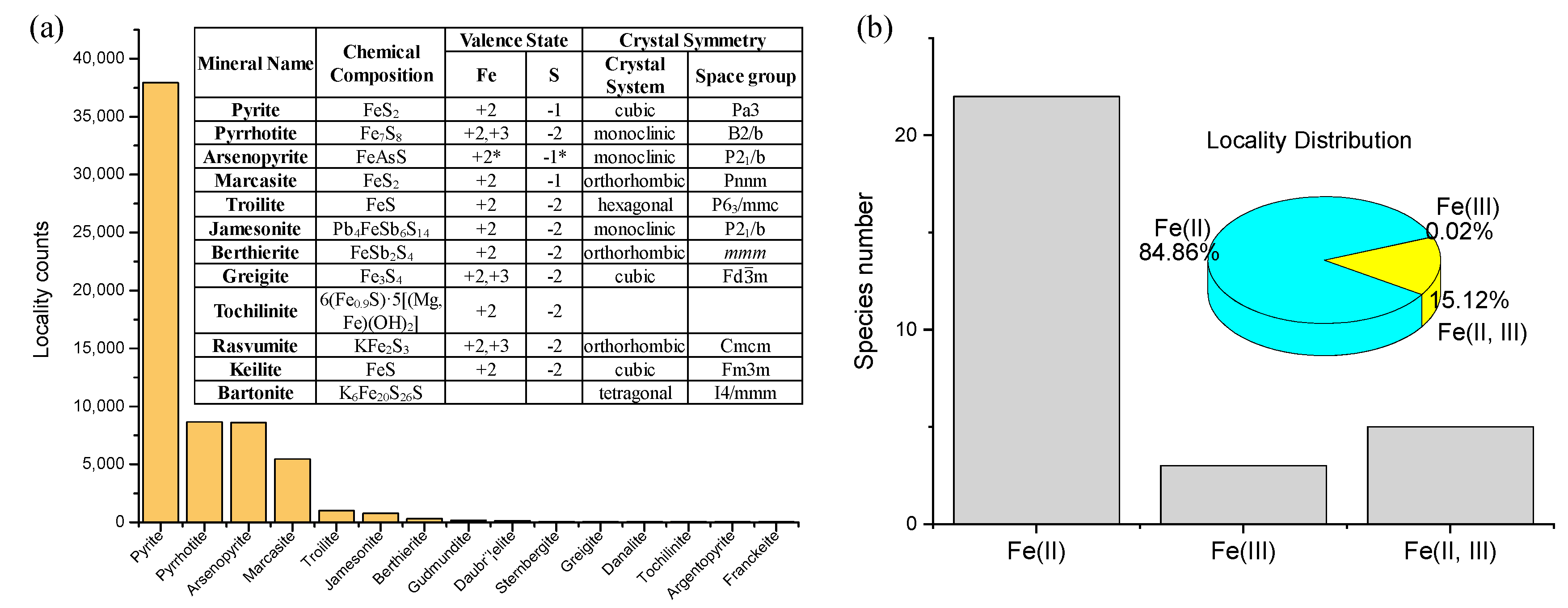
- Crystal structure and valence state of dominating elements. These two properties, particularly the valence state, control the electronic characteristics of the mineral. Valence state information is available for 89% of the localities of metal sulfides in the database (http://rruff.info/ima/).
3.1. Distribution of Transition Metal Sulfides
3.2. Single-Metal Sulfides
3.3. Binary- and Ternary-Metal Sulfides
3.4. X-ray Amorphous Metal Sulfides
4. Impact of Metal Sulfide Chemical Diversity on Catalysis
4.1. CO2 Reduction
4.2. Ammonia Synthesis from Nitrate/Nitrite or Dinitrogen Reduction
4.3. Hydrogen Evolution and Oxidation Reactions
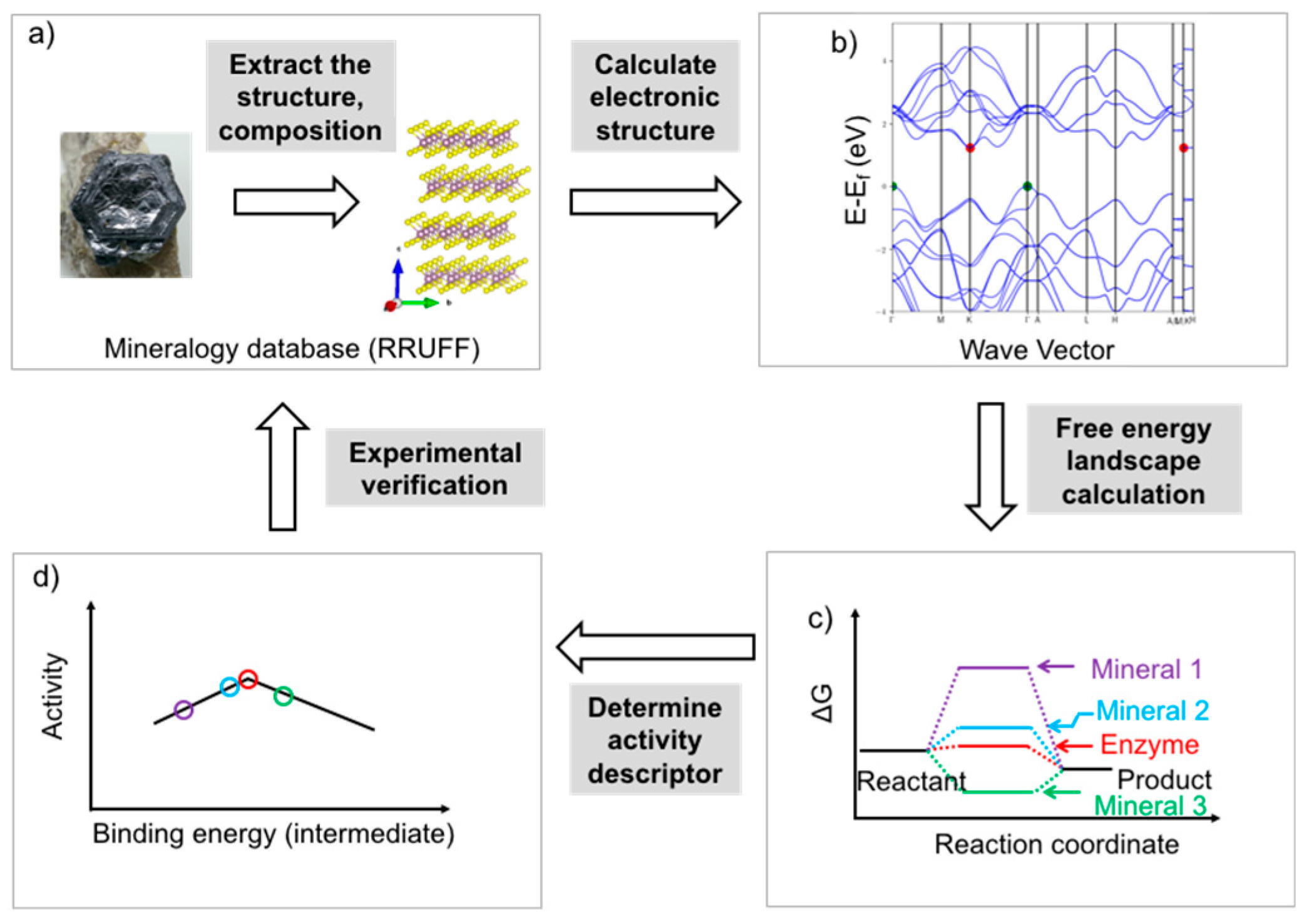
5. Rational Screening of Catalysts
6. Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kitadai, N.; Maruyama, S. Origins of building blocks of life: A review. Geosci. Front. 2018, 9, 1117–1153. [Google Scholar] [CrossRef]
- James Cleaves, H., II; Michalkova Scott, A.; Hill, F.C.; Leszczynski, J.; Sahai, N.; Hazen, R. Mineral–organic interfacial processes: Potential roles in the origins of life. Chem. Soc. Rev. 2012, 41, 5502. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Cody, G.D. Geochemical Connections to Primitive Metabolism. Elements 2005, 1, 139–143. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J.; Mellersh, A.R. On the Dissipation of Thermal and Chemical Energies on the Early Earth. In Natural and Laboratory-Simulated Thermal Geochemical Processes; Springer: Dordrecht, The Netherlands, 2003; pp. 325–388. [Google Scholar]
- Wächtershäuser, G. On the Chemistry and Evolution of the Pioneer Organism. Chem. Biodivers. 2007, 4, 584–602. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Before Enzymes and Templates: Theory of Surface Metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [PubMed]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. Lond. 1997, 154, 377–402. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J. The onset and early evolution of life. In Memoir 198: Evolution of Early Earth’s Atmosphere, Hydrosphere, and Biosphere—Constraints from Ore Deposits; Geological Society of America: Boulder, CO, USA, 2006; pp. 1–32. [Google Scholar]
- Lane, N.; Martin, W.F. The origin of membrane bioenergetics. Cell 2012, 151, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y.; Galperin, M.Y. On the origin of life in the Zinc world. 2. Validation of the hypothesis on the photosynthesizing zinc sulfide edifices as cradles of life on Earth. Biol. Direct 2009, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y. On the origin of life in the Zinc world: 1. Photosynthesizing, porous edifices built of hydrothermally precipitated zinc sulfide as cradles of life on Earth. Biol. Direct 2009, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nakamura, R.; Takai, K. Deep-Sea Hydrothermal Fields as Natural Power Plants. ChemElectroChem 2018, 5, 2162–2166. [Google Scholar] [CrossRef]
- Kitadai, N.; Nakamura, R.; Yamamoto, M.; Takai, K.; Li, Y.; Yamaguchi, A.; Gilbert, A.; Ueno, Y.; Yoshida, N.; Oono, Y. Geoelectrochemical CO production: Implications for the autotrophic origin of life. Sci. Adv. 2018, 4, eaao7265. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Yamamoto, M.; Takai, K.; Ishii, T.; Hashimoto, K.; Nakamura, R. Electrochemical CO2 Reduction by Ni-containing Iron Sulfides: How Is CO2 Electrochemically Reduced at Bisulfide-Bearing Deep-sea Hydrothermal Precipitates? Electrochim. Acta 2014, 141, 311–318. [Google Scholar] [CrossRef]
- Li, Y.; Yamaguchi, A.; Yamamoto, M.; Takai, K.; Nakamura, R. Molybdenum Sulfide: A Bioinspired Electrocatalyst for Dissimilatory Ammonia Synthesis with Geoelectrical Current. J. Phys. Chem. C 2017, 121, 2154–2164. [Google Scholar] [CrossRef]
- Nakamura, R.; Takashima, T.; Kato, S.; Takai, K.; Yamamoto, M.; Hashimoto, K. Electrical Current Generation across a Black Smoker Chimney. Angew. Chem. 2010, 122, 7858–7860. [Google Scholar] [CrossRef]
- Yamamoto, M.; Nakamura, R.; Oguri, K.; Kawagucci, S.; Suzuki, K.; Hashimoto, K.; Takai, K. Generation of Electricity and Illumination by an Environmental Fuel Cell in Deep-Sea Hydrothermal Vents. Angew. Chem. 2013, 125, 10958–10961. [Google Scholar] [CrossRef]
- Barge, L.M.; Kee, T.P.; Doloboff, I.J.; Hampton, J.M.P.; Ismail, M.; Pourkashanian, M.; Zeytounian, J.; Baum, M.M.; Moss, J.A.; Lin, C.-K.; et al. The fuel cell model of abiogenesis: a new approach to origin-of-life simulations. Astrobiology 2014, 14, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Barge, L.M.; Branscomb, E.; Brucato, J.R.; Cardoso, S.S.S.; Cartwright, J.H.E.; Danielache, S.O.; Galante, D.; Kee, T.P.; Miguel, Y.; Mojzsis, S.; et al. Thermodynamics, Disequilibrium, Evolution: Far-From-Equilibrium Geological and Chemical Considerations for Origin-Of-Life Research. Orig. Life Evol. Biospheres 2017, 47, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Barge, L.M.; Abedian, Y.; Russell, M.J.; Doloboff, I.J.; Cartwright, J.H.E.; Kidd, R.D.; Kanik, I. From Chemical Gardens to Fuel Cells: Generation of Electrical Potential and Current Across Self-Assembling Iron Mineral Membranes. Angew. Chem. 2015, 127, 8302–8305. [Google Scholar] [CrossRef]
- Sangster, D.F. Precambrian volcanogenic massive sulfide deposits in Canada: A review. Geol. Surv. Can. Pap. 1972, 72–22, 1–43. [Google Scholar]
- Hazen, R.M.; Papineau, D.; Bleeker, W.; Downs, R.T.; Ferry, J.M.; McCoy, T.J.; Sverjensky, D.A.; Yang, H. Mineral evolution. Am. Mineral. 2008, 93, 1693–1720. [Google Scholar] [CrossRef]
- Von Damm, K. Seafloor Hydrothermal Activity: Black Smoker Chemistry And Chimneys. Annu. Rev. Earth Planet. Sci. 1990, 18, 173–204. [Google Scholar] [CrossRef]
- Reed, M.H. Sulfide Mineral Precipitation from Hydrothermal Fluids. Rev. Mineral. Geochem. 2006, 61, 609–631. [Google Scholar] [CrossRef]
- Macleod, G.; McKeown, C.; Hall, A.J.; Russell, M.J. Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Orig. Life Evol. Biospheres 1994, 24, 19–41. [Google Scholar] [CrossRef]
- Dahl, T.W.; Wirth, S.B. Molybdenum isotope fractionation and speciation in a euxinic lake—Testing ways to discern isotope fractionation processes in a sulfidic setting. Chem. Geol. 2017, 460, 84–92. [Google Scholar] [CrossRef]
- Helz, G.R.; Bura-Nakić, E.; Mikac, N.; Ciglenečki, I. New model for molybdenum behavior in euxinic waters. Chem. Geol. 2011, 284, 323–332. [Google Scholar] [CrossRef]
- Erickson, B.E.; Helz, G.R. Molybdenum(VI) speciation in sulfidic waters: Stability and lability of thiomolybdates. Geochim. Cosmochim. Acta 2000, 64, 1149–1158. [Google Scholar] [CrossRef]
- Helz, G.R.R.; Miller, C.V.V.; Charnock, J.M.M.; Mosselmans, J.F.W.F.W.; Pattrick, R.A.D.; Garner, C.D.D.; Vaughan, D.J.J. Mechanism of molybdenum removal from the sea and its concentration in black shales: EXAFS evidence. Geochim. Cosmochim. Acta 1996, 60, 3631–3642. [Google Scholar] [CrossRef]
- Wilde, P.; Lyons, T.W.; Quinby-Hunt, M.S. Organic carbon proxies in black shales: molybdenum. Chem. Geol. 2004, 206, 167–176. [Google Scholar] [CrossRef]
- Kao, L.S.; Peacor, D.R.; Coveney, J.; Zhao, G.; Dungey, K.E.; Curtis, M.D.; Penner-Hahn, J.E. A C/MoS2mixed-layer phase (MoSC) occurring in metalliferous black shales from Southern China, and new data on jordisite. Am. Mineral. 2001, 86, 852–861. [Google Scholar] [CrossRef]
- Anbar, A.D. Oceans. Elements and evolution. Science 2008, 322, 1481–1483. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.B.; Wolfe-Simon, F.; Anbar, A.D. Coevolution of metal availability and nitrogen assimilation in cyanobacteria and algae. Geobiology 2009, 7, 100–123. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.K.; Jelen, B.I.; Giovannelli, D.; Raanan, H.; Falkowski, P.G. Metal availability and the expanding network of microbial metabolisms in the Archaean eon. Nat. Geosci. 2017, 10, 629–636. [Google Scholar] [CrossRef]
- Russell, M.J.; Daniel, R.M.; Hall, A.J. On the emergence of life via catalytic iron-sulphide membranes. Terra Nov. 1993, 5, 343–347. [Google Scholar] [CrossRef]
- Russell, M.J.; Daia, D.E.; Hall, A.J. The emergence of life from FeS bubbles at alkaline hot springs in an acid ocean. In Molecular Evolution and the Origin of Life; Wiegel, J., Michael, A.W.W., Eds.; Taylor & Francis: London, UK, 1998; pp. 77–126. [Google Scholar]
- Roldan, A.; Hollingsworth, N.; Roffey, A.; Islam, H.-U.; Goodall, J.B.M.; Catlow, C.R.A.; Darr, J.A.; Bras, W.; Sankar, G.; Holt, K.B.; et al. Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions. Chem. Commun. 2015, 51, 7501–7504. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M. Mineral surfaces and the prebiotic selection and organization of biomolecules. Am. Mineral. 2006, 91, 1715–1729. [Google Scholar] [CrossRef]
- Orgel, L.E. Polymerization on the rocks: Theoretical introduction. Orig. Life Evol. Biospheres 1998, 28, 227–234. [Google Scholar] [CrossRef]
- Shatynski, S.R. The thermochemistry of transition metal sulfides. Oxid. Met. 1977, 11, 307–320. [Google Scholar] [CrossRef]
- Moh, G.H. High-temperature metal sulfide chemistry. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 1978; pp. 107–151. ISBN 978-3-540-35680-6. [Google Scholar]
- Chianelli, R.R.; Daage, M.; Ledoux, M.J. Fundamental Studies of Transition-Metal Sulfide Catalytic Materials. Adv. Catal. 1994, 40, 177–232. [Google Scholar] [CrossRef]
- Chianelli, R.R. Periodic trends transition metal sulfide catalysis: Intuition and theory. Oil Gas Sci. Technol. 2006, 61, 503–513. [Google Scholar] [CrossRef]
- Summers, D.P.; Khare, B. Nitrogen Fixation on Early Mars and Other Terrestrial Planets: Experimental Demonstration of Abiotic Fixation Reactions to Nitrite and Nitrate. Astrobiology 2007, 7, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Summers, D.P. Ammonia Formation By The Reduction Of Nitrite/Nitrate By FeS: Ammonia Formation Under Acidic Conditions. Orig. Life Evol. Biospheres 2005, 35, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wachterhauser, G. Activated Acetic Acid by Carbon Fixation on (Fe, Ni) S under Primordial Conditions. Science 1997, 276, 245–247. [Google Scholar] [CrossRef]
- Huber, C.; Wächtershäuser, G. Α-Hydroxy and Α-Amino Acids Under Possible Hadean, Volcanic Origin-of-Life Conditions. Science 2006, 314, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Peptides by Activation of Amino Acids with CO on (Ni,Fe)S Surfaces: Implications for the Origin of Life. Science 1998, 281, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Primordial reductive amination revisited. Tetrahedron Lett. 2003, 44, 1695–1697. [Google Scholar] [CrossRef]
- Hazen, R.M.; Hystad, G.; Golden, J.J.; Hummer, D.R.; Liu, C.; Downs, R.T.; Morrison, S.M.; Ralph, J.; Grew, E.S. Cobalt mineral ecology. Am. Mineral. 2017, 102, 108–116. [Google Scholar] [CrossRef]
- Liu, C.; Hystad, G.; Golden, J.J.; Hummer, D.R.; Downs, R.T.; Morrison, S.M.; Ralph, J.P.; Hazen, R.M. Chromium mineral ecology. Am. Mineral. 2017, 102, 612–619. [Google Scholar] [CrossRef]
- Cody, G.D.; Boctor, N.Z.; Brandes, J.A.; Filley, T.R.; Hazen, R.M.; Yoder, H.S. Assaying the catalytic potential of transition metal sulfides for abiotic carbon fixation. Geochim. Cosmochim. Acta 2004, 68, 2185–2196. [Google Scholar] [CrossRef]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 0003. [Google Scholar] [CrossRef]
- Koper, M.T.M. Theory of the transition from sequential to concerted electrochemical proton-electron transfer. Phys. Chem. Chem. Phys. 2013, 15, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Koper, M.T.M. Theory of multiple proton–electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 2013, 4, 2710–2723. [Google Scholar] [CrossRef]
- Koper, M.T.M. Volcano Activity Relationships for Proton-Coupled Electron Transfer Reactions in Electrocatalysis. Top. Catal. 2015, 58, 1153–1158. [Google Scholar] [CrossRef]
- Nitschke, W.; McGlynn, S.E.; Milner-White, E.J.; Russell, M.J. On the antiquity of metalloenzymes and their substrates in bioenergetics. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Barge, L.M.; Bhartia, R.; Bocanegra, D.; Bracher, P.J.; Branscomb, E.; Kidd, R.; McGlynn, S.; Meier, D.H.; Nitschke, W.; et al. The drive to life on wet and icy worlds. Astrobiology 2014, 14, 308–343. [Google Scholar] [CrossRef] [PubMed]
- Heinen, W.; Lauwers, A.M. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig. Life Evol. Biospheres 1996, 26, 131–150. [Google Scholar] [CrossRef]
- Brandes, J.A.; Boctor, N.Z.; Cody, G.D.; Cooper, B.A.; Hazen, R.M.; Yoder, H.S. Abiotic nitrogen reduction on the early Earth. Nature 1998, 395, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Dörr, M.; Käßbohrer, J.; Grunert, R.; Kreisel, G.; Brand, W.A.; Werner, R.A.; Geilmann, H.; Apfel, C.; Robl, C.; Weigand, W. A Possible Prebiotic Formation of Ammonia from Dinitrogen on Iron Sulfide Surfaces. Angew. Chem. Int. Ed. 2003, 42, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Cody, G.D. Primordial Carbonylated Iron-Sulfur Compounds and the Synthesis of Pyruvate. Science 2000, 289, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Booth, B.G.; Chatt, J. The reactions of carbon monoxide and nitric oxide with tertiary phosphine complexes of iron (II), cobalt (II) and nickel (II). J. Chem. Soc. 1962, 2099–2106. [Google Scholar] [CrossRef]
- Butler, I.S.; Basolo, F.; Pearson, R.G. Kinetics Studies of Carbon Monoxide Insertion Reactions. Reactions of Cyclopentadienyl(methyl)iron Dicarbonyl and Cyclopentadienyl(methyl) Molybdenum Tricarbonyl with Phosphines and Phosphites. Inorg. Chem. 1967, 6, 2074–2079. [Google Scholar] [CrossRef]
- Calderazzo, F.; Cotton, F.A. Carbon Monoxide Insertion Reactions. I. The Carbonylation of Methyl Manganese Pentacarbonyl and Decarbonylation of Acetyl Manganese Pentacarbonyl. Inorg. Chem. 1962, 1, 30–36. [Google Scholar] [CrossRef]
- Novikov, Y.; Copley, S.D. Reactivity landscape of pyruvate under simulated hydrothermal vent conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 13283–13288. [Google Scholar] [CrossRef] [PubMed]
- Ang, R.; Khan, A.U.; Tsujii, N.; Takai, K.; Nakamura, R.; Mori, T. Thermoelectricity Generation and Electron-Magnon Scattering in a Natural Chalcopyrite Mineral from a Deep-Sea Hydrothermal Vent. Angew. Chem. Int. Ed. Engl. 2015, 54, 12909–12922. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Li, Y.; Takashima, T.; Hashimoto, K.; Nakamura, R. CO2 Reduction Using an Electrochemical Approach from Chemical, Biological, and Geological Aspects in the Ancient and Modern Earth; Springer: Cham, Switzerland, 2016; pp. 213–228. [Google Scholar]
- Trail, D.; Watson, E.B.; Tailby, N.D. The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 2011, 480, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Bullett, D.W. Electronic structure of 3d pyrite- and marcasite-type sulphides. J. Phys. C Solid State Phys. 1982, 15, 6163–6174. [Google Scholar] [CrossRef]
- Uhlig, I.; Szargan, R.; Nesbitt, H.; Laajalehto, K. Surface states and reactivity of pyrite and marcasite. Appl. Surf. Sci. 2001, 179, 222–229. [Google Scholar] [CrossRef]
- Snowball, I.F. Magnetic hysteresis properties of greigite (Fe3S4) and a new occurrence in Holocene sediments from Swedish Lappland. Phys. Earth Planet. Inter. 1991, 68, 32–40. [Google Scholar] [CrossRef]
- Rochette, P.; Fillion, G.; Mattéi, J.L.; Dekkers, M.J. Magnetic transition at 30–34 Kelvin in pyrrhotite: Insight into a widespread occurrence of this mineral in rocks. Earth Planet. Sci. Lett. 1990, 98, 319–328. [Google Scholar] [CrossRef]
- Boller, H. IUCr On the synthesis, crystal chemistry and magnetic properties of rasvumite and related compounds. Acta Crystallogr. Sect. A Found. Crystallogr. 2004, 60, s47. [Google Scholar] [CrossRef]
- Silva, J.C.M.; De Abreu, H.A.; Duarte, H.A. Electronic and structural properties of bulk arsenopyrite and its cleavage surfaces—A DFT study. RSC Adv. 2015, 5, 2013–2023. [Google Scholar] [CrossRef]
- Jones, R.A.; Nesbitt, H.W. XPS evidence for Fe and As oxidation states and electronic states in loellingite (FeAs2). Am. Mineral. 2002, 87, 1692–1698. [Google Scholar] [CrossRef]
- Fesseler, J.; Jeoung, J.H.; Dobbek, H. How the [NiFe4S4] Cluster of CO Dehydrogenase Activates CO2 and NCO-. Angew. Chem. Int. Ed. 2015, 54, 8560–8564. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, A.J.; Dekkers, M.J.; Zijderveld, J.D.A. Magnetic iron-nickel sulphides in the Pliocene and Pleistocene marine marls from the Vrica section (Calabria, Italy). Earth Planet. Sci. Lett. 1993, 115, 43–55. [Google Scholar] [CrossRef]
- Bostick, B.C.; Fendorf, S.; Helz, G.R. Differential adsorption of molybdate and tetrathiomolybdate on pyrite (FeS2). Environ. Sci. Technol. 2003, 37, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Tribovillard, N.; Riboulleau, A.; Lyons, T.; Baudin, F. Enhanced trapping of molybdenum by sulfurized marine organic matter of marine origin in Mesozoic limestones and shales. Chem. Geol. 2004, 213, 385–401. [Google Scholar] [CrossRef]
- Vorlicek, T.P.; Kahn, M.D.; Kasuya, Y.; Helz, G.R. Capture of molybdenum in pyrite-forming sediments: role of ligand-induced reduction by polysulfides. Geochim. Cosmochim. Acta 2004, 68, 547–556. [Google Scholar] [CrossRef]
- Luther, G.W.; Rickard, D.T. Metal sulfide cluster complexes and their biogeochemical importance in the environment. J. Nanopart. Res. 2005, 7, 389–407. [Google Scholar] [CrossRef]
- Guttenberg, N.; Virgo, N.; Chandru, K.; Scharf, C.; Mamajanov, I. Bulk measurements of messy chemistries are needed for a theory of the origins of life. Philos. Trans. A Math. Phys. Eng. Sci. 2017, 375, 20160347. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S. Messy biology and the origins of evolutionary innovations. Nat. Chem. Biol. 2010, 6, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Richardson, D.J.; Butt, J.N. Catalytic Protein Film Voltammetry from a Respiratory Nitrate Reductase Provides Evidence for Complex Electrochemical Modulation of Enzyme Activity. Biochemistry 2001, 40, 11294–11307. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Elucidating the mechanisms of coupled electron transfer and catalytic reactions by protein film voltammetry. Biochim. Biophys. Acta 2006, 1757, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, F.J.M.; Meijer, F.S.; Dekker, C.; Albracht, S.P.J.; Heering, H.A.; Lemay, S.G. Toward Single-Enzyme Molecule Electrochemistry: [NiFe]-Hydrogenase Protein Film Voltammetry at Nanoelectrodes. ACS Nano 2008, 2, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.; Yasuda, H. Transformation of Carbon Dioxide Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Dinh, C.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; García de Arquer, F.P.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Kikuchi, K.; Suzuki, S. Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. [Google Scholar] [CrossRef]
- Chan, K.; Tsai, C.; Hansen, H.A.; Nørskov, J.K. Molybdenum Sulfides and Selenides as Possible Electrocatalysts for CO2 Reduction. ChemCatChem. 2014, 6, 1899–1905. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Shi, C.; Hansen, H.A.; Lausche, A.C.; Nørskov, J.K. Trends in electrochemical CO2 reduction activity for open and close-packed metal surfaces. Phys. Chem. Chem. Phys. 2014, 16, 4720–4727. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B.A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; et al. Robust carbon dioxide reduction on molybdenum disulphide edges. Nat. Commun. 2014, 5, 4470. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Li, Y.; Ooka, H.; Go, Y.K.; Jin, F.; Kim, S.H.; Nakamura, R. Selective Electrocatalytic Reduction of Nitrite to Dinitrogen Based on Decoupled Proton-Electron Transfer. J. Am. Chem. Soc. 2018, 140, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, J.J.G. How Biology Handles Nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Perathoner, S.; Ampelli, C.; Mebrahtu, C.; Su, D.; Centi, G. Room-Temperature Electrocatalytic Synthesis of NH3 from H2O and N2 in a Gas-liquid-Solid Three-Phase Reactor. ACS Sustain. Chem. Eng. 2017, 5, 7393–7400. [Google Scholar] [CrossRef]
- Chen, S.; Perathoner, S.; Ampelli, C.; Mebrahtu, C.; Su, D.; Centi, G. Electrocatalytic Synthesis of Ammonia at Room Temperature and Atmospheric Pressure from Water and Nitrogen on a Carbon-Nanotube-Based Electrocatalyst. Angew. Chem. Int. Ed. 2017, 56, 2699–2703. [Google Scholar] [CrossRef] [PubMed]
- Kordali, V.; Kyriacou, G.; Lambrou, C. Electrochemical synthesis of ammonia at atmospheric pressure and low temperature in a solid polymer electrolyte cell. Chem. Commun. 2000, 1673–1674. [Google Scholar] [CrossRef]
- Bao, D.; Zhang, Q.; Meng, F.-L.; Zhong, H.-X.; Shi, M.-M.; Zhang, Y.; Yan, J.-M.; Jiang, Q.; Zhang, X.-B. Electrochemical Reduction of N2 under Ambient Conditions for Artificial N2 Fixation and Renewable Energy Storage Using N2 /NH3 Cycle. Adv. Mater. 2017, 29, 1604799. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Bao, D.; Shi, M.-M.; Wulan, B.-R.; Yan, J.-M.; Jiang, Q. Amorphizing of Au Nanoparticles by CeO x -RGO Hybrid Support towards Highly Efficient Electrocatalyst for N2 Reduction under Ambient Conditions. Adv. Mater. 2017, 29, 1700001. [Google Scholar] [CrossRef] [PubMed]
- Yandulov, D.V. Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Acc. Chem. Res. 2005, 38, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Ji, X.; Ren, X.; Cui, G.; Li, L.; Xie, F.; Wang, H.; Li, B.; Sun, X. MoO3 nanosheets for efficient electrocatalytic N2 fixation to NH3. J. Mater. Chem. A 2018, 6, 12974–12977. [Google Scholar] [CrossRef]
- Zhang, L.; Ji, X.; Ren, X.; Ma, Y.; Shi, X.; Tian, Z.; Asiri, A.M.; Chen, L.; Tang, B.; Sun, X. Electrochemical Ammonia Synthesis via Nitrogen Reduction Reaction on a MoS2 Catalyst: Theoretical and Experimental Studies. Adv. Mater. 2018, 30, 1800191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ji, X.; Ren, X.; Luo, Y.; Shi, X.; Asiri, A.M.; Zheng, B.; Sun, X. Efficient Electrochemical N2 Reduction to NH3 on MoN Nanosheets Array under Ambient Conditions. ACS Sustain. Chem. Eng. 2018, 6, 9550–9554. [Google Scholar] [CrossRef]
- Di Giovanni, C.; Wang, W.A.; Nowak, S.; Grenèche, J.M.; Lecoq, H.; Mouton, L.; Giraud, M.; Tard, C. Bioinspired iron sulfide nanoparticles for cheap and long-lived electrocatalytic molecular hydrogen evolution in neutral water. ACS Catal. 2014, 4, 681–687. [Google Scholar] [CrossRef]
- Kornienko, N.; Resasco, J.; Becknell, N.; Jiang, C.M.; Liu, Y.S.; Nie, K.; Sun, X.; Guo, J.; Leone, S.R.; Yang, P. Operando Spectroscopic Analysis of an Amorphous Cobalt Sulfide Hydrogen Evolution Electrocatalyst. J. Am. Chem. Soc. 2015, 137, 7448–7455. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Ting, L.R.L.; Neo, P.H.L.; Zhang, Y.-J.; Peterson, A.A.; Yeo, B.S. Operando Raman Spectroscopy of Amorphous Molybdenum Sulfide (MoSx) during the Electrochemical Hydrogen Evolution Reaction: Identification of Sulfur Atoms as Catalytically Active Sites for H + Reduction. ACS Catal. 2016, 6, 7790–7798. [Google Scholar] [CrossRef]
- Ting, L.R.L.; Deng, Y.; Ma, L.; Zhang, Y.-J.; Peterson, A.A.; Yeo, B.S. Catalytic Activities of Sulfur Atoms in Amorphous Molybdenum Sulfide for the Electrochemical Hydrogen Evolution Reaction. ACS Catal. 2016, 6, 861–867. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, H.; Li, S.; Wang, R.; Sun, X.; Zhou, M.; Zhou, J.; Lou, X.W.D.; Xie, Y. Defect-rich MoS2 ultrathin nanosheets with additional active edge sites for enhanced electrocatalytic hydrogen evolution. Adv. Mater. 2013, 25, 5807–5820. [Google Scholar] [CrossRef] [PubMed]
- Merki, D.; Fierro, S.; Vrubel, H.; Hu, X. Amorphous molybdenum sulfide films as catalysts for electrochemical hydrogen production in water. Chem. Sci. 2011, 2, 1262–1267. [Google Scholar] [CrossRef]
- Lukowski, M.A.; Daniel, A.S.; Meng, F.; Forticaux, A.; Li, L.; Jin, S. Enhanced Hydrogen Evolution Catalysis from Enhanced Hydrogen Evolution Catalysis from Chemically Exfoliated Metallic MoS2 Nanosheets. J. Am. Chem. Soc. 2013, 135, 10274–10277. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.F.; Bonde, J.; Zhang, J.; Ooi, B.L.; Andersson, K.; Ulstrup, J.; Chorkendorff, I. Hydrogen evolution on supported incomplete cubane-type [Mo3S4]4+ electrocatalysts. J. Phys. Chem. C 2008, 112, 17492–17498. [Google Scholar] [CrossRef]
- Voiry, D.; Yamaguchi, H.; Li, J.; Silva, R.; Alves, D.C.B.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 2013, 12, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Voiry, D.; Ahn, S.J.; Kang, D.; Kim, A.Y.; Chhowalla, M.; Shin, H.S. Two-dimensional hybrid nanosheets of tungsten disulfide and reduced graphene oxide as catalysts for enhanced hydrogen evolution. Angew. Chem. Int. Ed. 2013, 52, 13751–13754. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Li, G.; Wang, Z.; Zhu, H.; Zhang, T.; Xiao, S.; Guo, W.; Yang, S. Metallic Iron–Nickel Sulfide Ultrathin Nanosheets As a Highly Active Electrocatalyst for Hydrogen Evolution Reaction in Acidic Media. J. Am. Chem. Soc. 2015, 137, 11900–11903. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Gong, M.; Chou, H.L.; Pan, C.J.; Chen, H.A.; Wu, Y.; Lin, M.C.; Guan, M.; Yang, J.; Chen, C.W.; et al. Highly active and stable hybrid catalyst of cobalt-doped FeS2 nanosheets-carbon nanotubes for hydrogen evolution reaction. J. Am. Chem. Soc. 2015, 137, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.F.; Song, J.; Li, K.; Tahir, M.; Wang, Y.T.; Pan, L.; Wang, L.; Zhang, X.; Zou, J.J. Hollow Cobalt-Based Bimetallic Sulfide Polyhedra for Efficient All-pH-Value Electrochemical and Photocatalytic Hydrogen Evolution. J. Am. Chem. Soc. 2016, 138, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Faber, M.S.; Lukowski, M.A.; Ding, Q.; Kaiser, N.S.; Jin, S. Earth-abundant metal pyrites (FeS2, CoS2, NiS2, and their alloys) for highly efficient hydrogen evolution and polysulfide reduction electrocatalysis. J. Phys. Chem. C 2014, 118, 21347–21356. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Chan, K.; Nørskov, J.K.; Abild-Pedersen, F. Rational design of MoS2 catalysts: tuning the structure and activity via transition metal doping. Catal. Sci. Technol. 2015, 5, 246–253. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, J.; Li, S.; Grote, F.; Zhang, X.; Zhang, H.; Wang, R. Controllable Disorder Engineering in Oxygen-Incorporated MoS2 Ultrathin Nanosheets for Efficient Hydrogen Evolution. J. Am. Chem. Soc. 2013, 135, 17881–17888. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhuo, J.; Du, K.; Chen, B.; Zhu, Z.; Shao, Y.; Li, M. Electrochemically fabricated polypyrrole and MoSx copolymer films as a highly active hydrogen evolution electrocatalyst. Adv. Mater. 2014, 26, 3761–3766. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Dou, K.; Kaun, C.; Kuang, Q.; Yang, S. MoSe nanosheets and their graphene hybrids: Synthesis, characterization and hydrogen evolution reaction studies. Mater. Chem. A 2014, 2, 360–364. [Google Scholar] [CrossRef]
- Li, D.J.; Maiti, U.N.; Lim, J.; Choi, D.S.; Lee, W.J.; Oh, Y.; Lee, G.Y.; Kim, S.O. Molybdenum sulfide/N-doped CNT forest hybrid catalysts for high-performance hydrogen evolution reaction. Nano Lett. 2014, 14, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Lassalle-Kaiser, B.; Merki, D.; Vrubel, H.; Gul, S.; Yachandra, V.K.; Hu, X.; Yano, J. Evidence from in Situ X-ray Absorption Spectroscopy for the Involvement of Terminal Disulfide in the Reduction of Protons by an Amorphous Molybdenum Sulfide Electrocatalyst. J. Am. Chem. Soc. 2015, 137, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Casalongue, H.G.S.; Benck, J.D.; Tsai, C.; Karlsson, R.K.B.; Kaya, S.; Ng, M.L.; Pettersson, L.G.M.; Abild-Pedersen, F.; Nørskov, J.K.; Ogasawara, H.; et al. Operando Characterization of an Amorphous Molybdenum Sulfide Nanoparticle Catalyst during the Hydrogen Evolution Reaction. J. Phys. Chem. C 2014, 118, 29252–29259. [Google Scholar] [CrossRef]
- Tran, P.D.; Tran, T.V.; Orio, M.; Torelli, S.; Truong, Q.D.; Nayuki, K.; Sasaki, Y.; Chiam, S.Y.; Yi, R.; Honma, I.; et al. Coordination polymer structure and revisited hydrogen evolution catalytic mechanism for amorphous molybdenum sulfide. Nat. Mater. 2016, 15, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.F.; Jorgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendorff, I. Identification of Active Edge Sites for Electrochemical H2 Evolution from MoS2 Nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jorgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef] [PubMed]
- Karunadasa, H.I.; Montalvo, E.; Sun, Y.; Majda, M.; Long, J.R.; Chang, C.J. A Molecular MoS2 Edge Site Mimic for Catalytic Hydrogen Generation. Science 2012, 335, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, A.; Kemmegne-Mbouguen, J.C.; Floquet, S.; Marrot, J.; Fize, J.; Artero, V.; David, O.; Magnier, E.; Pegot, B.; Cadot, E. Tuning the electrocatalytic hydrogen evolution reaction promoted by [Mo2O2S2]-based molybdenum cycles in aqueous medium. Dalton Trans. 2013, 42, 4848–4858. [Google Scholar] [CrossRef] [PubMed]
- Kibsgaard, J.; Jaramillo, T.F.; Besenbacher, F. Building an appropriate active-site motif into a hydrogen-evolution catalyst with thiomolybdate [Mo3S13]2− clusters. Nat. Chem. 2014, 6, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.A.; Saha, A.; Raghavachari, K. Bond Activation and Hydrogen Evolution from Water through Reactions with M3S4 (M = Mo, W) and W3S3 Anionic Clusters. J. Phys. Chem. A 2017, 121, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nakamura, R. Structural change of molybdenum sulfide facilitates the electrocatalytic hydrogen evolution reaction at neutral pH as revealed by in situ Raman spectroscopy. Chin. J. Catal. 2018, 39, 401–406. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasché, F.; Herranz, J.; Gasteiger, H.A. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef]
- Vasić, D.D.; Pašti, I.A.; Mentus, S.V. DFT study of platinum and palladium overlayers on tungsten carbide: Structure and electrocatalytic activity toward hydrogen oxidation/evolution reaction. Int. J. Hydrogen Energy 2013, 38, 5009–5018. [Google Scholar] [CrossRef]
- Kiros, Y.; Schwartz, S. Long-term hydrogen oxidation catalysts in alkaline fuel cells. J. Power Sources 2000, 87, 101–105. [Google Scholar] [CrossRef]
- Zhuang, Z.; Giles, S.A.; Zheng, J.; Jenness, G.R.; Caratzoulas, S.; Vlachos, D.G.; Yan, Y. Nickel supported on nitrogen-doped carbon nanotubes as hydrogen oxidation reaction catalyst in alkaline electrolyte. Nat. Commun. 2016, 7, 10141. [Google Scholar] [CrossRef] [PubMed]
- Mund, K.; Richter, G.; von Sturm, F. Titanium-Containing Raney Nickel Catalyst for Hydrogen Electrodes in Alkaline Fuel Cell Systems. J. Electrochem. Soc. 1977, 124, 1–6. [Google Scholar] [CrossRef]
- Sheng, W.; Bivens, A.P.; Myint, M.; Zhuang, Z.; Forest, R.V.; Fang, Q.; Chen, J.G.; Yan, Y. Non-precious metal electrocatalysts with high activity for hydrogen oxidation reaction in alkaline electrolytes. Energy Environ. Sci. 2014, 7, 1719–1724. [Google Scholar] [CrossRef]
- Wilson, A.D.; Newell, R.H.; McNevin, M.J.; Muckerman, J.T.; Rakowski DuBois, M.; DuBois, D.L. Hydrogen Oxidation and Production Using Nickel-Based Molecular Catalysts with Positioned Proton Relays. J. Am. Chem. Soc. 2006, 128, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.H.; Peterson, A.A.; Nørskov, J.K. Insights into C-C Coupling in CO2 Electroreduction on Copper Electrodes. ChemCatChem 2013, 5, 737–742. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T.M. Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes. Angew. Chem. Int. Ed. 2013, 52, 7282–7285. [Google Scholar] [CrossRef] [PubMed]
- Gurudayal, G.; Bullock, J.; Srankó, D.F.; Towle, C.M.; Lum, Y.; Hettick, M.; Scott, M.C.; Javey, A.; Ager, J. Efficient solar-driven electrochemical CO2 reduction to hydrocarbons and oxygenates. Energy Environ. Sci. 2017, 10, 2222–2230. [Google Scholar] [CrossRef]
- He, J.; Dettelbach, K.E.; Salvatore, D.A.; Li, T.; Berlinguette, C.P. High-Throughput Synthesis of Mixed-Metal Electrocatalysts for C2 Reduction. Angew. Chem. Int. Ed. 2017, 56, 6068–6072. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Zhuang, Z.; Gao, M.; Zheng, J.; Chen, J.G.; Yan, Y. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6, 5848. [Google Scholar] [CrossRef] [PubMed]
- Skúlason, E.; Tripkovic, V.; Björketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jónsson, H.; Nørskov, J.K. Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Howalt, J.G.; Bligaard, T.; Rossmeisl, J.; Vegge, T. DFT based study of transition metal nano-clusters for electrochemical NH3 production. Phys. Chem. Chem. Phys. 2013, 15, 7785–7795. [Google Scholar] [CrossRef] [PubMed]
- Skúlason, E.; Bligaard, T.; Gudmundsdóttir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jónsson, H.; Nørskov, J.K. A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 2012, 14, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Abghoui, Y.; Garden, A.L.; Hlynsson, V.F.; Björgvinsdóttir, S.; Ólafsdóttir, H.; Skúlason, E. Enabling electrochemical reduction of nitrogen to ammonia at ambient conditions through rational catalyst design. Phys. Chem. Chem. Phys. 2015, 17, 4909–4918. [Google Scholar] [CrossRef] [PubMed]
- van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Koper, M.T.M.; Calle-Vallejo, F. Bond-making and breaking between carbon, nitrogen, and oxygen in electrocatalysis. J. Am. Chem. Soc. 2014, 136, 15694–15701. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Ma, X.; Li, Z.; Achenie, L.E.K.; Xin, H. Machine-learning-augmented chemisorption model for CO2 electroreduction catalyst screening. J. Phys. Chem. Lett. 2016, 6, 3528–3533. [Google Scholar] [CrossRef] [PubMed]
- Ulissi, Z.W.; Tang, M.T.; Xiao, J.; Liu, X.; Torelli, D.A.; Karamd, M.; Cummins, K.; Hahn, C.; Lewis, N.S.; Jaramillo, T.F.; et al. Machine-learning methods enable exhaustive searches for active bimetallic facets and reveal active site motifs for CO2 reduction. ACS Catal. 2017, 7, 6600–6608. [Google Scholar] [CrossRef]
- Hong, W.T.; Welsch, R.E.; Shao-Horn, Y. Descriptors of oxygen-evolution activity for oxides: A statistical evaluation. J. Phys. Chem. C 2016, 120, 78–86. [Google Scholar] [CrossRef]
- Ooka, H.; Hashimoto, K.; Nakamura, R. Design strategy of multi-electron transfer catalysts based on a bioinformatic analysis of oxygen evolution and reduction enzymes. Mol. Inf. 2018, 37, 1700139. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, I.K.; Enemark, J.H. EPR Studies of Oxo–Molybdenum(V) Complexes with Sulfur Donor Ligands: Implications for the Molybdenum Center of Sulfite Oxidase. Inorg. Chem. 1996, 35, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Inscore, F.E.; McNaughton, R.; Westcott, B.L.; Helton, M.E.; Jones, R.; Dhawan, I.K.; Enemark, J.H.; Kirk, M.L. Spectroscopic Evidence for a Unique Bonding Interaction in Oxo-Molybdenum Dithiolate Complexes: Implications for σ Electron Transfer Pathways in the Pyranopterin Dithiolate Centers of Enzymes. Inorg. Chem. 1999, 38, 1401–1410. [Google Scholar] [CrossRef]
- Roldan, A.; Santos-Carballal, D.; de Leeuw, N.H. A comparative DFT study of the mechanical and electronic properties of greigite Fe3S4 and magnetite Fe3O4. J. Chem. Phys. 2013, 138, 204712. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.J.; Tossell, J.A. Electronic structures of sulfide minerals-Theory and experiment. Phys. Chem. Miner. 1983, 9, 253–262. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Kitadai, N.; Nakamura, R. Chemical Diversity of Metal Sulfide Minerals and Its Implications for the Origin of Life. Life 2018, 8, 46. https://doi.org/10.3390/life8040046
Li Y, Kitadai N, Nakamura R. Chemical Diversity of Metal Sulfide Minerals and Its Implications for the Origin of Life. Life. 2018; 8(4):46. https://doi.org/10.3390/life8040046
Chicago/Turabian StyleLi, Yamei, Norio Kitadai, and Ryuhei Nakamura. 2018. "Chemical Diversity of Metal Sulfide Minerals and Its Implications for the Origin of Life" Life 8, no. 4: 46. https://doi.org/10.3390/life8040046
APA StyleLi, Y., Kitadai, N., & Nakamura, R. (2018). Chemical Diversity of Metal Sulfide Minerals and Its Implications for the Origin of Life. Life, 8(4), 46. https://doi.org/10.3390/life8040046