
Fitness Landscapes of Functional RNAs

Abstract
:1. Introduction

2. Whole Genotype–Fitness Map
- (1)
- There are multiple solutions to being a GTP aptamer. The fact that there is neither a common sequence motif nor a common structural motif that would tie together the fitness peaks suggests that there are multiple, independent solutions to a GTP aptamer. Thus the sequence space can be populated with different aptamers that have nothing in common except that they have the very same function.
- (2)
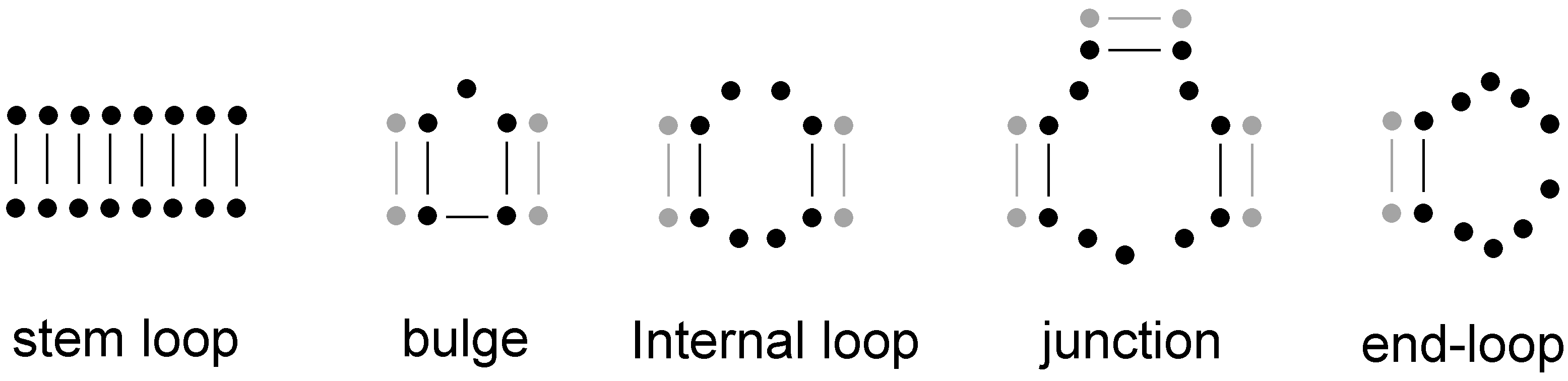
- There are uncommon structural solutions as well. In order to assess how frequent or infrequent the structures reported in S14 of the paper by Jiménez et al. [43] were, we folded 1 million random sequences of length 24 and estimated the frequency of the common structures from this sample. We know that this is a tiny fraction of the 2.814 × 1014 unique 24mers, but the common structures should be well represented even in this small sample. The structural repertoire of 24mers is rather limited. The structural space is dominated by the unstructured structure with roughly 18% of the sequences folding into this structure. Four of the peaks were found to be unstructured according to the Vienna Package. We cannot exclude the possibility of such a structure to bind the ligand, but it is more likely that an induced fit mechanism determines the final structure, or that the structure is stabilized by interactions that cannot be predicted by the employed 2D folding algorithm. Most of the other peaks were also folding into quite common structures (Table 1). A common structure is defined as those structures that are formed by more sequences than the average structure [44], i.e., Nc > 4L / SL, where Nc is the number of sequences folding into a common structure, SL is the number of distinct structures of length L. Most of the sequences fold into one of the common structures [44]. For example, more than 90% of the 25 nt long sequences comprising only G and C nucleotides fold into a common structure. Consequently, while there could be many rare structures, very few sequences fold into them. However, there was one peak (m20j22) with a structure that we have not found in our sample of structures for 10 million sequences. We can safely assume that this structure is uncommon. While we expect that evolution will mostly (if not always) find the common structures, it is reassuring to know that uncommon structures can also have function, just they are unlikely to be found by evolution. Aptamers and ribozymes we know are all folding into common structures not because only common structures can have function, but because they are the ones that can be reached by evolution.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure 1 | Peaks folding into this structure 2 | Frequency in our sample 3 | Frequency in our larger sample 4 |
|---|---|---|---|
| ........................ | m18j13, m14j12, m04j04, m02j01 | 18.7% | 18.8% |
| ((((....)))) | m19j09 | 3.4% | 3.4% |
| (((......))) | m15j18, m10j11, m07j07 | 2.4% | 2.5% |
| ((((...)))) | m16j20 | 1.8% | 1.9% |
| ((((.........))) | m17j08 | 0.8% | 0.8% |
| (((..........))) | m01j03 | 0.6% | 0.6% |
| ((((.......)))) | m03j02 | 0.4% | 0.4% |
| ((.((....)).)) | m05j10 | 0.3% | 0.3% |
| (((((.(.....).))))) | m06j06 | 0.05% | 0.006% |
| ((.....))...(....) | m20j22 | not found in our sample | not found in our sample |
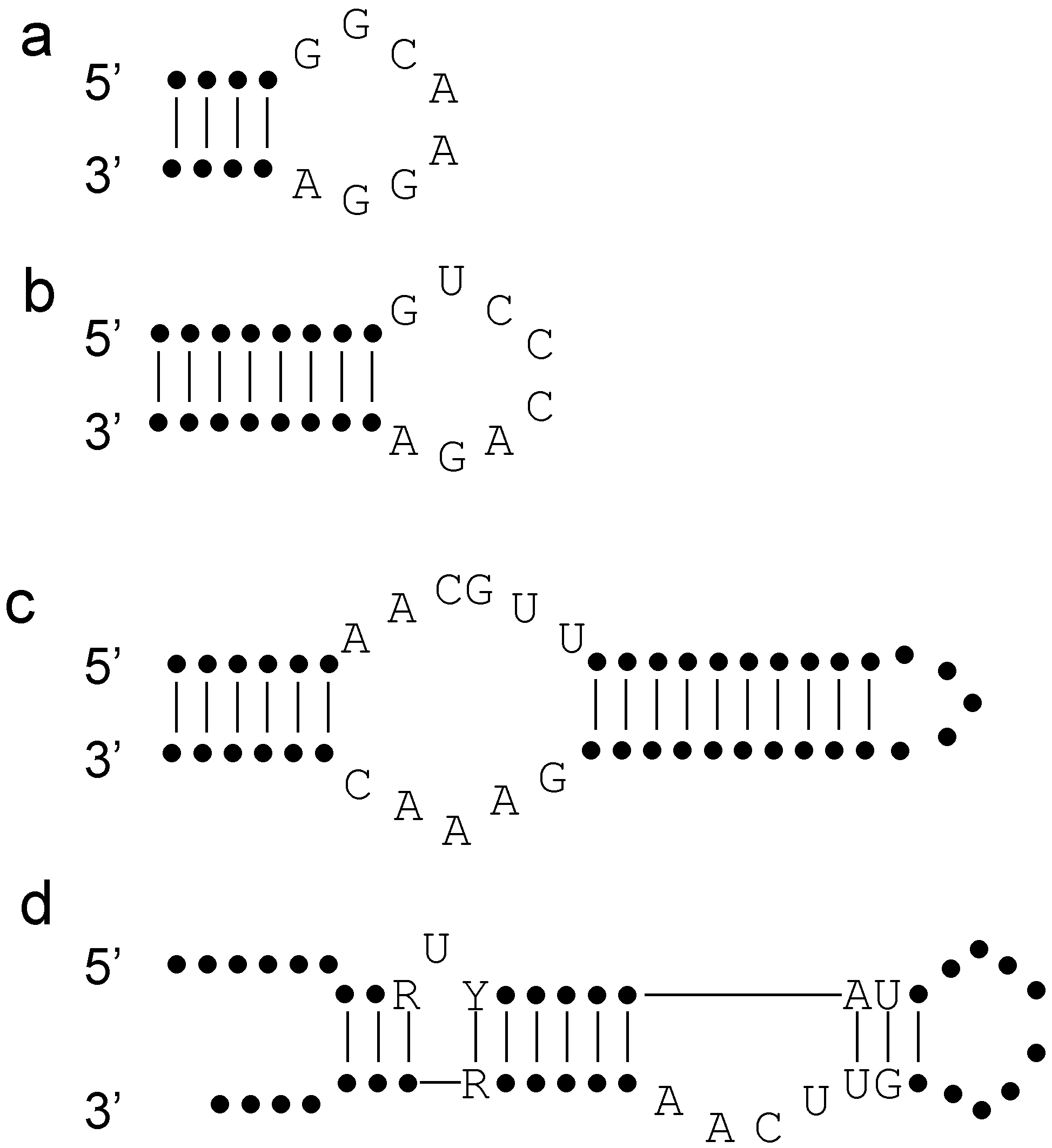
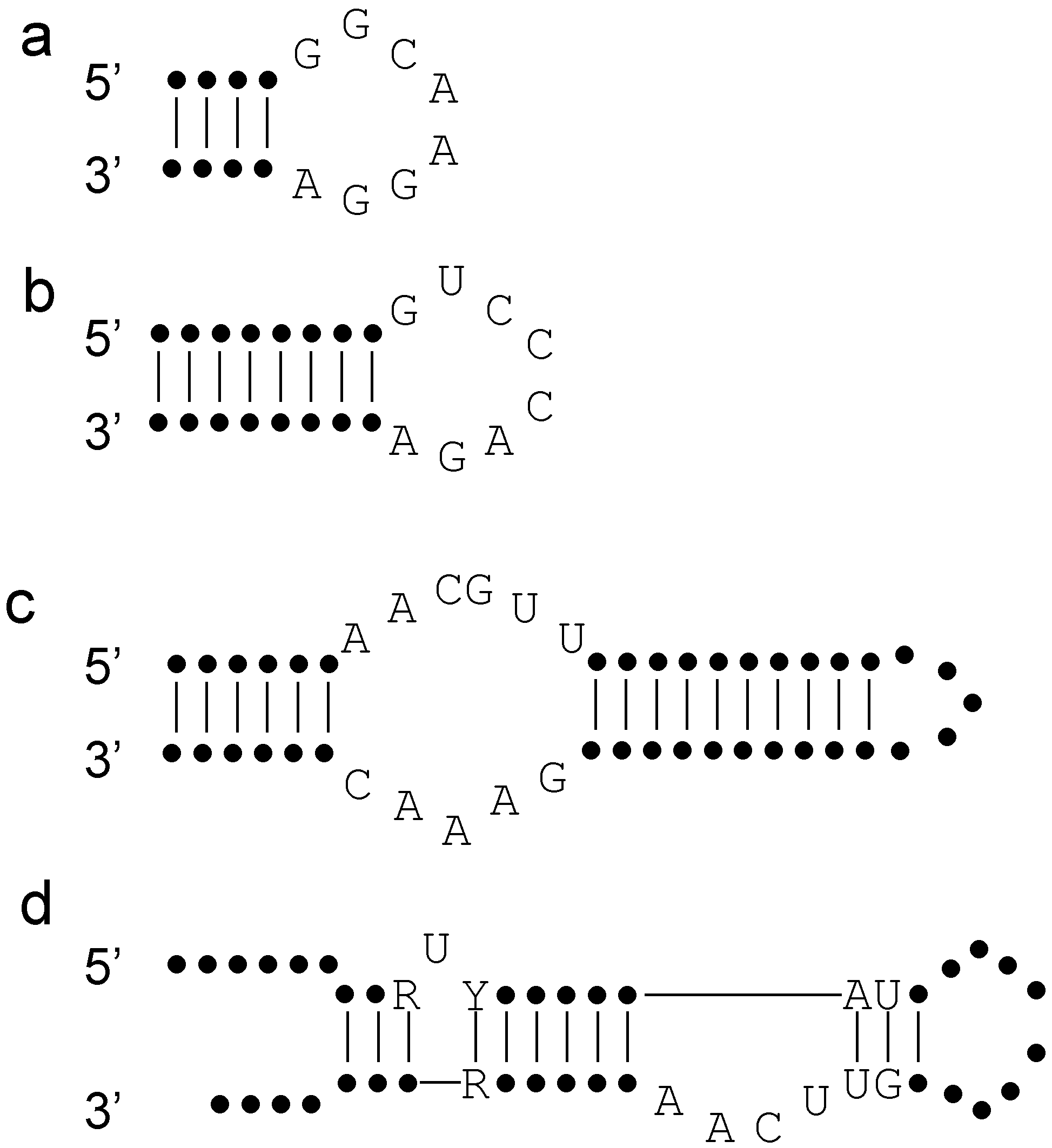
3. Lessons from Aptamers
4. Lessons from Ribozymes
| Mutation of the Class II Ligase | Activity from [77] | Activity from [78] |
|---|---|---|
| G1A | 0.00173 | 3.80 × 10−4 |
| G2A | 0.03043 | 1.10 × 10−1 |
| G2C | 0.00599 | 3.50 × 10−2 |
| G2U | 0.03584 | 1.30 × 10−1 |
| A3G | 0.00147 | 6.30 × 10−4 |
| A3C | 0.00108 | 8.00 × 10−5 |
| A3U | 0.00134 | 1.60 × 10−4 |
| G49A | 0.01048 | 2.10 × 10−2 |
| G47A | 0.00111 | 2.00 × 10−5 |
| G47C | 0.00089 | 1.90 × 10−5 |
| G47U | 0.00108 | 9.50 × 10−6 |
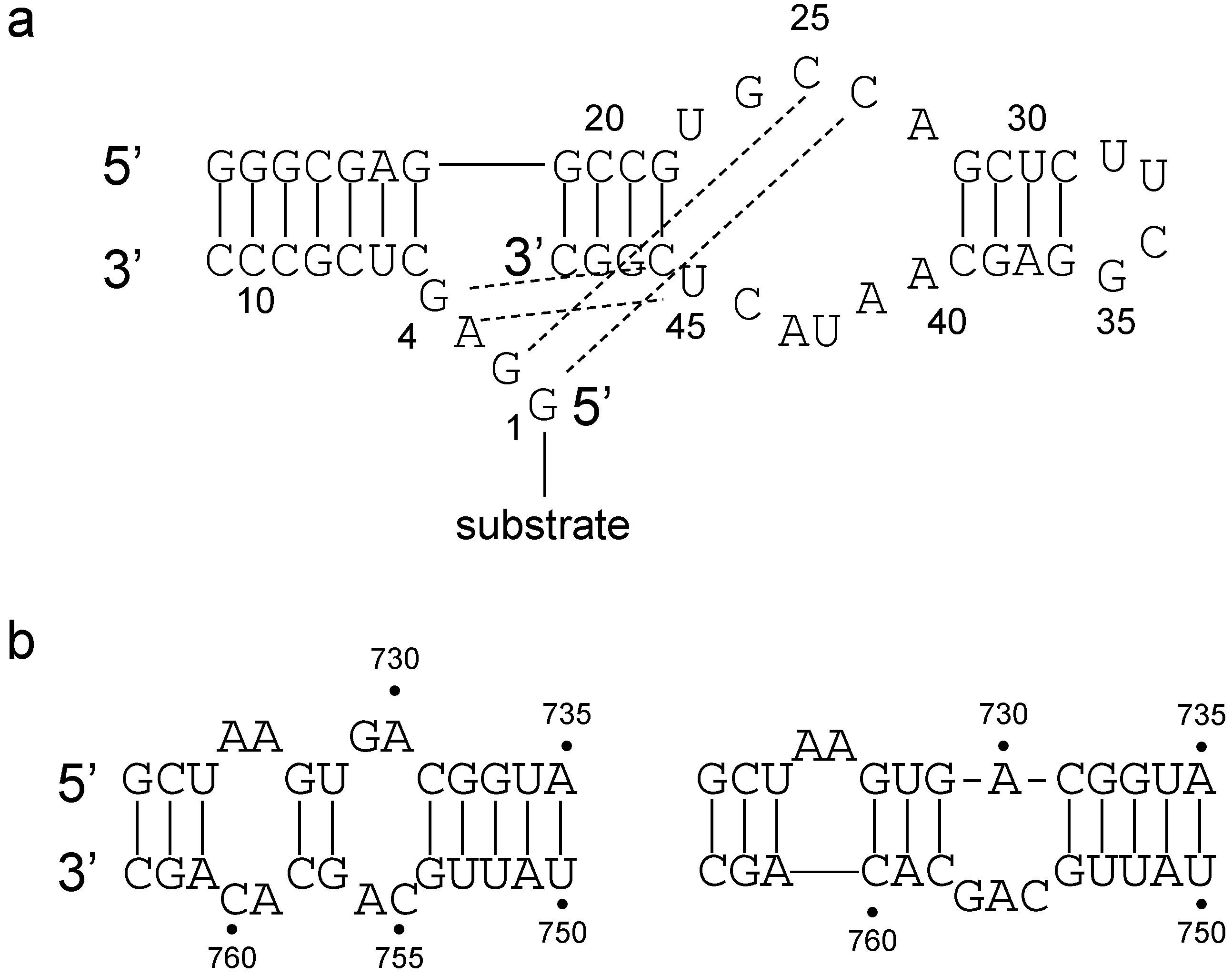
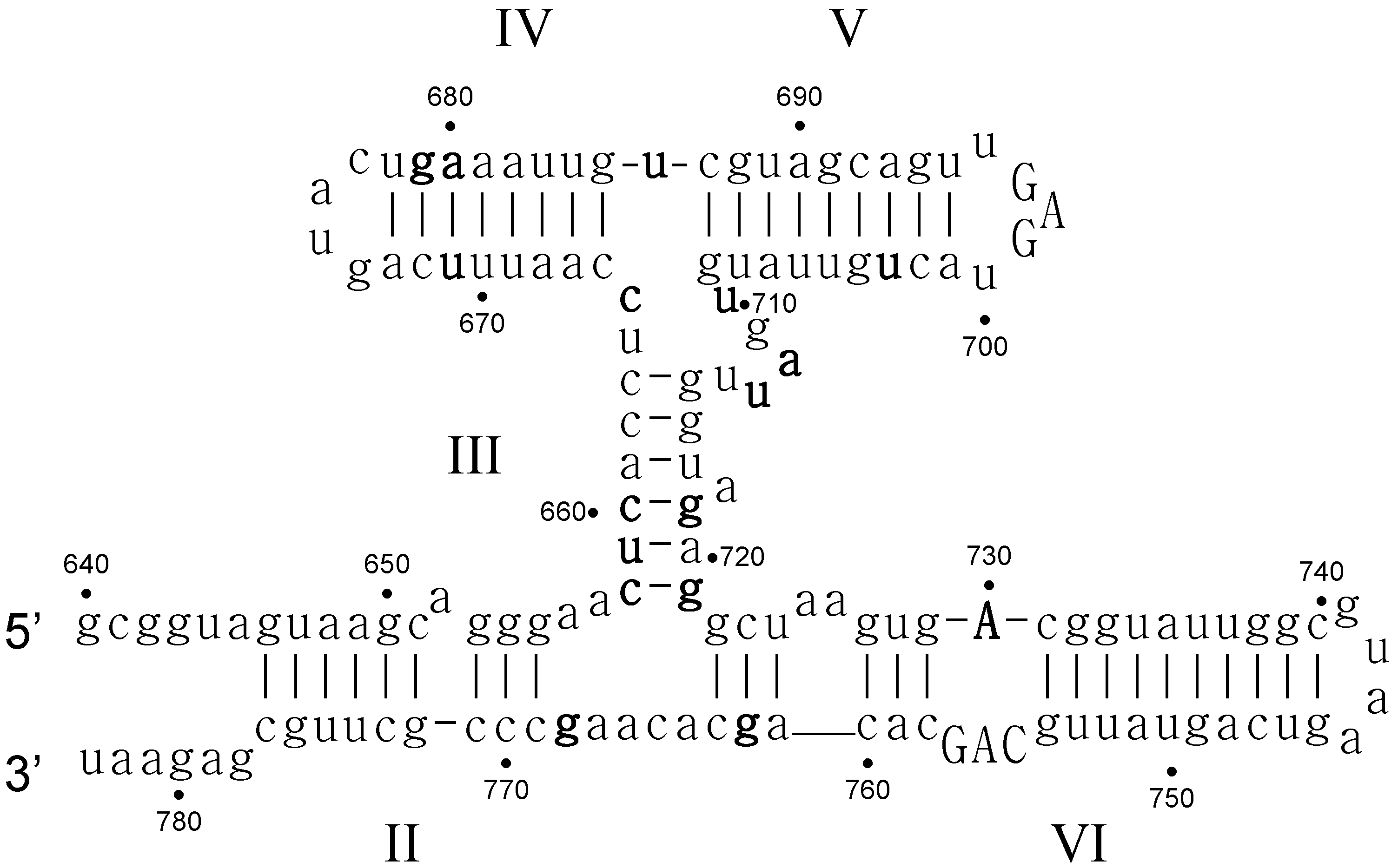
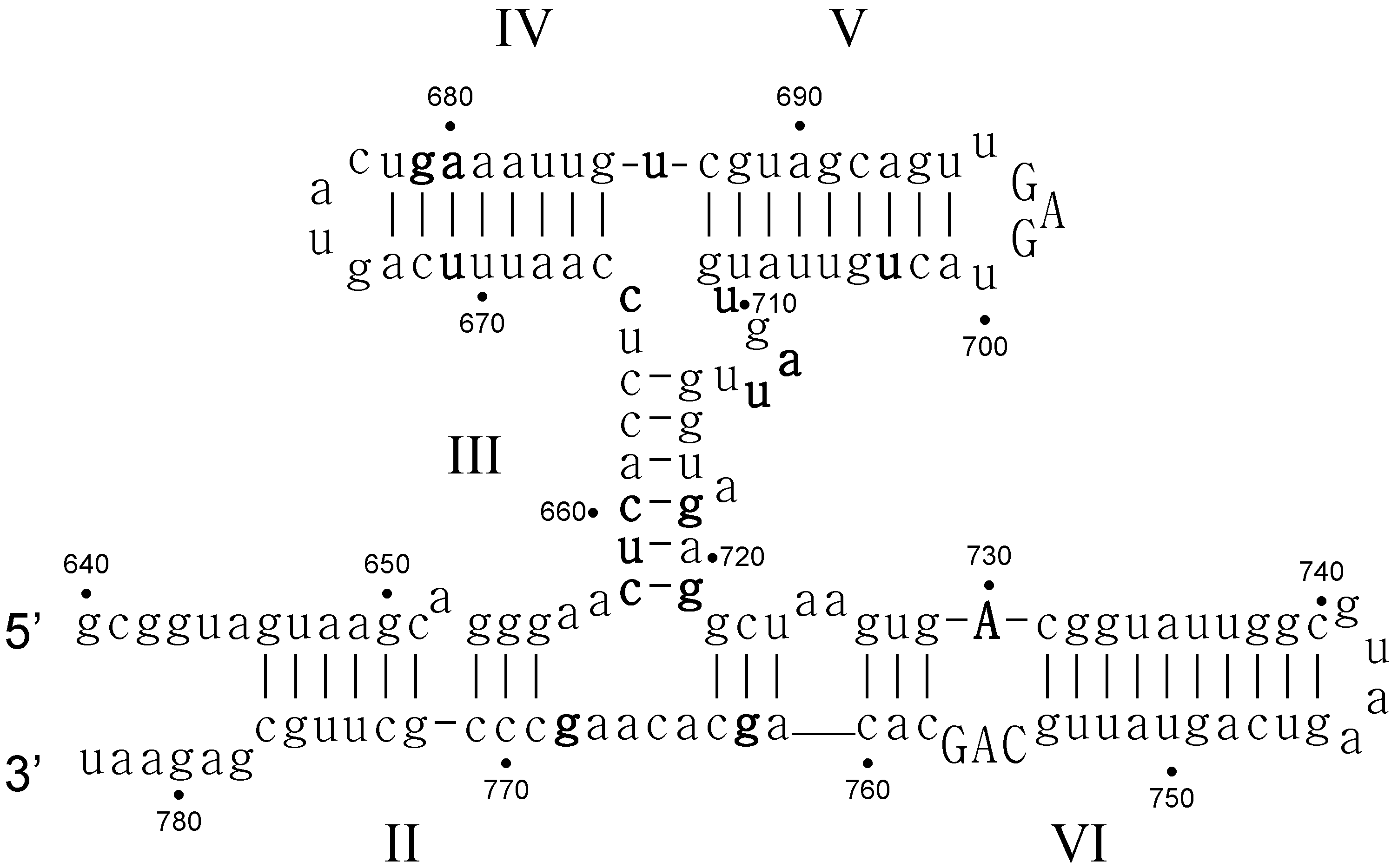
- Critical structure contains functional critical sites. Figure 2 shows a number of such structural elements, and the end loop of region V and the internal loop of region VI of the VS ribozyme are further examples.
- Connecting structure contains no functional critical sites, but it has structural critical sites, and is important for the positioning of the functional critical sites. Region III is such a structure: it connects the substrate binding region V and the active site region VI.
- Neutral structure is the part of the ribozyme that can be freely changed, even removed. Region IV and II, and the distal part of region VI are examples of such structures.
- Forbidden structure is one that would decrease or abolish activity if present. Examples are the inverted bulges in region II, III and IV of the VS ribozyme. The information content of absence is often overlooked [100]. For a general fitness landscape we also need to know what can be there and what cannot.
5. Double Mutations and the Question of Epistasis
6. Far from the Wild-Type Region of the Landscape
7. Summary
- There are multiple solutions to the same functionality.
- Functionality often depends on small motifs, and the rest of the aptamer need only not interfere with the motif or to allow the required stacking of motif parts.
- Even very rare structures can have a function, though evolution will rarely, if ever, find them.
- There are critical sites where substitutions are not tolerated. These sites can be either functional critical or structural critical sites/units. Change in a functional critical site does not change the structure of the RNA, but abolishes activity. Change in a structural unit (a single nucleotide or a base-pair) abolishes activity through change in the structure.
- Structural elements or bases not appearing at certain places are as informative as permitted structures. While these structural elements, by definition, are not part of the structure, they show the limit of structural changes. We call these features forbidden structures.
- Apart from critical and forbidden structures, a structure can be characterized by uncovering connecting structures and neutral structures too. Connecting structures link the structural critical sites, but their exact forms are not prescribed. Neutral structures can be changed (to some extent) freely without affecting activity.
- RNA structure is even important for peptides, as the structure of the mRNA or RNA viruses can influence the replication, expression and activity of the translated peptide even when the resultant peptides would have the very same sequence (i.e., there are only synonymous changes).
Supplementary Files
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Wright, S. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. In Proceedings of the Sixth International Congress on Genetics, New York, NY, USA, 1932; pp. 355–366.
- Lafontaine, D.A.; Norman, D.G.; Lilley, D.M.J. The structure and active site of the varkund satellite ribozyme. Biochem. Soc. Trans. 2002, 30, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Fedor, M. Structure and function of the hairpin ribozyme. J. Mol. Biol. 2000, 297, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N. Assessing the likelihood of recurrence during RNA evolution in vitro. Artif. Life 2004, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G. RNA secondary structure: Physical and computational aspects. Q. Rev. Biophys. 2000, 33, 199–253. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zucker, M.; Turner, H. Expanded sequence dependence of thermodynamic parameters provides robust prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
- Deigan, K.E.; Li, T.W.; Mathews, D.H.; Weeks, K.M. Accurate shape-directed RNA structure determination. Proc. Natl. Acad. Sci. USA 2009, 106, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Haslinger, C.; Stadler, P.F. RNA structure with pseudo-knots: Graph-theoretical and combinatorial properties. Bull. Math. Biol. 1999, 61, 437–467. [Google Scholar] [CrossRef] [PubMed]
- Schuster, P.; Fontana, W.; Stadler, P.F.; Hofacker, I.L. From sequences to shapes and back: A case study in RNA secondary structures. Proc. R. Soc. Lond. B 1994, 255, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.; Honer zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.; Hofacker, I. Viennarna package 2.0. Algorithms Mol. Biol. 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Andronescu, M.; Bereg, V.; Hoos, H.H.; Condon, A. RNA strand: The RNA secondary structure and statistical analysis database. BMC Bioinform. 2008, 9, 340–340. [Google Scholar] [CrossRef] [PubMed]
- Doshi, K.J.; Cannone, J.J.; Cobaugh, C.W.; Gutell, R.R. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinform. 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowell, R.D.; Eddy, S.R. Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinform. 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.-Q.; Townsend, H.L.; Jha, B.K.; Paranjape, J.M.; Silverman, R.H.; Barton, D.J. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J. Virol. 2007, 81, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 2014, 505, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire hiv-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Chen, R.; Yun, R.; Rossi, S.L.; Plante, K.S.; Guerbois, M.; Forrester, N.; Perng, G.C.; Sreekumar, E.; Leal, G.; et al. Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in aedes albopictus mosquitoes. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Luksza, M.; Lassig, M. A predictive fitness model for influenza. Nature 2014, 507, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Marton, S.; Reyes-Darias, J.A.; Sánchez-Luque, F.J.; Romero-López, C.; Berzal-Herranz, A. In vitro and ex vivo selection procedures for identifying potentially therapeutic DNA and RNA molecules. Molecules 2010, 15, 4610–4638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri, S.; Kashani-Sabet, M. Ribozymes in the age of molecular therapeutics. Curr. Mol. Med. 2004, 4, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Thiel, K.W.; Giangrande, P.H. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides 2009, 19, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Bunka, D.H.J.; Platonova, O.; Stockley, P.G. Development of aptamer therapeutics. Curr. Opin. Pharmacol. 2010, 10, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Santosh, B.; Yadava, P.K. Nucleic acid aptamers: Research tools in disease diagnostics and therapeutics. BioMed Res. Int. 2014, 2014, 540451. [Google Scholar] [CrossRef] [PubMed]
- Bunka, D.H.J.; Stockley, P.G. Aptamers come of age—At last. Nat. Rev. Microbiol. 2006, 4, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Shum, K.-T.; Zhou, J.; Rossi, J. Aptamer-based therapeutics: New approaches to combat human viral diseases. Pharmaceuticals 2013, 6, 1507–1542. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, Y.; Hong, H.; Zhang, Y.; Cai, W.; Fang, D. Aptamers as therapeutics in cardiovascular diseases. Curr. Med. Chem. 2011, 18, 4169–4174. [Google Scholar] [CrossRef] [PubMed]
- Strehlitz, B.; Reinemann, C.; Linkorn, S.; Stoltenburg, R. Aptamers for pharmaceuticals and their application in environmental analytics. Bioanal. Rev. 2012, 4, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Reinemann, C.; Strehlitz, B. Aptamer-modified nanoparticles and their use in cancer diagnostics and treatment. Swiss Med. Wkly. 2014, 144, w13908. [Google Scholar] [CrossRef] [PubMed]
- Aravind, A.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Aptamer-conjugated polymeric nanoparticles for targeted cancer therapy. Drug Deliv. Transl. Res. 2012, 2, 418–436. [Google Scholar] [CrossRef] [PubMed]
- Barbas, A.S.; Mi, J.; Clary, B.M.; White, R.R. Aptamer applications for targeted cancer therapy. Future Oncol. 2010, 6, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Kun, Á.; Szilágyi, A.; Könnyű, B.; Boza, G.; Zachár, I.; Szathmáry, E. The dynamics of the RNA world: Insights and challenges. Ann. N. Y. Acad. Sci. 2015, 1341, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Colegrave, N.; Buckling, A. Microbial experiments on adaptive landscapes. BioEssays 2005, 27, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.; Hartl, D.L. Adaptive landscapes and protein evolution. Proc. Natl. Acad. Sci. USA 2010, 107, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Sanjuán, R. RNA viruses as complex adaptive systems. Biosystems 2005, 81, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Athavale, S.S.; Spicer, B.; Chen, I.A. Experimental fitness landscapes to understand the molecular evolution of RNA-based life. Curr. Opin. Chem. Biol. 2014, 22, 35–39. [Google Scholar] [CrossRef] [PubMed]
- De Visser, J.A.G.M.; Krug, J. Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 2014, 15, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. Selex—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage t4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.I.; Xulvi-Brunet, R.; Campbell, G.W.; Turk-MacLeod, R.; Chen, I.A. Comprehensive experimental fitness landscape and evolutionary network for small RNA. Proc. Natl. Acad. Sci. USA 2013, 110, 14984–14989. [Google Scholar] [CrossRef] [PubMed]
- Schuster, P. Genotypes with phenotypes: Adventures in an RNA toy world. Biophys. Chem. 1997, 66, 75–110. [Google Scholar] [CrossRef]
- Bayrac, A.T.; Sefah, K.; Parekh, P.; Bayrac, C.; Gulbakan, B.; Oktem, H.A.; Tan, W. In vitro selection of DNA aptamers to glioblastoma multiforme. ACS Chem. Neurosci. 2011, 2, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, K.; Li, Y. Diverse evolutionary trajectories characterize a community of RNA-cleaving deoxyribozymes: A case study into the population dynamics of in vitro selection. J. Mol. Evol. 2005, 61, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Ameta, S.; Winz, M.-L.; Previti, C.; Jäschke, A. Next-generation sequencing reveals how RNA catalysts evolve from random space. Nucleic Acids Res. 2014, 42, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Xiao, Y.; Nie, J.; Stewart, R.; Csordas, A.T.; Oh, S.S.; Thomson, J.A.; Soh, H.T. Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 15373–15378. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Bair, T.; Peek, A.S.; Liu, X.; Dassie, J.; Stockdale, K.R.; Behlke, M.A.; Miller, F.J., Jr.; Giangrande, P.H. Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS ONE 2012, 7, e43836. [Google Scholar] [CrossRef] [PubMed]
- Musafia, B.; Oren-Banaroya, R.; Noiman, S. Designing anti-influenza aptamers: Novel quantitative structure activity relationship approach gives insights into aptamer—Virus interaction. PLoS ONE 2014, 9, e97696. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.O.; Tok, J.B.H.; Tarasow, T.M. Massively parallel interrogation of aptamer sequence, structure and function. PLoS ONE 2008, 3, e2720. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Luque, F.J.; Stich, M.; Manrubia, S.; Briones, C.; Berzal-Herranz, A. Efficient hiv-1 inhibition by a 16 nt-long RNA aptamer designed by combining in vitro selection and in silico optimisation strategies. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Ducongé, F.; Toulmé, J.J. In vitro selection identifies key determinants for loop-loop interactions: RNA aptamers selective for the tar RNA element of hiv-1. RNA 1999, 5, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Ditzler, M.A.; Lange, M.J.; Bose, D.; Bottoms, C.A.; Virkler, K.F.; Sawyer, A.W.; Whatley, A.S.; Spollen, W.; Givan, S.A.; Burke, D.H. High-throughput sequence analysis reveals structural diversity and improved potency among RNA inhibitors of hiv reverse transcriptase. Nucleic Acids Res. 2013, 41, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Whatley, A.S.; Ditzler, M.A.; Lange, M.J.; Biondi, E.; Sawyer, A.W.; Chang, J.L.; Franken, J.D.; Burke, D.H. Potent inhibition of HIV-1 reverse transcriptase and replication by nonpseudoknot, “ucaa-motif” RNA aptamers. Mol. Ther. Nucleic Acids 2013, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Chumachenko, N.V.; Novikov, Y.; Yarus, M. Rapid and simple ribozymic aminoacylation using three conserved nucleotides. J. Am. Chem. Soc. 2009, 131, 5257–5263. [Google Scholar] [CrossRef] [PubMed]
- Illangasekare, M.; Yarus, M. A tiny RNA that catalyzes both aminoacyl-tRNA and peptidyl-RNA synthesis. RNA 1999, 5, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Firnberg, E.; Labonte, J.W.; Gray, J.J.; Ostermeier, M. A comprehensive, high-resolution map of a gene’s fitness landscape. Mol. Biol. Evol. 2014, 31, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Cech, T.R. The chemical repertoire of natural ribozymes. Nature 2002, 418, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Sharmeen, L.; Kuo, M.Y.P.; Dinner-Gottlieb, G.; Taylor, J. Antigenomic RNA of human hepatitis delta viruses can undergo self-cleavage. J. Virol. 1988, 62, 2674–2679. [Google Scholar] [PubMed]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease p is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Perrotta, A.T.; Been, M.D. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature 1991, 350, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P.; Szostak, J.W. Isolation of a new ribozyme from a large pool of random sequences. Science 1993, 261, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.K.; Unrau, P.J.; Lawrence, M.S.; Glasen, M.E.; Bartel, D.P. RNA-catalyzed RNA polymerization: Accurate and general RNA-templated primer extension. Science 2001, 292, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Zaher, H.S.; Unrau, P.J. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA 2007, 13, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Attwater, J.; Wochner, A.; Holliger, P. In-ice evolution of RNA polymerase ribozyme activity. Nat. Chem. 2013, 5, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Wochner, A.; Attwater, J.; Coulson, A.; Holliger, P. Ribozyme-catalyzed transcription of an active ribozyme. Science 2011, 332, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C.; Nahvi, A.; Roth, A.; Collins, J.A.; Breaker, R.R. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 2004, 428, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Weinberg, Z.; Chen, A.G.Y.; Kim, P.B.; Ames, T.D.; Breaker, R.R. A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat. Chem. Biol. 2014, 10, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Kruger, K.; Grabowski, P.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Peebles, C.L.; Perlman, P.S.; Mecklenburg, K.L.; Pertillo, M.L.; Tabor, J.H.; Jarrell, K.A.; Cheng, H.-L. A self-splicing RNA excises an intron lariat. Cell 1986, 44, 213–223. [Google Scholar] [CrossRef]
- Saville, B.J.; Collins, R.A. A site-specific self-cleavage reaction performed by a novel RNA in neurospora mitochondria. Cell 1990, 61, 685–696. [Google Scholar] [CrossRef]
- Hampel, A.; Tritz, R.R. RNA catalytic properties of the minimum (-)strsv sequences. Biochemistry 1989, 28, 4929–4933. [Google Scholar] [CrossRef] [PubMed]
- Forster, A.C.; Symons, R.H. Self-cleavage of plus and minus RNAs of a virusoid and a structural model for the active site. Cell 1987, 49, 211–220. [Google Scholar] [CrossRef]
- Seelig, B.; Jäschke, A. A small catalytic RNA motif with diels-alderase activity. Chem. Biol. 1999, 6, 167–176. [Google Scholar] [CrossRef]
- Ekland, E.H.; Szostak, J.W.; Bartel, D.P. Structurally complex and highly active RNA ligases derived from random RNA sequences. Science 1995, 269, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.N.; Ferré-D’Amaré, A.R. Rapid construction of empirical RNA fitness landscapes. Science 2010, 330, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.N.; Ferré-D’Amaré, A.R. Structure-guided engineering of the regioselectivity of RNA ligase ribozymes. J. Am. Chem. Soc. 2009, 131, 3532–3540. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatchefte Chem. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Keiper, S.; Bebenroth, D.; Seelig, B.; Westhof, E.; Jaschke, A. Architecture of a diels-alderase ribozyme with a preformed catalytic pocket. Chem. Biol. 2004, 11, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Keiper, S.; Malinina, L.; Tereshko, V.; Skripkin, E.; Hobartner, C.; Polonskaia, A.; Phan, A.T.; Wombacher, R.; Micura, R.; et al. Structural basis for diels-alder ribozyme-catalyzed carbon-carbon bond formation. Nat. Struct. Mol. Biol. 2005, 12, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Beattie, T.L.; Olive, J.E.; Collins, R.A. A secondary-structure model for the self-cleaving region of the Neurospora VS RNA. Proc. Natl. Acad. Sci. USA 1995, 92, 4686–4690. [Google Scholar] [CrossRef] [PubMed]
- Flinders, J.; Dieckmann, T. The solution structure of the VS ribozyme active site loop reveals a dynamic “hot-spot”. J. Mol. Biol. 2004, 341, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Kun, Á.; Mauro, S.; Szathmáry, E. Real ribozymes suggest a relaxed error threshold. Nat. Genet. 2005, 37, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Kun, Á.; Maurel, M.-C.; Santos, M.; Szathmáry, E. Fitness landscapes, error thresholds, and cofactors in aptamer evolution. In The Aptamer Handbook; Klussmann, S., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 54–92. [Google Scholar]
- Campbell, D.O.; Bouchard, P.; Desjardins, G.; Legault, P. NMR structure of varkud satellite ribozyme stem-loop v in the presence of magnesium ions and localization of metal-binding sites. Biochemistry 2006, 45, 10591–10605. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.O.; Legault, P. Nuclear magnetic resonance structure of the varkud satellite ribozyme stem-loop v RNA and magnesium-ion binding from chemical-shift mapping. Biochemistry 2005, 44, 4157–4170. [Google Scholar] [CrossRef] [PubMed]
- Michiels, P.J.; Schouten, C.H.; Hilbers, C.W.; Heus, H.A. Structure of the ribozyme substrate hairpin of Neurospora VS RNA: A close look at the cleavage site. RNA 2000, 6, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Mitchell, G.T.; Gendron, P.; Major, F.; Andersen, A.A.; Collins, R.A.; Legault, P. Nmr structure of the active conformation of the varkud satellite ribozyme cleavage site. Proc. Natl. Acad. Sci. USA 2003, 100, 7003–7008. [Google Scholar] [CrossRef] [PubMed]
- Flinders, J.; Dieckmann, T. A ph controlled conformational switch in the cleavage site of the VS ribozyme substrate RNA. J. Mol. Biol. 2001, 308, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, P.; Lacroix-Labonté, J.; Desjardins, G.; Lampron, P.; Lisi, V.; Lemieux, S.; Major, F.; Legault, P. Role of slv in sli substrate recognition by the Neurospora VS ribozyme. RNA 2008, 14, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, P.; Legault, P. Structural insights into substrate recognition by the neurospora varkud satellite ribozyme: Importance of u-turns at the kissing-loop junction. Biochemistry 2013, 53, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, P.; Legault, P. A remarkably stable kissing-loop interaction defines substrate recognition by the Neurospora Varkud Satellite ribozyme. RNA 2014, 20, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, G.; Bonneau, E.; Girard, N.; Boisbouvier, J.; Legault, P. Nmr structure of the a730 loop of the Neurospora VS ribozyme: Insights into the formation of the active site. Nucleic Acids Res. 2011, 39, 4427–4437. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.A.; Norman, D.G.; Lilley, D.M.J. The global structure of the VS ribozyme. EMBO J. 2002, 21, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, T.; Collins, R.A. Smaller, faster ribozymes reveal the catalytic core of Neurospora VS RNA. J. Mol. Biol. 1998, 277, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lacroix-Labonté, J.; Girard, N.; Lemieux, S.; Legault, P. Helix-length compensation studies reveal the adaptability of the VS ribozyme architecture. Nucleic Acids Res. 2012, 40, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, D.A.; Wilson, T.J.; Norman, D.G.; Lilley, D.M.J. The a730 loop is an important component of the active site of the VS ribozyme. J. Mol. Biol. 2001, 312, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, E.; Legault, P. Nuclear Magnetic Resonance Structure of the III–IV–V Three-Way Junction from the Varkud Satellite Ribozyme and Identification of Magnesium-Binding Sites Using Paramagnetic Relaxation Enhancement. Biochemistry 2014, 53, 6264–6275. [Google Scholar] [CrossRef] [PubMed]
- Jakó, É.; Ittzés, P.; Szenes, Á.; Kun, Á.; Szathmáry, E.; Pál, G. In silico detection of tRNA sequence features characteristic to aminoacyl-tRNA synthetase class membership. Nucleic Acids Res. 2007, 35, 5593–5609. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; McLeod, A.C.; Lilley, D.M.J. A guanine nucleobase important for catalysis by the VS ribozyme. EMBO J. 2007, 26, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Solé, R.V.; Sardanyés, J. Simple genomes, complex interactions: Epistasis in RNA virus. Chaos: Interdiscip. J. Nonlinear Sci. 2010, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreich, D.M.; Watson, R.A.; Chao, L. Perspective: Sign epistasis and genetic constraint on evolutionary trajectories. Evolution 2005, 59, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, L.; Arribas, M.; Lazaro, E. Evolution at increased error rate leads to the coexistence of multiple adaptive pathways in an RNA virus. BMC Evolut. Biol. 2013, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreich, D.M.; Delaney, N.F.; DePristo, M.A.; Hartl, D.L. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 2006, 312, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Salverda, M.L.M.; de Visser, J.A.G.M.; Barlow, M. Natural evolution of tem-1 β-lactamase: Experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010. [Google Scholar] [CrossRef] [PubMed]
- De Visser, J.; Arjan, G.M.; Park, S.-C.; Krug, J. Exploring the effect of sex on empirical fitness landscapes. Am. Nat. 2009, 174, S15–S30. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.J.; Wagner, A. Environmental change exposes beneficial epistatic interactions in a catalytic RNA. Proc. R. Soc. Lond. B 2012, 279, 3418–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, N.; Joyce, G.F. Evolution in vitro: Analysis of a lineage of ribozymes. Curr. Biol. 1993, 3, 723–734. [Google Scholar] [CrossRef]
- Lalic, J.; Elena, S.F. Magnitude and sign epistasis among deleterious mutations in a positive-sense plant RNA virus. Heredity 2012, 109, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, E.A.; Bartel, D.P. New catalytic structures from an existing ribozyme. Nat. Struct. Mol. Biol. 2005, 12, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Curtis, E.A.; Bartel, D.P. Synthetic shuffling and in vitro selection reveal the rugged adaptive fitness landscape of a kinase ribozyme. RNA 2013, 19, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Huynen, M.A. Exploring phenotype space through neutral evolution. J. Mol. Evol. 1996, 43, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Aita, T.; Toyota, H.; Husimi, Y.; Urabe, I.; Yomo, T. Experimental rugged fitness landscape in protein sequence space. PLoS ONE 2006, 1, e96. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kun, Á.; Szathmáry, E. Fitness Landscapes of Functional RNAs. Life 2015, 5, 1497-1517. https://doi.org/10.3390/life5031497
Kun Á, Szathmáry E. Fitness Landscapes of Functional RNAs. Life. 2015; 5(3):1497-1517. https://doi.org/10.3390/life5031497
Chicago/Turabian StyleKun, Ádám, and Eörs Szathmáry. 2015. "Fitness Landscapes of Functional RNAs" Life 5, no. 3: 1497-1517. https://doi.org/10.3390/life5031497
APA StyleKun, Á., & Szathmáry, E. (2015). Fitness Landscapes of Functional RNAs. Life, 5(3), 1497-1517. https://doi.org/10.3390/life5031497