
The Origin and Evolution of Ribonucleotide Reduction


Abstract
:
1. Introduction
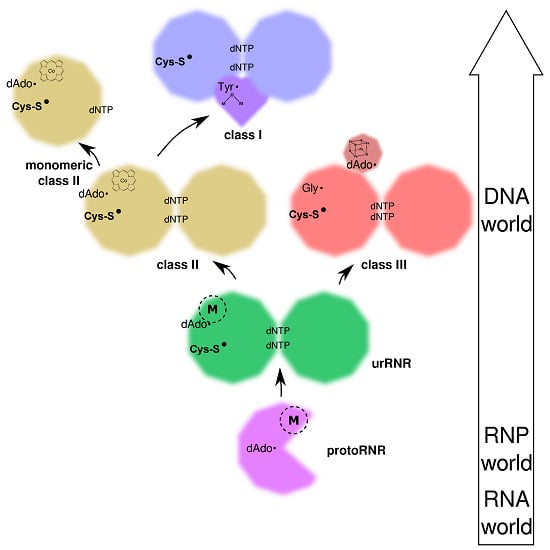
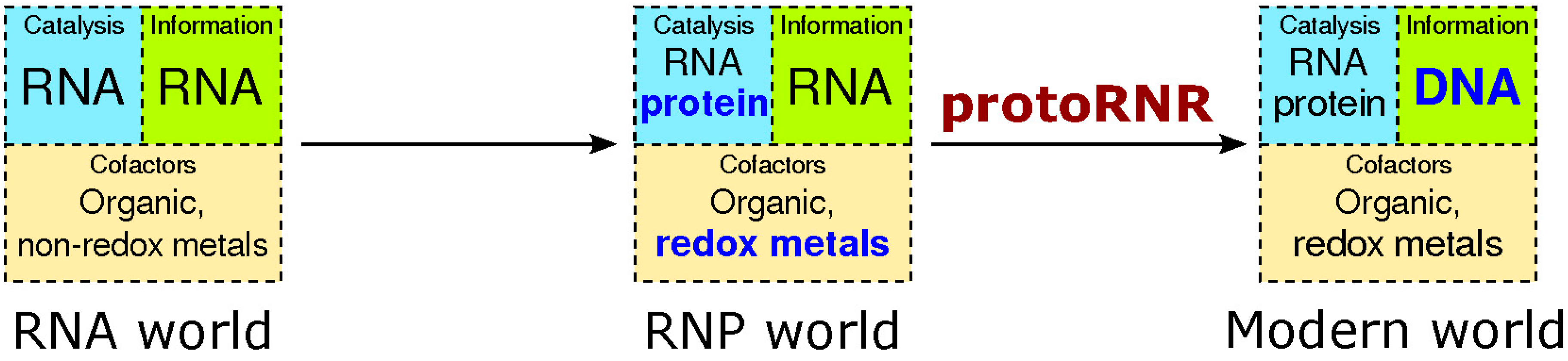
2. Origin of Ribonucleotide Reduction
2.1. The RNP World
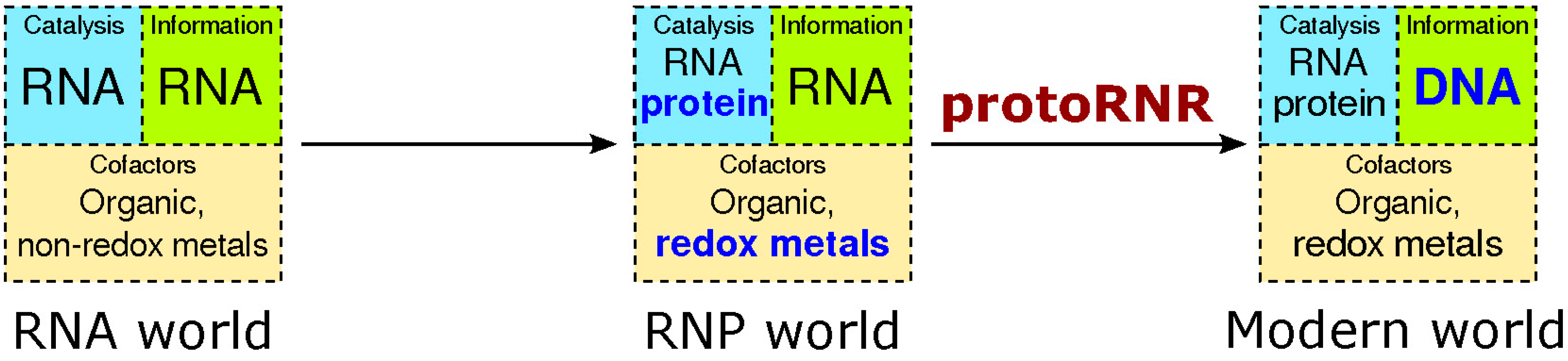
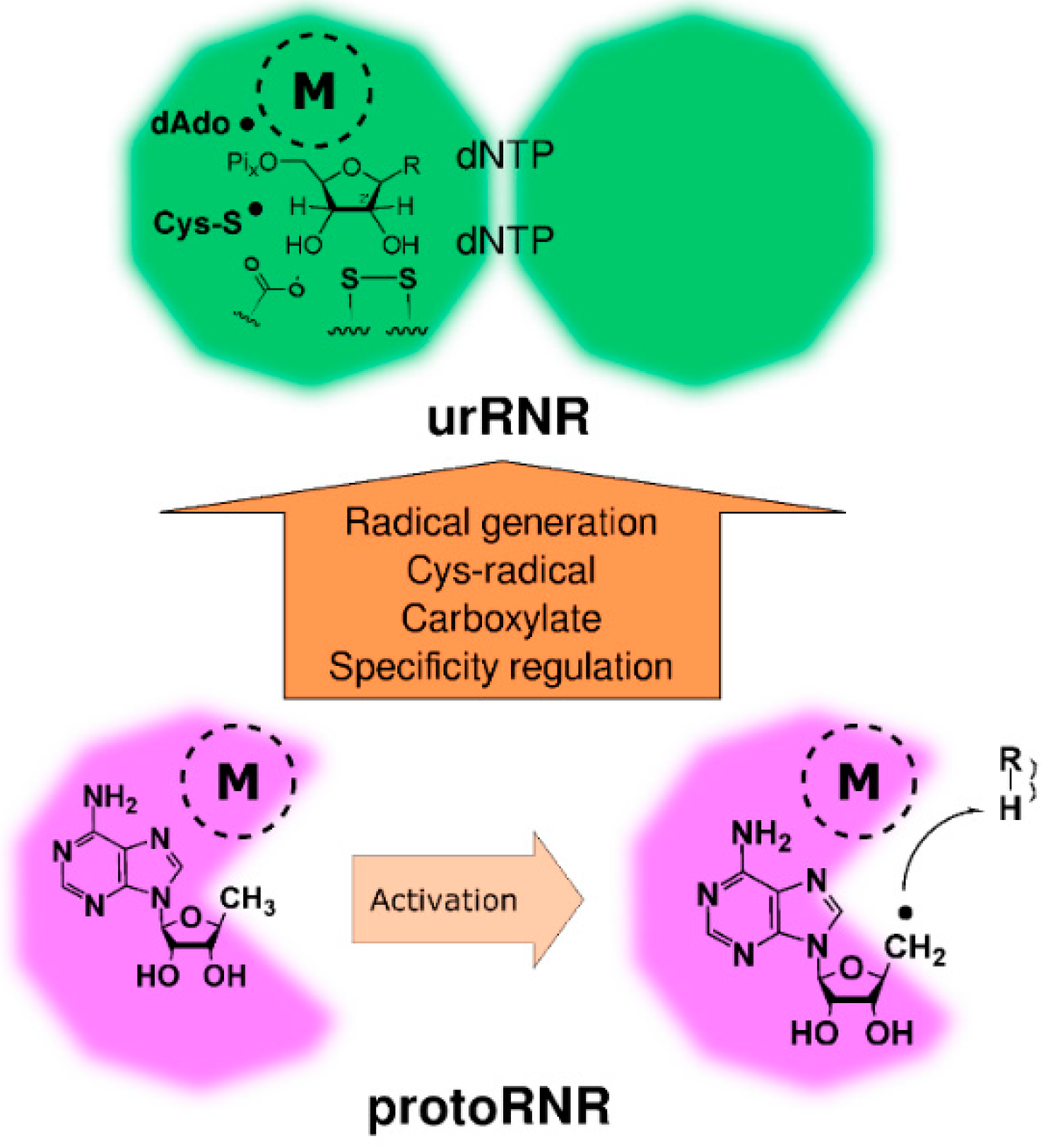
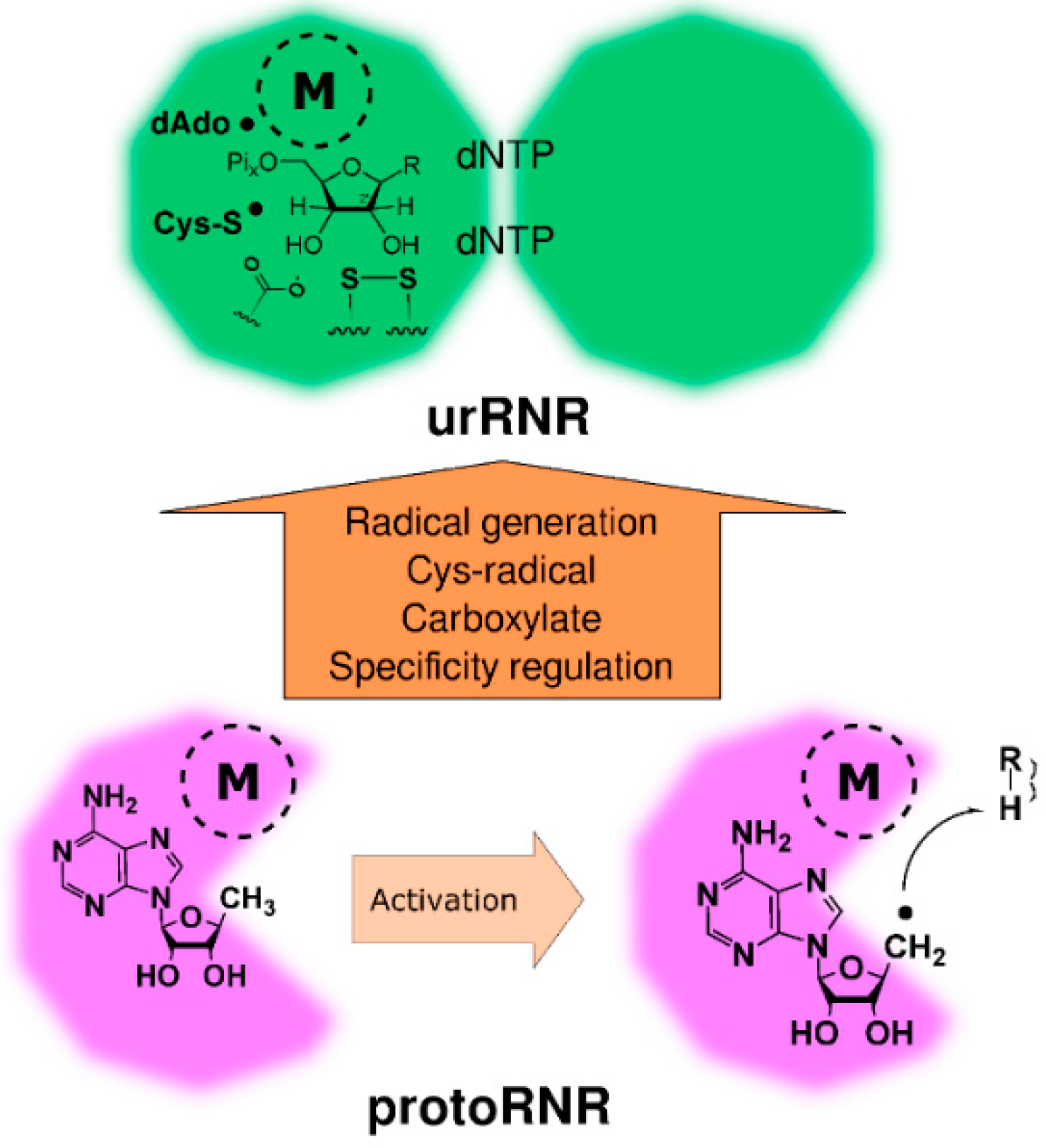
2.2. The ProtoRNR
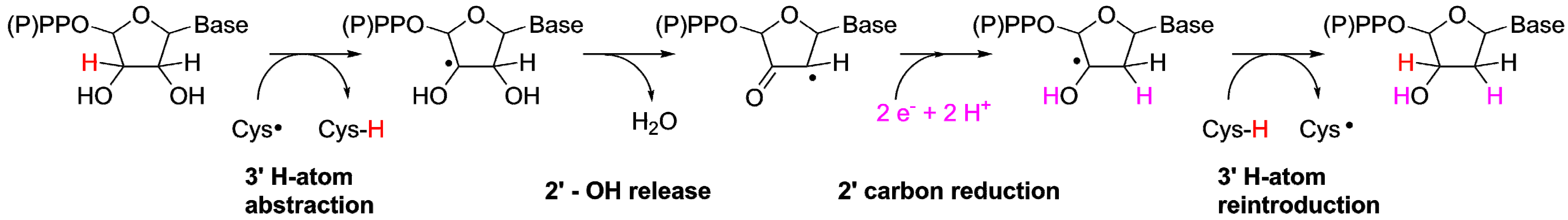
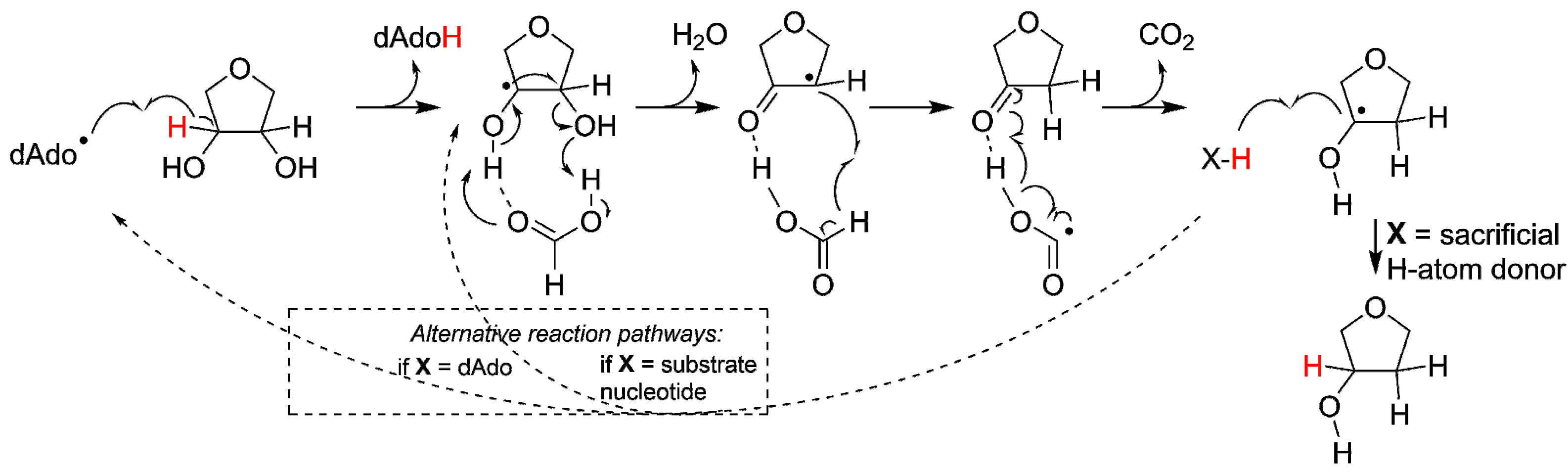
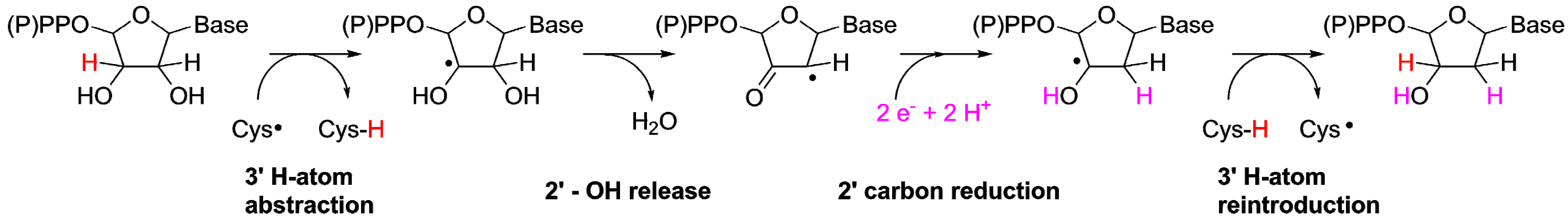
2.2.1. Reaction Mechanism of the ProtoRNR
2.2.2. The Three-Dimensional Structure of the ProtoRNR Protein
2.2.3. The ProtoRNR: An Unspecific Metal-Catalyzed Radical Enzyme
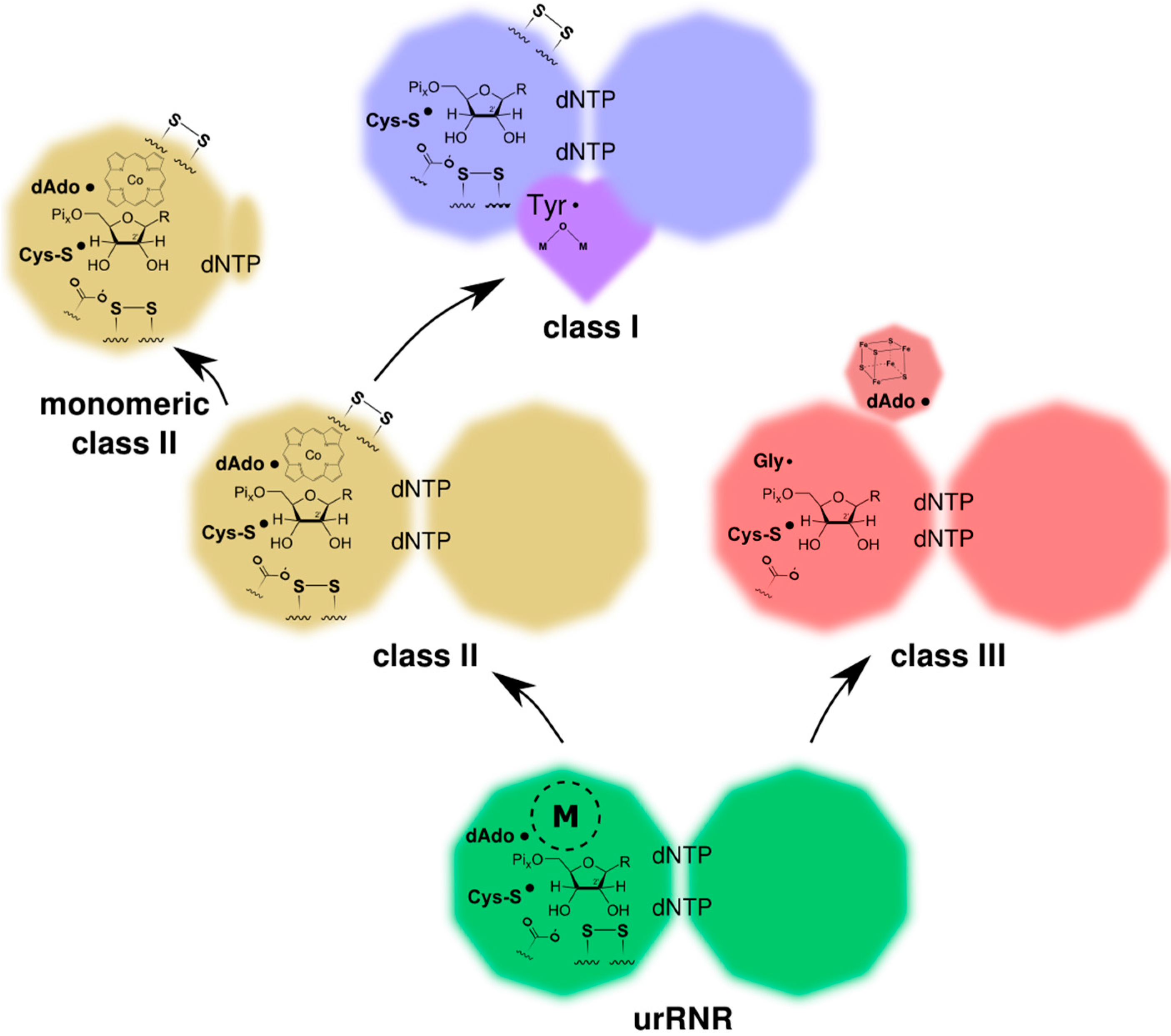
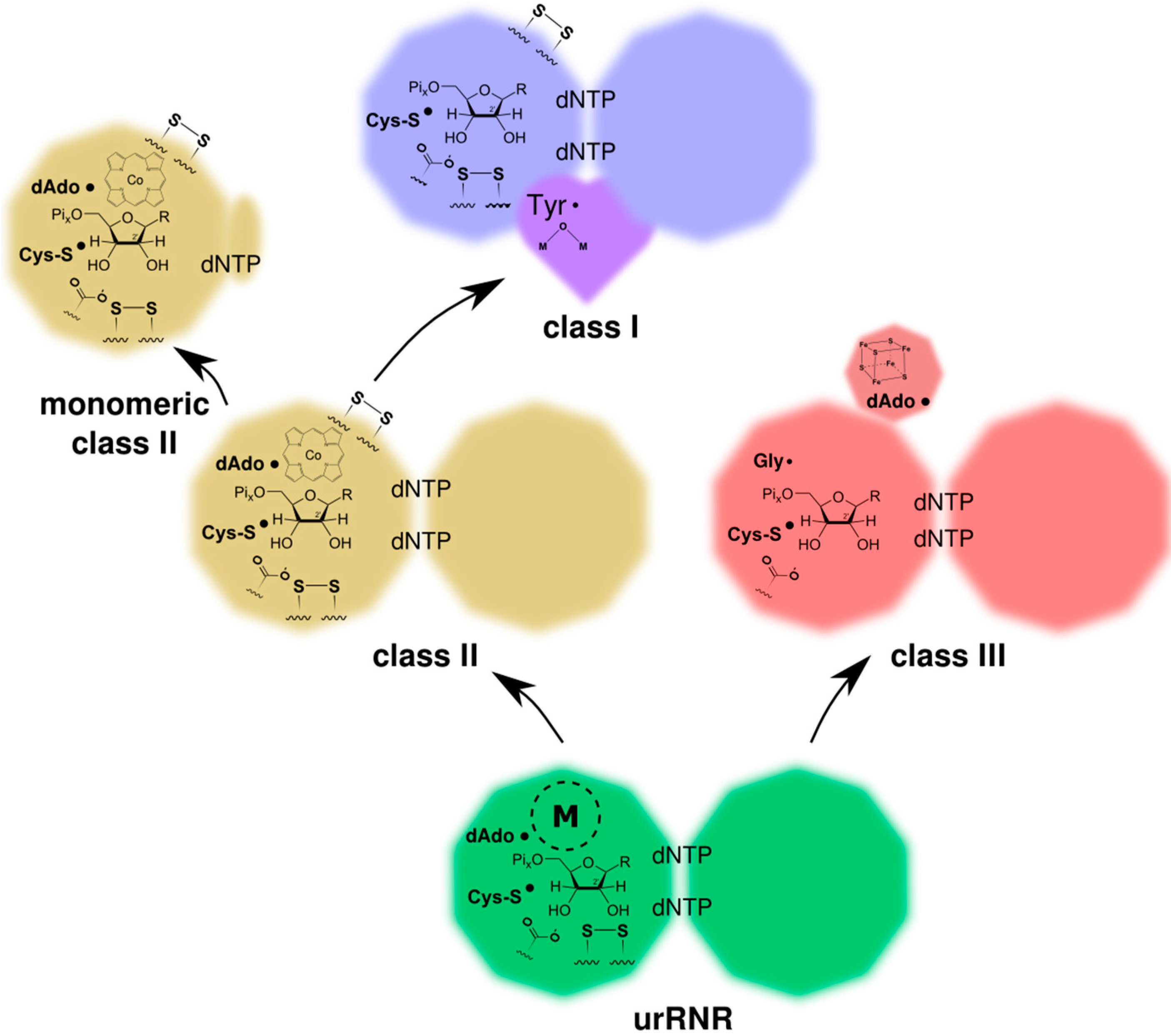
3. Origin of the UrRNR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Class I | Class II | Class III | urRNR |
|---|---|---|---|---|
| Fold | 10-stranded β/α barrel | 10-stranded β/α barrel | 10-stranded β/α barrel | 10-stranded β/α barrel |
| Substrate | NDP | Either NDP or NTP | NTP | NTP? |
| Radical generation | Dimetal-oxo center in separate subunit | AdoCbl (B12) in enzyme | AdoMet in separate subunit | Metal center in enzyme + dAdo•? |
| Protein storage radical | Tyrosine in separate subunit | None (AdoCbl regenerated) | Glycine in enzyme | None? |
| Cysteinyl radical | Yes | Yes | Yes (1) | Likely |
| Electron and proton-donating cysteine (2) | Yes | Yes | Yes | Yes |
| Primary reductant | Cysteine pair | Cysteine pair | Cysteine plus formate (3) | Cysteine pair? |
| Terminal reductant | Thioredoxin, glutaredoxin acting on C-terminal disulfide | Thioredoxin, glutaredoxin acting on C-terminal disulfide | Formate or thioredoxin acting on a disulfide (3) | ? |
| Base | Glutamate | Glutamate | Formate, glutamate (4) | Carboxylate? |
| Quaternary structure | Homodimer formed between helices A and B (5) | Homodimer formed between helices A and B (5,6) | Homodimer formed between helices A and B (5) | Homodimer formed between helices A and B (5) |
| Allosteric substrate specificity regulation | Nucleotide binding in dimer interface | Nucleotide binding in dimer interface (6) | Nucleotide binding in dimer interface | Nucleotide binding in dimer interface? |
| Allosteric activity regulation | 47% with ATP-cone (7) | 7% with ATP-cone (7) | 76% with ATP-cone (7) | Likely not |
- (2) The electron and proton-donating cysteine is one of the partners in the cysteine pair working as primary reductant that is only present in class I and II, but see note (3).
- (5) The dimer geometry is different between class I and II on the one hand and class III RNR on the other, see Section 3.5 and Figure 7.
- (6) A monomeric form with an inserted domain mimicking the dimer interface exists [39] (Section 4.3.1).
- (7) Activity regulation has only been found in conjunction with an N-terminal ATP-cone. A few RNRs lacking activity regulation due to non-functional ATP-cones are not discriminated by the HMMER profile (Pfam PF03477, see Table 2).
3.1. Substrate Phosphorylation Level
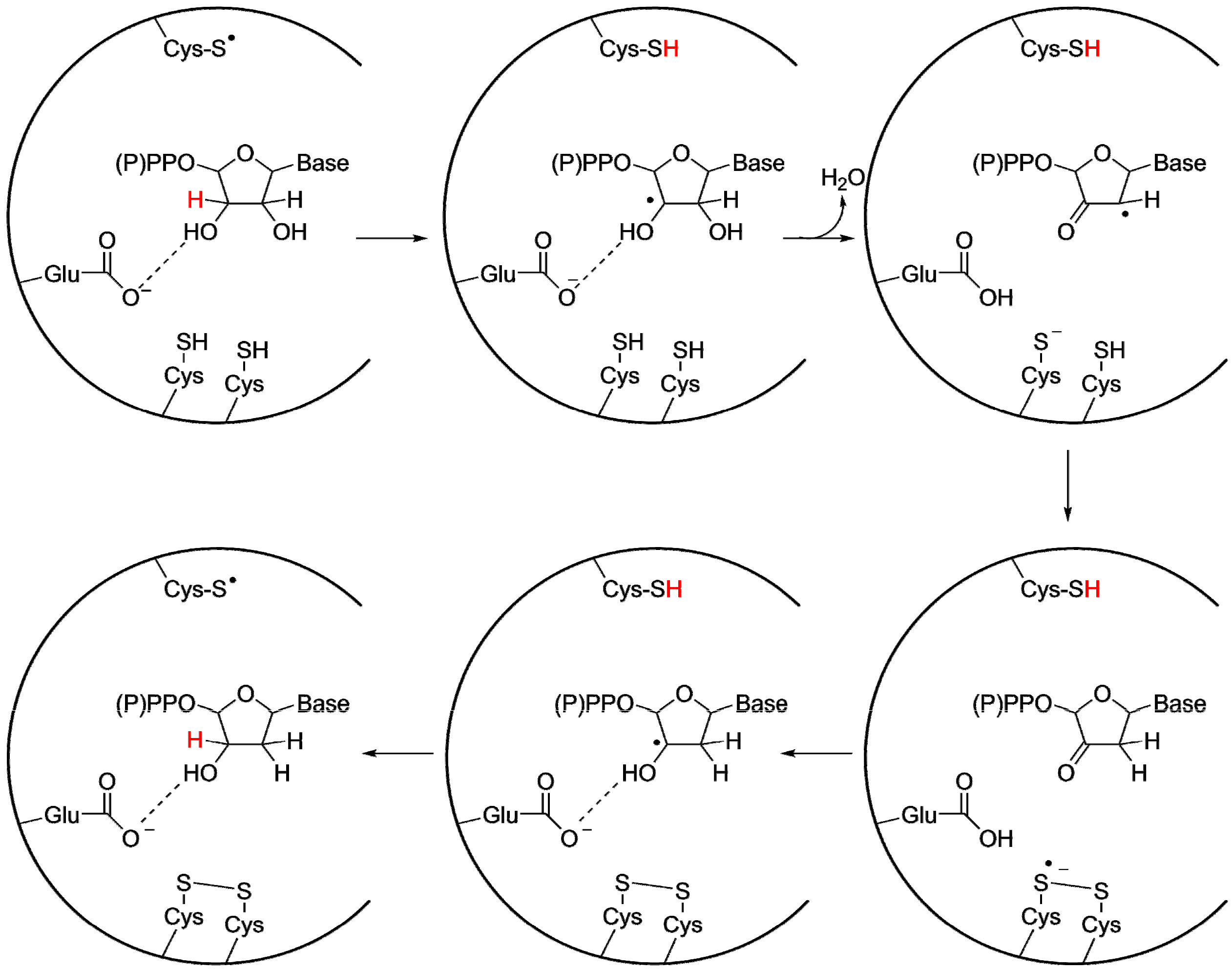
3.2. A Highly Conserved Reaction Mechanism
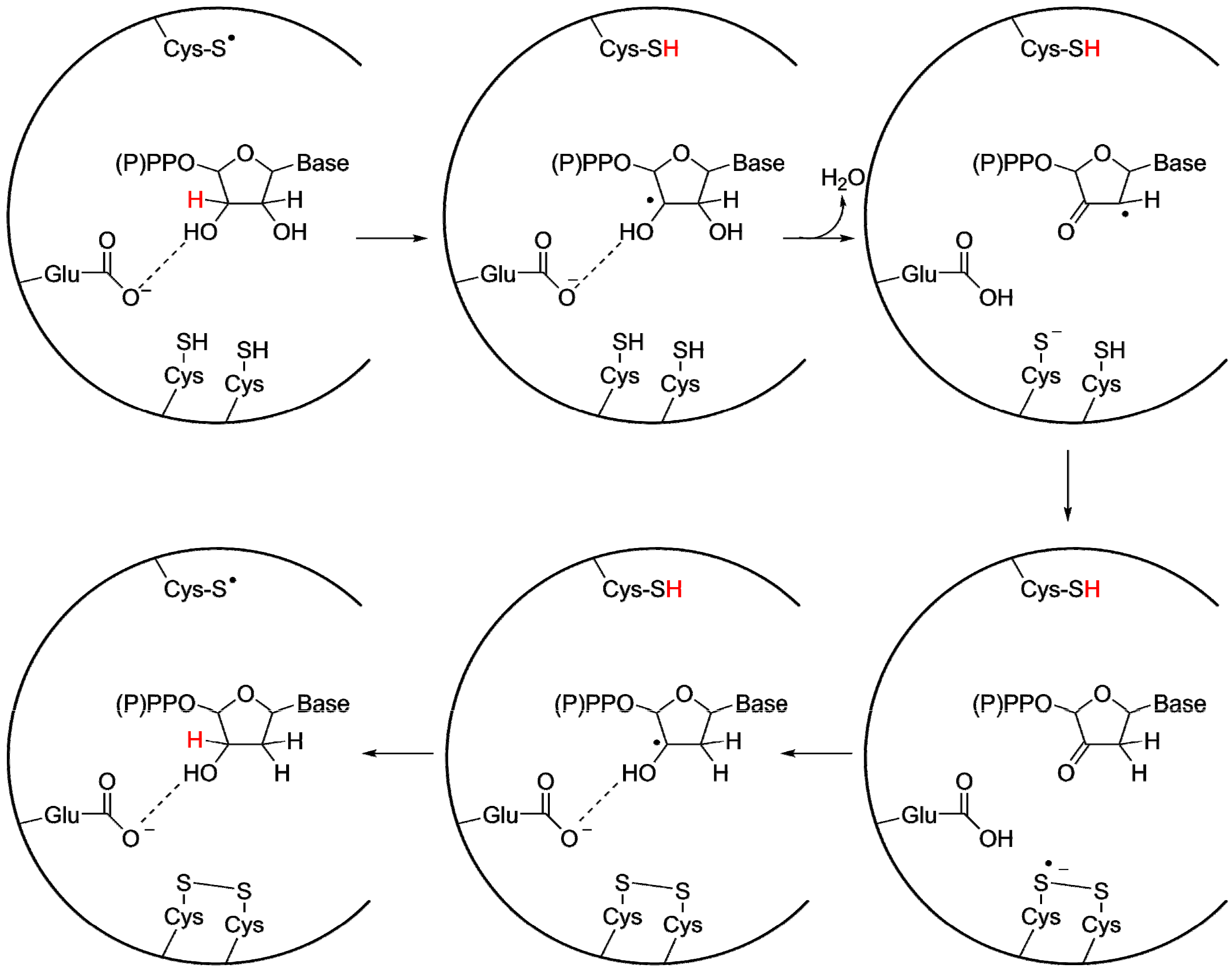
Differences Between the Class I/II and the Class III Reaction Mechanisms
3.3. Radical-Generation in the UrRNR
3.4. The Origin of the 10-Stranded β/α Barrel
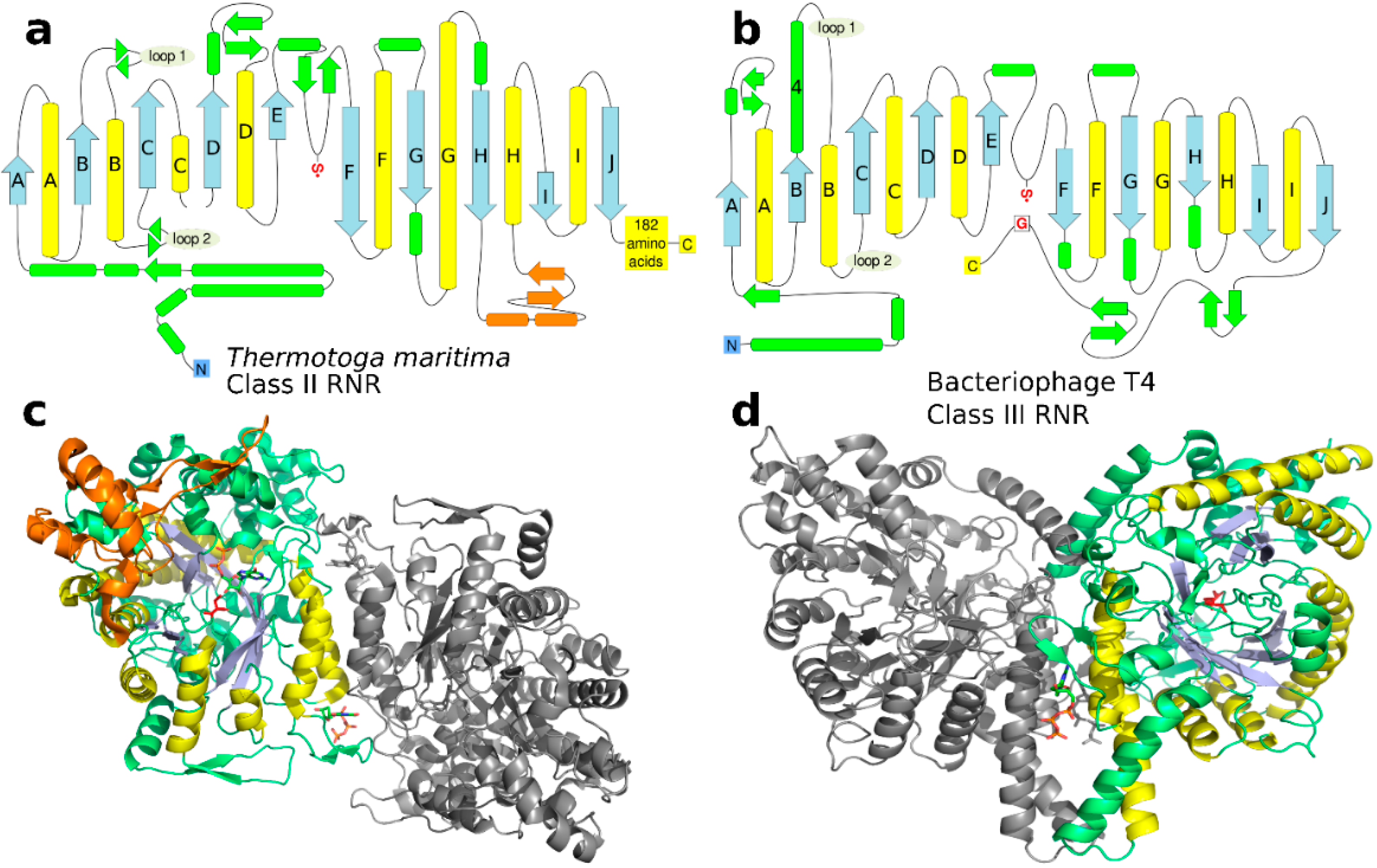
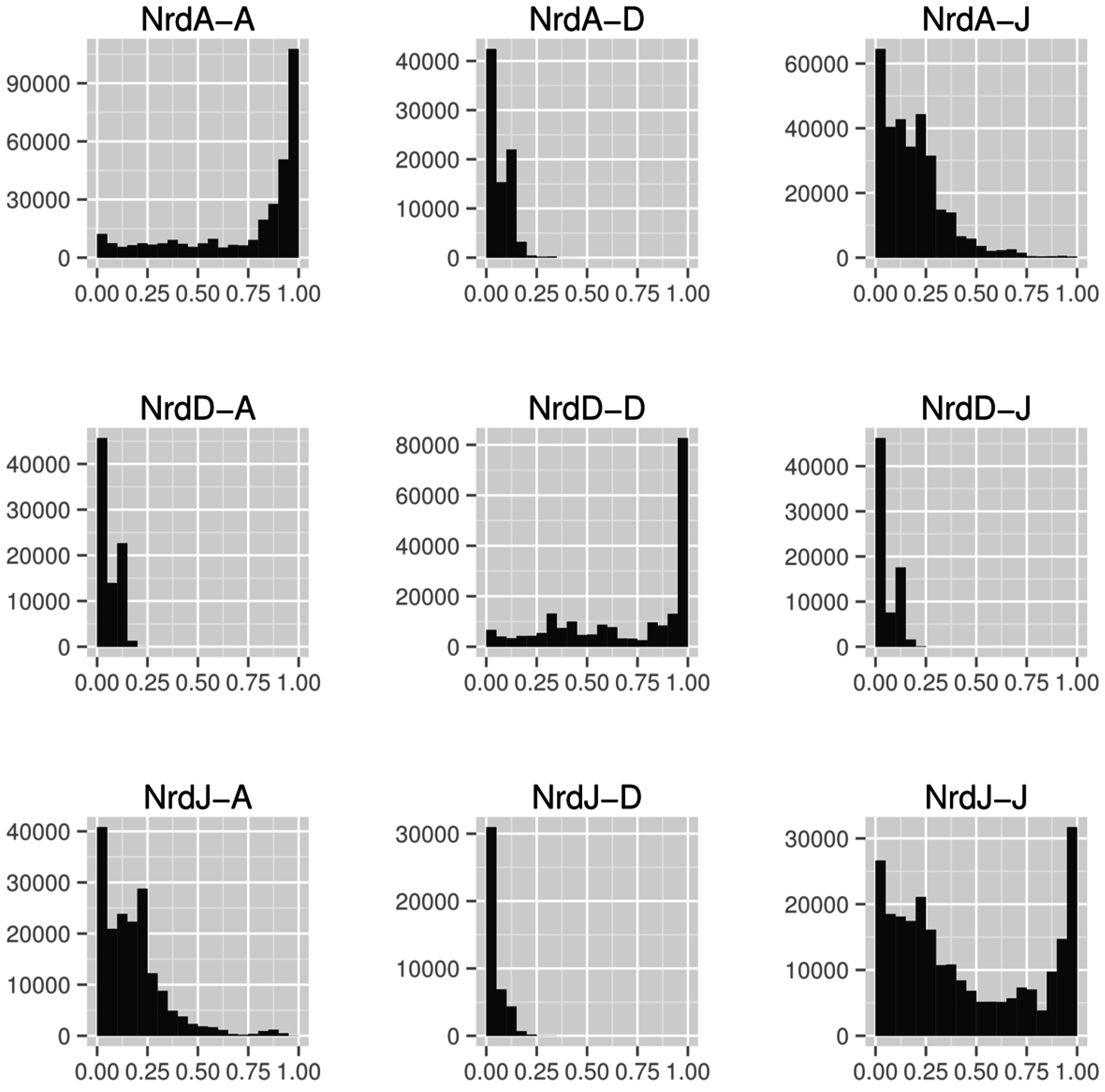
3.5. Specificity Regulation—Ancestral or Convergent?
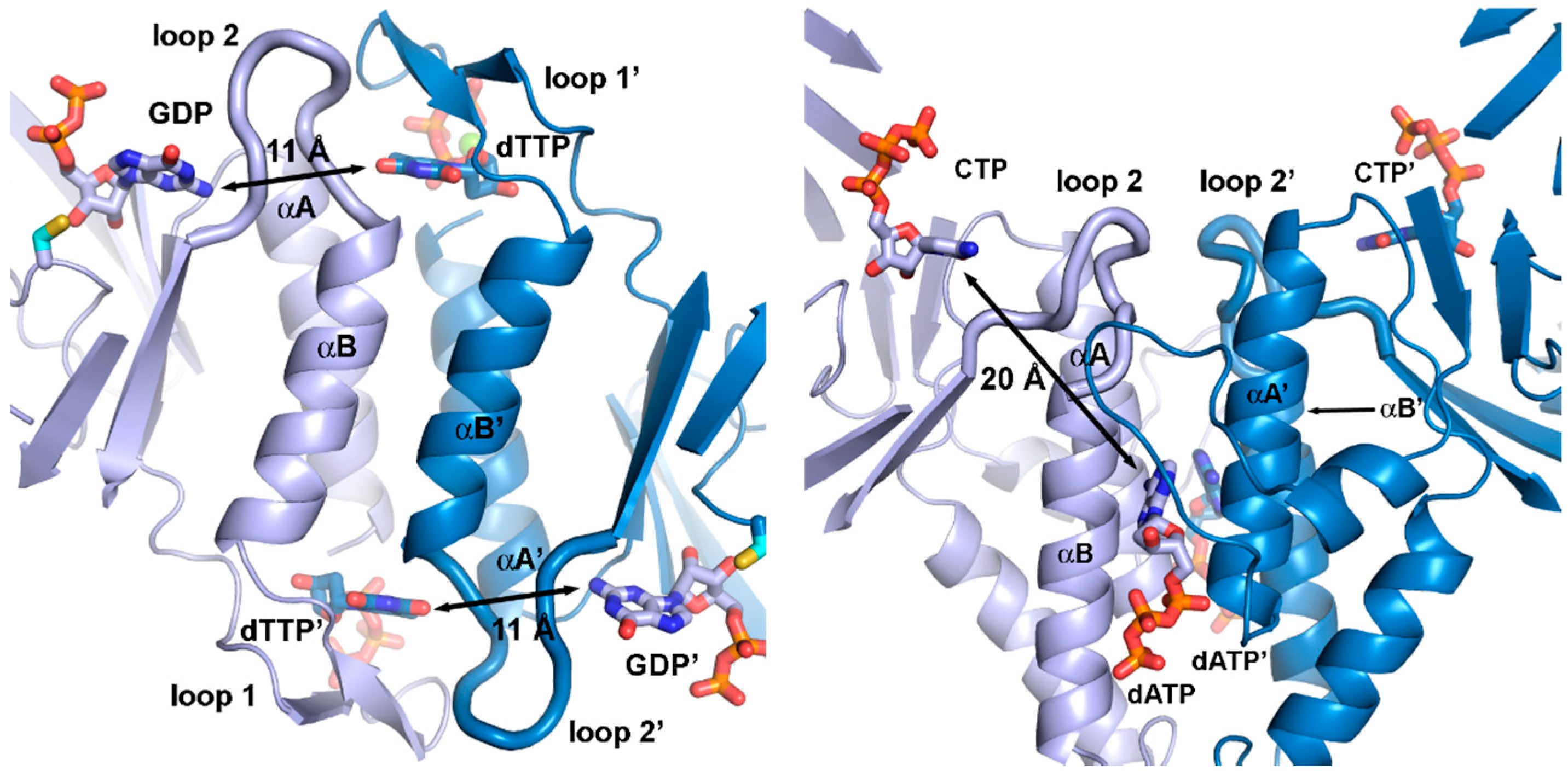
3.6. Activity Regulation—Ancestral and Lost or Multiple Origins?
| RNR class | Frequency of number of ATP-cones (%) | Number of proteins | |||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | ||
| I (NrdA/E) | 53 | 33 | 13 | 1 | 4186 |
| II (NrdJ) | 93 | 6 | 1 | 0 | 1800 |
| III (NrdD) | 24 | 70 | 6 | 0 | 2426 |
4. Birth of the Three Classes of RNR
4.1. Ancestral and Derived Characteristics of the Three RNR Classes
4.2. RNR R1/PFL Structural Phylogeny
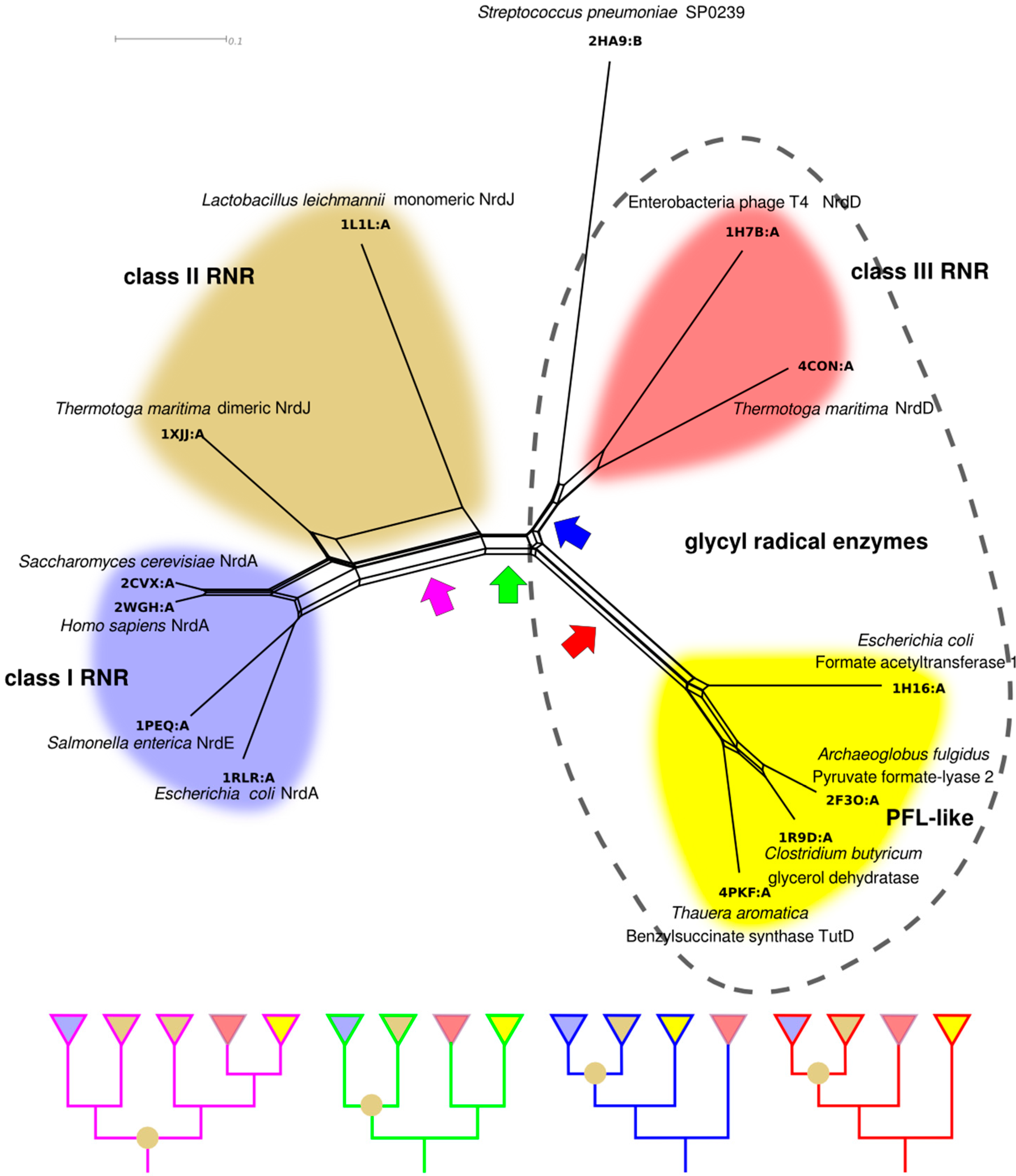
4.3. Selection for the Three Classes and the RNR Repertoire in the Tree of Life
| Domain | Nr genomes | Class I (NrdA/E) | Class I (NrdB/F) | Class II (NrdJ) | Class III (NrdD) |
|---|---|---|---|---|---|
| Archaea | 117 | 10 | 10 | 72 | 90 |
| Bacteria | 2318 | 2119 | 2159 | 1555 | 861 |
| Eukaryotes | 76 | 110 | 129 | 3 | 7 |
| Viruses | 87 | 70 | 51 | 33 | 17 |
4.3.1. Origin of Class II RNR
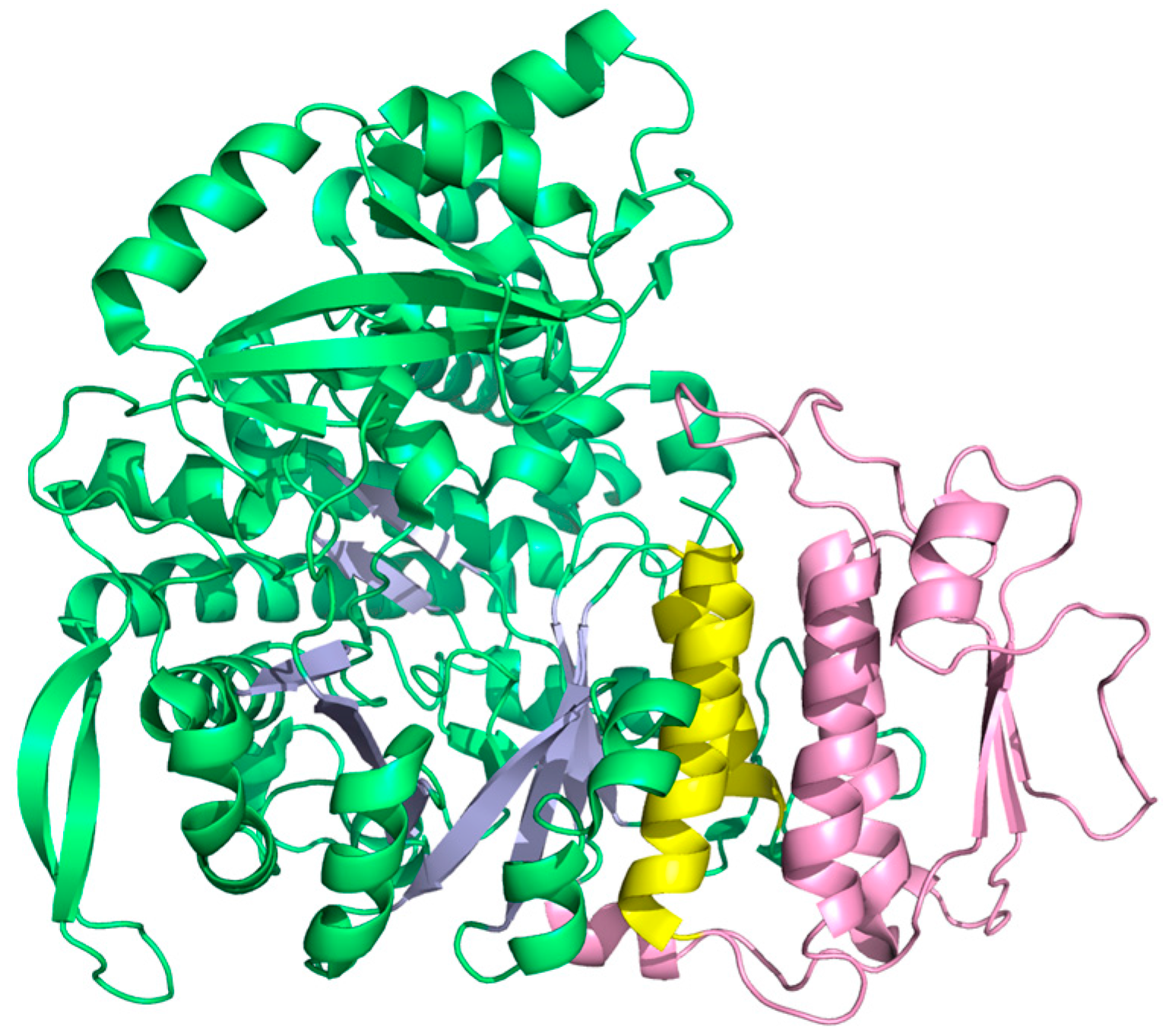
4.3.2. Origin of Class I RNR
4.3.3. Origin of Class III RNR and the Glycyl Radical Enzymes
5. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| AdoCbl | adenosylcobalamin |
| AdoMet | S-adenosylmethionine |
| dAdo• | 5'-deoxyadenosyl radical |
| dNDP | deoxyribonucleoside diphosphate |
| dNTP | deoxyribonucleoside triphosphate |
| GRE | glycyl radical enzyme |
| NDP | ribonucleoside diphosphate |
| NTP | ribonucleoside triphosphate |
| NrdA | class I RNR catalytic subunit |
| NrdB | class I RNR radical generating subunit |
| NrdD | class III RNR catalytic subunit |
| NrdE | subclass Ib RNR catalytic subunit |
| NrdF | subclass Ib RNR radical generating subunit |
| NrdG | class III RNR activase |
| NrdJ | class II RNR |
| PFL | pyruvate formate lyase |
| RNP | RNA+protein |
| RNR | ribonucleotide reductase |
Conflicts of Interest
References
- Torrents, E.; Sahlin, M.; Sjöberg, B.-M. The ribonucleotide reductase family—genetics and genomics. In Ribonucleotide reductase; Andersson, K.K., Ed.; NovaScience Publishers: Hauppauge, NY, USA, 2008; pp. 17–78. [Google Scholar]
- Burton, A.S.; Lehman, N. DNA before proteins? Recent discoveries in nucleic acid catalysis strengthen the case. Astrobiology 2009, 9, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.M. On alternative biological scenarios for the evolutionary transitions to DNA and biological protein synthesis. In Origins of Life: The Primal Self-Organization; Egel, R., Lankenau, D.-H., Mulkidjanian, A.Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 209–223. [Google Scholar]
- Poole, A.M.; Horinouchi, N.; Catchpole, R.J.; Si, D.; Hibi, M.; Tanaka, K.; Ogawa, J. The case for an early biological origin of DNA. J. Mol. Evol. 2014, 79, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Neveu, M.; Kim, H.-J.; Benner, S.A. The “strong” RNA world hypothesis: Fifty years old. Astrobiology 2013, 13, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.M.; Logan, D.T. Modern mRNA proofreading and repair: Clues that the last universal common ancestor possessed an RNA genome? Mol. Biol. Evol. 2005, 22, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. The origin of genetic information: Viruses as models. Gene 1993, 135, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.; Jeffares, D.; Penny, D. Early evolution: Prokaryotes, the new kids on the block. BioEssays 1999, 21, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Raffaelli, N. Nicotinamide coenzyme synthesis: A case of ribonucleotide emergence or a byproduct of the RNA world? In Origins of Life: The Primal Self-Organization; Egel, R., Lankenau, D.-H., Mulkidjanian, A.Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 185–208. [Google Scholar]
- Yarus, M. How many catalytic RNAs? Ions and the Cheshire cat conjecture. FASEB J. 1993, 7, 31–39. [Google Scholar] [PubMed]
- Benner, S.A.; Ellington, A.D.; Tauer, A. Modern metabolism as a palimpsest of the RNA world. Proc. Natl. Acad. Sci. USA 1989, 86, 7054–7058. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. From RNA to DNA, why so many ribonucleotide reductases? Science 1993, 260, 1773–1777. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.; Chou, I.-C.; Okafor, C.D.; Bowman, J.C.; O’Neill, E.B.; Athavale, S.S.; Petrov, A.S.; Hud, N.V.; Wartell, R.M.; Harvey, S.C.; et al. RNA with iron(II) as a cofactor catalyses electron transfer. Nat. Chem. 2013, 5, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Hamm, M.L.; Nikolic, D.; van Breemen, R.B.; Piccirilli, J.A. Unconventional origin of metal ion rescue in the hammerhead ribozyme reaction: Mn2+-assisted redox conversion of 2’-Mercaptocytidine to cytidine. J. Am. Chem. Soc. 2000, 122, 12069–12078. [Google Scholar] [CrossRef]
- Sigel, R.K.O.; Pyle, A.M. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem. Rev. 2007, 107, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anollés, G.; Yafremava, L.S.; Gee, H.; Caetano-Anollés, D.; Kim, H.S.; Mittenthal, J.E. The origin and evolution of modern metabolism. Int. J. Biochem. Cell Biol. 2009, 41, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Sofia, H.J.; Chen, G.; Hetzler, B.G.; Reyes-Spindola, J.F.; Miller, N.E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: Functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Broderick, J.B.; Duffus, B.R.; Duschene, K.S.; Shepard, E.M. Radical S-adenosylmethionine enzymes. Chem. Rev. 2014, 114, 4229–4317. [Google Scholar] [CrossRef] [PubMed]
- Booker, S.J. Anaerobic functionalization of unactivated C–H bonds. Curr. Opin. Chem. Biol. 2009, 13, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.L.; Borovik, A. Lessons from nature: Unraveling biological CH bond activation. Curr. Opin. Chem. Biol. 2009, 13, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Lawrence, J.; Bobik, T. COBALAMIN (COENZYME B12): Synthesis and biological significance. Annu. Rev. Microbiol. 1996, 50, 137–181. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Malkin, R.; Rabinowitz, J.C. The reconstitution of clostridial ferredoxin. Biochem. Biophys. Res. Commun. 1966, 23, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.M.; Smith, K.M.; Guilard, R. The Porphyrin Handbook; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Eschenmoser, A. Vitamin B12: Experiments concerning the origin of its molecular structure. Angew. Chem. Int. Ed. 1988, 27, 5–39. [Google Scholar] [CrossRef]
- Anbar, A.D. Elements and evolution. Science 2008, 322, 1481–1483. [Google Scholar] [CrossRef] [PubMed]
- Hol, W.G.J.; van Duijnen, P.T.; Berendsen, H.J.C. The α-helix dipole and the properties of proteins. Nature 1978, 273, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Mulliez, E.; Ollagnier, S.; Fontecave, M.; Eliasson, R.; Reichard, P. Formate is the hydrogen donor for the anaerobic ribonucleotide reductase from Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 8759–8762. [Google Scholar] [CrossRef] [PubMed]
- Hioe, J.; Zipse, H. Hydrogen transfer in SAM-mediated enzymatic radical reactions. Chem. Eur. J. 2012, 18, 16463–16472. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, L. Spore photoproduct lyase: The known, the controversial, and the unknown. J. Biol. Chem. 2014. [Google Scholar] [CrossRef]
- Mancia, F.; Keep, N.H.; Nakagawa, A.; Leadlay, P.F.; McSweeney, S.; Rasmussen, B.; Secke, P.B.; Diat, O.; Evans, P.R. How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 å resolution. Structure 1996, 4, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.G.; Chen, L.; Ji, H.F.; Chen, Z.H.; Yang, F.R.; Wang, L.; Qu, G.; Jiang, Y.Y.; Ji, C.; Zhang, H.Y. Characters of very ancient proteins. Biochem. Biophys. Res. Commun. 2008, 366, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Orengo, C.A.; Thornton, J.M. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002, 321, 741–765. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Nicolet, Y.; Fontecilla-Camps, J.C. The RNA world and the origin of life: An ancient protein fold links metal-based gas reactions with the RNA world. J. Cosmol. 2010, 10, 3243–3257. [Google Scholar]
- Farías-Rico, J.A.; Schmidt, S.; Höcker, B. Evolutionary relationship of two ancient protein superfolds. Nat. Chem. Biol. 2014, 10, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Nepomnyachiy, S.; Ben-Tal, N.; Kolodny, R. Global view of the protein universe. Proc. Natl. Acad. Sci. USA 2014, 111, 11691–11696. [Google Scholar] [CrossRef] [PubMed]
- Logan, D.T.; Andersson, J.; Sjöberg, B.-M.; Nordlund, P. A glycyl radical site in the crystal structure of a class III ribonucleotide reductase. Science 1999, 283, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Sintchak, M.D.; Arjara, G.; Kellogg, B.A.; Stubbe, J.; Drennan, C.L. The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nat. Struct. Mol. Biol. 2002, 9, 293–300. [Google Scholar] [CrossRef]
- Uhlin, U.; Eklund, H. Structure of ribonucleotide reductase protein R1. Nature 1994, 370, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.M.; Logan, D.T.; Sjöberg, B.-M. The evolution of the ribonucleotide reductases: Much Ado about oxygen. J. Mol. Evol. 2002, 55, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. The evolution of ribonucleotide reduction. Trends Biochem. Sci. 1997, 22, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Stubbe, J.; Ge, J.; Yee, C.S. The evolution of ribonucleotide reduction revisited. Trends Biochem. Sci. 2001, 26, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Torrents, E.; Aloy, P.; Gibert, I.; Rodríguez-Trelles, F. Ribonucleotide reductases: Divergent evolution of an ancient enzyme. J. Mol. Evol. 2002, 55, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Aurelius, O.; Johansson, R.; Bågenholm, V.; Lundin, D.; Tholander, F.; Balhuizen, A.; Beck, T.; Sahlin, M.; Sjöberg, B.-M.; Mulliez, E.; et al. Thermotoga maritima class III ribonucleotide reductase lacks a radical cysteine pre-positioned in the active site. PLoS One 2015. submitted for publication. [Google Scholar]
- Wei, Y.; Funk, M.A.; Rosado, L.A.; Baek, J.; Drennan, C.L.; Stubbe, J. The class III ribonucleotide reductase from Neisseria bacilliformis can utilize thioredoxin as a reductant. Proc. Natl. Acad. Sci. USA 2014, 111, E3756–E3765. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Mathies, G.; Yokoyama, K.; Chen, J.; Griffin, R.G.; Stubbe, J. A chemically competent thiosulfuranyl radical on the Escherichia coli class III ribonucleotide reductase. J. Am. Chem. Soc. 2014, 136, 9001–9013. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Uhlin, U.; Ramaswamy, S.; Ekberg, M.; Regnström, K.; Sjöberg, B.-M.; Eklund, H. Binding of allosteric effectors to ribonucleotide reductase protein R1: Reduction of active-site cysteines promotes substrate binding. Structure 1997, 5, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Larsson, K.-M.; Jordan, A.; Eliasson, R.; Reichard, P.; Logan, D.T.; Nordlund, P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat. Struct. Mol. Biol. 2004, 11, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Faber, C.; Uchiki, T.; Fairman, J.W.; Racca, J.; Dealwis, C. Structures of eukaryotic ribonucleotide reductase I provide insights into dNTP regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 4022–4027. [Google Scholar] [CrossRef] [PubMed]
- Persson, A.L.; Eriksson, M.; Katterle, B.; Pötsch, S.; Sahlin, M.; Sjöberg, B.-M. A new mechanism-based radical intermediate in a mutant R1 protein affecting the catalytically essential Glu441 in Escherichia coli ribonucleotide reductase. J. Biol. Chem. 1997, 272, 31533–31541. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C.C.; Bennati, M.; Obias, H.V.; Bar, G.; Griffin, R.G.; Stubbe, J. High-field EPR detection of a disulfide radical anion in the reduction of cytidine 5'-diphosphate by the E441Q R1 mutant of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. USA 1999, 96, 8979–8984. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-B.; Pelmenschikov, V.; Gräslund, A.; Siegbahn, P.E.M. Density functional calculations on class III ribonucleotide reductase: Substrate reaction Mechanism with two formates. J. Phys. Chem. B 2004, 108, 2056–2065. [Google Scholar] [CrossRef]
- Cotruvo, J.A.; Stubbe, J. Class I ribonucleotide reductases: Metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 2011, 80, 733–767. [Google Scholar] [CrossRef] [PubMed]
- Larsson, K.-M.; Andersson, J.; Sjöberg, B.-M.; Nordlund, P.; Logan, D.T. Structural basis for allosteric substrate specificity regulation in anaerobic ribonucleotide reductases. Structure 2001, 9, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Torrents, E.; Buist, G.; Liu, A.; Eliasson, R.; Kok, J.; Gibert, I.; Gräslund, A.; Reichard, P. The anaerobic (class III) ribonucleotide reductase from Lactococcus lactis. J. Biol. Chem. 2000, 275, 2463–2471. [Google Scholar] [CrossRef] [PubMed]
- Aberg, A.; Hahne, S.; Karlsson, M.; Larsson, A.; Ormö, M.; Ahgren, A.; Sjöberg, B.-M. Evidence for two different classes of redox-active cysteines in ribonucleotide reductase of Escherichia coli. J. Biol. Chem. 1989, 264, 12249–12252. [Google Scholar] [PubMed]
- Mao, S.S.; Holler, T.P.; Yu, G.X.; Bollinger, J.M.; Booker, S.; Johnston, M.I.; Stubbe, J. A model for the role of multiple cysteine residues involved in ribonucleotide reduction: amazing and still confusing. Biochemistry 1992, 31, 9733–9743. [Google Scholar] [CrossRef] [PubMed]
- Padovani, D.; Mulliez, E.; Fontecave, M. Activation of class III ribonucleotide reductase by thioredoxin. J. Biol. Chem. 2001, 276, 9587–9589. [Google Scholar] [CrossRef] [PubMed]
- Licht, S.S.; Booker, S.; Stubbe, J. Studies on the catalysis of carbon−cobalt bond homolysis by ribonucleoside triphosphate reductase: Evidence for concerted carbon−cobalt bond homolysis and thiyl radical formation. Biochemistry 1999, 38, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.G.; Szöke, A.; Blaustein, J.; O’Rourke, S.M.; Robertson, M.P. RNA catalysis, thermodynamics and the origin of life. Life 2014, 4, 131–141. [Google Scholar] [CrossRef] [PubMed]
- White, H.B. Evolution of coenzymes and the origin of pyridine nucleotides. In The Pyridine Nucleotide Coenzymes; Everse, J., Ed.; Elsevier: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Frey, M.; Rothe, M.; Wagner, A.F.; Knappe, J. Adenosylmethionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S hydrogen atom. Studies of [2H]glycine-substituted enzyme and peptides homologous to the glycine 734 site. J. Biol. Chem. 1994, 269, 12432–12437. [Google Scholar] [PubMed]
- Torrents, E.; Eliasson, R.; Wolpher, H.; Gräslund, A.; Reichard, P. The anaerobic ribonucleotide reductase from Lactococcus lactis interactions between the two proteins NrdD and NrdG. J. Biol. Chem. 2001, 276, 33488–33494. [Google Scholar] [CrossRef] [PubMed]
- Lundin, D.; Gribaldo, S.; Torrents, E.; Sjöberg, B.-M.; Poole, A.M. Ribonucleotide reduction—horizontal transfer of a required function spans all three domains. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Eklund, H.; Fontecave, M. Glycyl radical enzymes: A conservative structural basis for radicals. Structure 1999, 7, R257–R262. [Google Scholar] [CrossRef] [PubMed]
- Selmer, T.; Pierik, A.J.; Heider, J. New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol. Chem. 2005, 386, 981–988. [Google Scholar] [CrossRef] [PubMed]
- King, D.S.; Reichard, P. Mass spectrometric determination of the radical scission site in the anaerobic ribonucleotide reductase of Escherichia coli. Biochem. Biophys. Res. Commun. 1995, 206, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.F.; Frey, M.; Neugebauer, F.A.; Schäfer, W.; Knappe, J. The free radical in pyruvate formate-lyase is located on glycine-734. Proc. Natl. Acad. Sci. USA 1992, 89, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Henn-Sax, M.; Höcker, B.; Wilmanns, M.; Sterner, R. Divergent evolution of (βα)8-barrel enzymes. Biol. Chem. 2005, 382, 1315–1320. [Google Scholar]
- Mathews, C.K. Deoxyribonucleotides as genetic and metabolic regulators. FASEB J. 2014, 28, 3832–3840. [Google Scholar] [CrossRef]
- Hofer, A.; Crona, M.; Logan, D.T.; Sjöberg, B.-M. DNA building blocks: Keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 2011, 47, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Venkateswara, R.J.; Crona, M.; Rofougaran, R.; Lundin, D.; Johansson, S.; Brännström, K.; Sjöberg, B.-M.; Hofer, A. Pseudomonas aeruginosa class I ribonucleotide reductase represents a novel mechanism of overall activity regulation. J. Biol. Chem. 2015. submitted for publication. [Google Scholar]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Bujnicki, J.M. Phylogeny of the restriction endonuclease-like superfamily inferred from comparison of protein structures. J. Mol. Evol. 2000, 50, 39–44. [Google Scholar] [PubMed]
- Breitling, R.; Laubner, D.; Adamski, J. Structure-based phylogenetic analysis of short-chain alcohol dehydrogenases and reclassification of the 17beta-hydroxysteroid dehydrogenase family. Mol. Biol. Evol. 2001, 18, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Garau, G.; Guilmi, A.M.D.; Hall, B.G. Structure-based phylogeny of the metallo-β-lactamases. Antimicrob. Agents Chemother. 2005, 49, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Lundin, D.; Poole, A.M.; Sjöberg, B.-M.; Högbom, M. Use of structural phylogenetic networks for classification of the ferritin-like superfamily. J. Biol. Chem. 2012, 287, 20565–20575. [Google Scholar] [CrossRef] [PubMed]
- Uberto, R.; Moomaw, E.W. Protein similarity networks reveal relationships among sequence, structure, and function within the cupin superfamily. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Khafif, M.; Cottret, L.; Balagué, C.; Raffaele, S. Identification and phylogenetic analyses of VASt, an uncharacterized protein domain associated with lipid-binding domains in Eukaryotes. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef]
- Cheng, H.; Schaeffer, R.D.; Liao, Y.; Kinch, L.N.; Pei, J.; Shi, S.; Kim, B.-H.; Grishin, N.V. ECOD: An Evolutionary Classification of Protein Domains. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef]
- Uppsten, M.; Färnegårdh, M.; Jordan, A.; Eliasson, R.; Eklund, H.; Uhlin, U. Structure of the large subunit of class Ib ribonucleotide reductase from Salmonella typhimurium and its complexes with allosteric effectors. J. Mol. Biol. 2003, 330, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Fairman, J.W.; Wijerathna, S.R.; Ahmad, M.F.; Xu, H.; Nakano, R.; Jha, S.; Prendergast, J.; Welin, R.M.; Flodin, S.; Roos, A.; et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011, 18, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Fritz-Wolf, K.; Kabsch, W.; Knappe, J.; Schultz, S.; Volker Wagner, A.F. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat. Struct. Mol. Biol. 1999, 6, 969–975. [Google Scholar] [CrossRef]
- Lehtiö, L.; Günter Grossmann, J.; Kokona, B.; Fairman, R.; Goldman, A. Crystal structure of a glycyl radical enzyme from Archaeoglobus fulgidus. J. Mol. Biol. 2006, 357, 221–235. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.R.; Raynaud, C.; Croux, C.; Girbal, L.; Soucaille, P.; Lanzilotta, W.N. Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: Preliminary biochemical and structural characterization. Biochemistry 2004, 43, 4635–4645. [Google Scholar] [CrossRef] [PubMed]
- Funk, M.A.; Judd, E.T.; Marsh, E.N.G.; Elliott, S.J.; Drennan, C.L. Structures of benzylsuccinate synthase elucidate roles of accessory subunits in glycyl radical enzyme activation and activity. Proc. Natl. Acad. Sci. USA 2014, 111, 10161–10166. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Moulton, V. Neighbor-net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Malod-Dognin, N.; Pržulj, N. GR-Align: Fast and flexible alignment of protein 3D structures using graphlet degree similarity. Bioinformatics 2014. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Monod, J.; Wyman, J.; Changeux, J.-P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E.; Némethy, G.; Filmer, D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 1966, 5, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Corcoran, E.E.; Hattangadi, N.A.; Zhang, J.V.; Gere, S.A.; Hellinga, H.W. The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors. Proc. Natl. Acad. Sci. USA 1997, 94, 4366–4371. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Mitsuya, M.; Nishimura, T.; Eiki, J.; Nagata, Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 2004, 12, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Cotruvo, J.A.; Stubbe, J. An active dimanganese(III)−tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase. Biochemistry 2010, 49, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Voevodskaya, N.; Lendzian, F.; Ehrenberg, A.; Gräslund, A. High catalytic activity achieved with a mixed manganese–iron site in protein R2 of Chlamydia ribonucleotide reductase. FEBS Lett. 2007, 581, 3351–3355. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Yun, D.; Saleh, L.; Barr, E.W.; Xing, G.; Hoffart, L.M.; Maslak, M.-A.; Krebs, C.; Bollinger, J.M. A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science 2007, 316, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.C. The Ferritin-like superfamily: Evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim. Biophys. Acta 2010, 1800, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Merkx, M.; Kopp, D.A.; Sazinsky, M.H.; Blazyk, J.L.; Müller, J.; Lippard, S.J. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: A tale of two irons and three proteins. Angew. Chem. Int. Ed. 2001, 40, 2782–2807. [Google Scholar] [CrossRef]
- Warren, M.J.; Raux, E.; Schubert, H.L.; Escalante-Semerena, J.C. The biosynthesis of adenosylcobalamin (vitamin B12). Nat. Prod. Rep. 2002, 19, 390–412. [Google Scholar] [CrossRef] [PubMed]
- Högbom, M.; Stenmark, P.; Voevodskaya, N.; McClarty, G.; Gräslund, A.; Nordlund, P. The radical site in chlamydial ribonucleotide reductase defines a new R2 subclass. Science 2004, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Voevodskaya, N.; Lendzian, F.; Gräslund, A. A stable FeIII–FeIV replacement of tyrosyl radical in a class I ribonucleotide reductase. Biochem. Biophys. Res. Commun. 2005, 330, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Voevodskaya, N.; Narvaez, A.-J.; Domkin, V.; Torrents, E.; Thelander, L.; Gräslund, A. Chlamydial ribonucleotide reductase: Tyrosyl radical function in catalysis replaced by the FeIII-FeIV cluster. Proc. Natl. Acad. Sci. USA 2006, 103, 9850–9854. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Thauer, R.K.; Shima, S. Methane as fuel for anaerobic microorganisms. Ann. N. Y. Acad. Sci. 2008, 1125, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Crable, B.R.; Plugge, C.M.; McInerney, M.J.; Stams, A.J.M. Formate formation and formate conversion in biological fuels production. Enzyme Res. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Leonhartsberger, S.; Korsa, I.; Bock, A. The molecular biology of formate metabolism in enterobacteria. J. Mol. Microbiol. Biotechnol. 2002, 4, 269–276. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundin, D.; Berggren, G.; Logan, D.T.; Sjöberg, B.-M. The Origin and Evolution of Ribonucleotide Reduction. Life 2015, 5, 604-636. https://doi.org/10.3390/life5010604
Lundin D, Berggren G, Logan DT, Sjöberg B-M. The Origin and Evolution of Ribonucleotide Reduction. Life. 2015; 5(1):604-636. https://doi.org/10.3390/life5010604
Chicago/Turabian StyleLundin, Daniel, Gustav Berggren, Derek T. Logan, and Britt-Marie Sjöberg. 2015. "The Origin and Evolution of Ribonucleotide Reduction" Life 5, no. 1: 604-636. https://doi.org/10.3390/life5010604
APA StyleLundin, D., Berggren, G., Logan, D. T., & Sjöberg, B.-M. (2015). The Origin and Evolution of Ribonucleotide Reduction. Life, 5(1), 604-636. https://doi.org/10.3390/life5010604