1. Introduction
Cerebral aneurysms are focal dilatations of the cerebral arterial wall that can lead to serious neurological complications, including subarachnoid hemorrhage and elevated mortality risk upon rupture. The pathogenesis of cerebral aneurysms involves several interrelated mechanisms, including hemodynamic stress, chronic inflammation, vascular smooth muscle cell degeneration, and, most notably, endothelial dysfunction [
1,
2]. Endothelial cells are essential for maintaining vascular integrity, providing an antithrombotic barrier, and modulating inflammatory responses. Loss of endothelial function compromises the structural stability of the vascular wall, thereby promoting aneurysm development. Among the mechanisms contributing to endothelial dysfunction, apoptosis is a major contributor to endothelial cell loss and weakening of the vessel wall. Apoptosis represents a key cellular response to oxidative and inflammatory stress and has been observed at elevated levels in the walls of ruptured cerebral aneurysms, often accompanied by Extracellular Matrix (ECM) degradation [
3]. This pathological remodeling reduces the mechanical strength of the aneurysm wall, increasing the likelihood of rupture.
Ang II is a potent vasoconstrictor known for its role in blood pressure regulation, but it also contributes to vascular pathology through its proinflammatory, pro-oxidative, and pro-apoptotic properties [
4]. Ang II stimulates the generation of ROS via NADPH oxidase activation, which initiates mitochondrial apoptotic pathways in endothelial cells [
5]. Furthermore, Ang II disrupts endothelial integrity by upregulating proinflammatory cytokines such as IL-6 and TNF-α, as well as adhesion molecules including Vascular Cell Adhesion Molecule-1 and Intercellular Adhesion Molecule-1, thereby promoting vascular inflammation and endothelial barrier breakdown [
6].
Although CL is primarily known as a centrally acting α2-adrenergic receptor agonist, it has recently gained attention for its antioxidative and anti-inflammatory effects within the peripheral vascular system. Studies have shown that CL modulates endothelial stress responses by attenuating sympathetic nervous system activity and reducing the production of ROS [
7]. Additionally, CL has been reported to suppress apoptotic signaling pathways by maintaining mitochondrial membrane potential, thereby enhancing endothelial cell survival [
8]. Despite these promising effects, the potential protective role of CL on cerebral endothelial cells—particularly in the context of Ang II-induced oxidative stress, inflammation, and apoptosis—remains inadequately explored.
This study aimed to evaluate the protective effects of CL against endothelial cell dysfunction and apoptosis induced by Ang II in an in vitro model that mimics early molecular events associated with CA pathogenesis. Specifically, the effects of CL on endothelial cell viability, oxidative stress (ROS and NO levels), proinflammatory cytokine release, and apoptosis-related gene expression were examined. By targeting key pathways involved in vascular inflammation and degeneration, the study sought to explore CL’s potential as a pharmacological agent for mitigating endothelial damage and thereby contributing to aneurysm prevention.
In the present study, we employed an in vitro model of endothelial dysfunction using HBMECs exposed to Ang II to simulate the early pathological processes associated with in vitro endothelial injury model simulating cerebral aneurysm-related pathophysiology formation. Although this cellular model does not fully recapitulate the complex hemodynamic and structural changes seen in true aneurysms, it effectively mimics key features of the endothelial injury, oxidative stress, and proinflammatory signaling that are known contributors to aneurysmal wall degeneration. As such, it provides a controlled and reproducible platform for evaluating pharmacological interventions targeting vascular inflammation and oxidative damage in a context relevant to neurovascular pathology.
2. Methods
2.1. Cell Culture
Immortalized human brain microvascular endothelial cells (HBMECs; ATCC® CRL-3245™) were obtained from the American Type Culture Collection (ATCC®, Manassas, VA, USA) and used in this study. The cells were cultured in Endothelial Cell Growth Medium (ECGM; PromoCell GmbH, Heidelberg, Germany) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. Cultures were maintained at 37 °C in a humidified incubator with 5% CO2. Once the cells reached approximately 80% confluency, they were subcultured using 0.25% trypsin-EDTA. All experiments were conducted using cells at passages 3 to 5.
2.2. Establishment of an In Vitro Endothelial Injury Model Relevant to Cerebral Aneurysm Pathophysiology and Treatment with CL
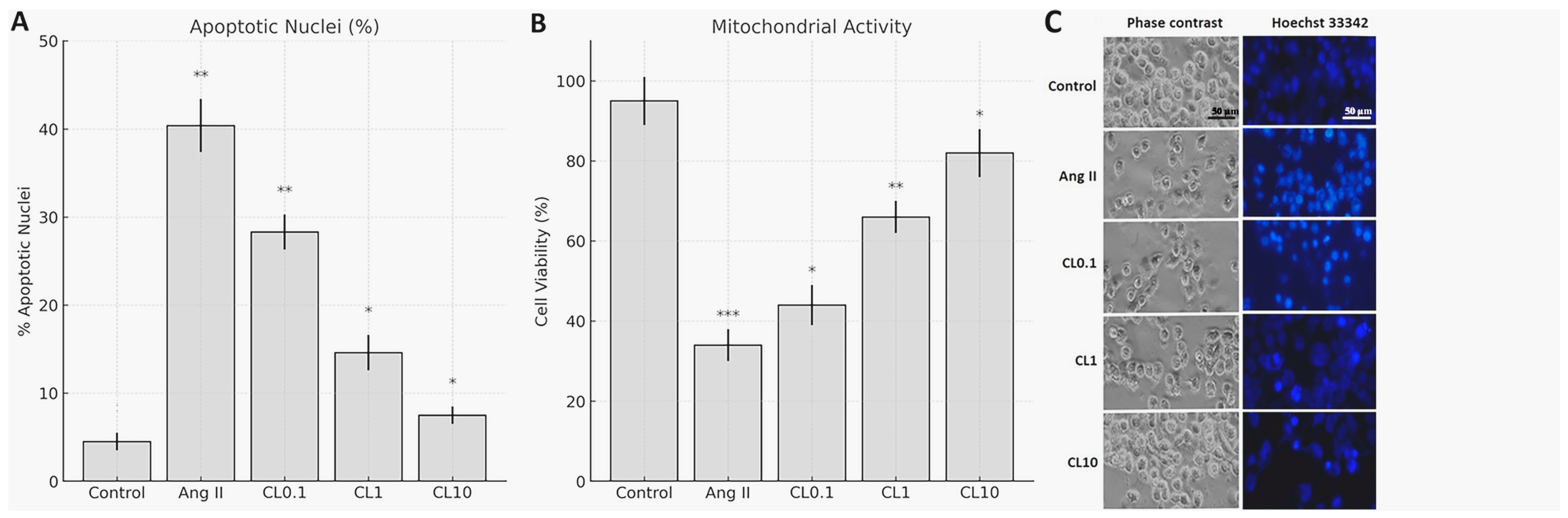
To establish endothelial dysfunction, HBMECs were treated with 1 µM Ang II (Ang II; Sigma-Aldrich, St. Louis, MO, USA) for 24 h. CL (Sigma-Aldrich) was dissolved in sterile distilled water according to the manufacturer’s instructions and diluted to final concentrations of 0.1 µM, 1 µM, and 10 µM. These concentrations were selected based on previous studies demonstrating CL’s endothelial protective effects within this range [
7,
8], pilot experiments confirming non-toxicity (viability > 90%) and concentration-dependent efficacy in HBMECs (
Figure 1), and clinical relevance, as 0.1–10 µM spans subclinical to supraphysiological ranges while avoiding cytotoxicity observed at higher concentrations (>20 µM). CL was administered to the cells 2 h before Ang II exposure. To ensure that observed effects were due to CL itself and not the vehicle, the control group received the same volume of sterile distilled water as the vehicle control. The experimental groups consisted of (i) control (vehicle only), (ii) Ang II only, (iii) Ang II + CL 0.1 µM (CL0.1), (iv) Ang II + CL 1 µM (CL1), and (v) Ang II + CL 10 µM (CL10).
Although plasma concentrations of Ang II in vivo are typically in the picomolar to nanomolar range, the concentration of 1 µM was selected based on prior studies in endothelial cell cultures, where it consistently induces robust oxidative and inflammatory responses [
4,
5]. This relatively high dose accounts for the lack of in vivo clearance mechanisms and mimics localized vascular Ang II accumulation observed in pathophysiological states such as cerebral aneurysms or hypertension [
9].
2.3. Cell Viability Test
Cells were seeded into 96-well plates at a density of 1 × 10
3 cells per well. Following seeding, the plates were incubated in a humidified CO
2 incubator at 37 °C for 24 h to allow for cell attachment. After incubation, cells were treated with 1 µM Ang II alone or in combination with CL at concentrations of Ang II + CL 0.1 µM (CL0.1), Ang II + CL 1 µM (CL1), and Ang II + CL 10 µM (CL10) for an additional 24 h. Following treatment, the culture medium was removed, and the MTT assay was performed as described by Mosmann [
10]. Briefly, 20 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) solution was added to each well, and the volume was adjusted to 100 µL with ECGM. Plates were incubated for 2 h at 37 °C in the dark. After incubation, absorbance was measured at 490 nm using a microplate reader, with a reference wavelength of 630 nm. For apoptotic nuclear morphology assessment, cells were stained with Hoechst 33342 (5 μg/mL) for 10 min at 37 °C. Apoptotic cells (condensed/fragmented nuclei) were quantified under fluorescence microscopy (20× magnification). Three random fields per well were analyzed, and the apoptotic index was calculated as (apoptotic nuclei/total nuclei) × 100.
2.4. Determination of ROS and NO Levels
2.4.1. Determination of ROS Levels
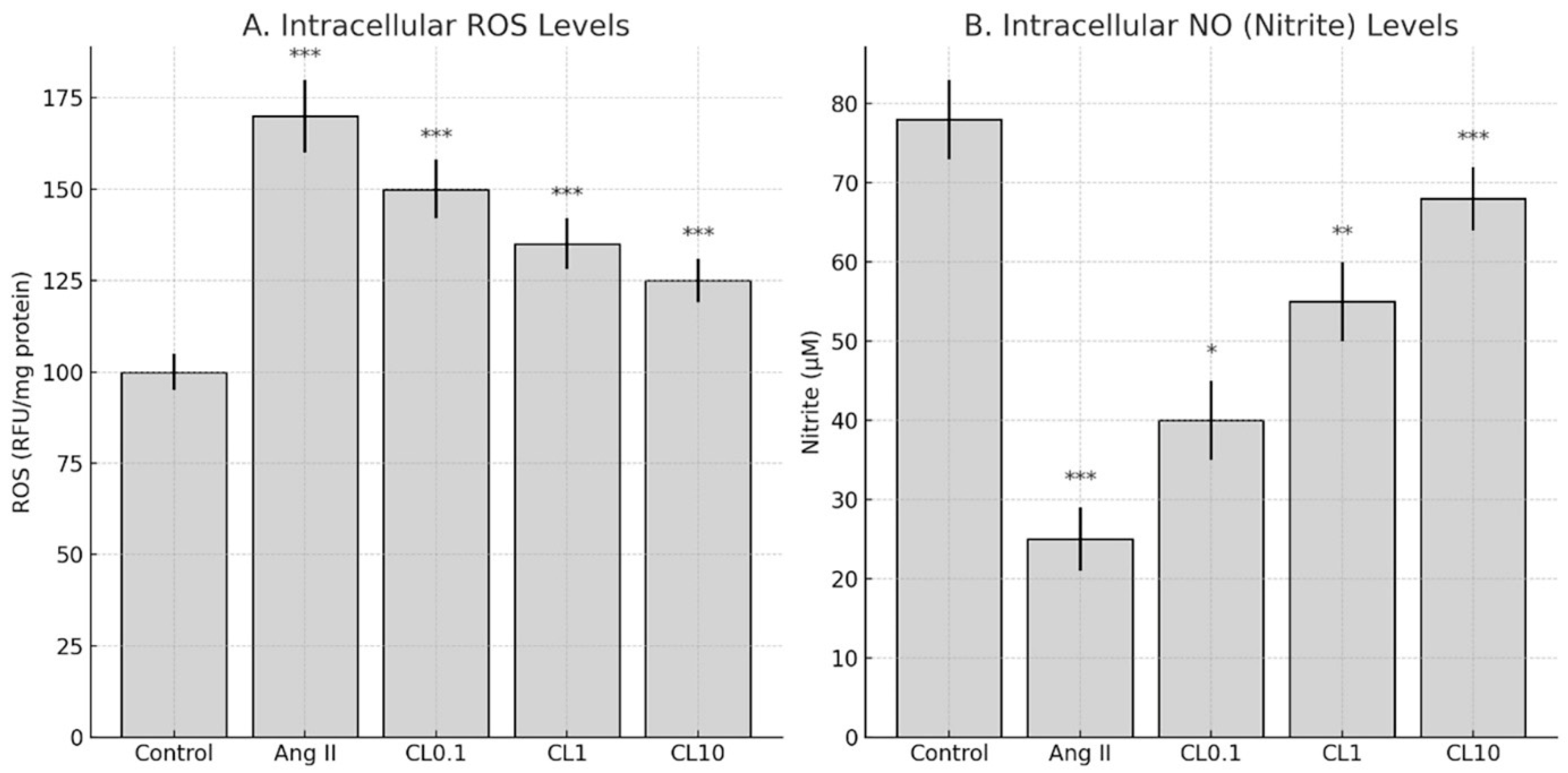
Intracellular ROS levels were measured using DCFDA Cellular ROS Detection Assay Kit (Sigma Aldrich, Cat: 287810), following the manufacturer’s protocol. After treatment with Ang II and CL, cells were incubated with 25 µM DCFDA at 37 °C in the dark for 45 min. Subsequently, the cells were washed with phosphate-buffered saline (PBS), and fluorescence intensity was measured at 485 nm excitation and 535 nm emission wavelengths using a fluorescence plate reader (BioTek, Synergy H1, BioTek Instruments, Inc., Winooski, VT, USA). ROS levels were normalized to protein content and expressed as relative fluorescence units per mg protein (RFU/mg).
2.4.2. Determination of NO Levels
NO production was determined indirectly by measuring nitrite accumulation using the Griess Reagent Assay (Thermo Scientific, Waltham, MA, USA) [
11]. After treatment, 50 µL of cell culture supernatant was mixed with an equal volume of Griess reagent (1% sulfanilamide, 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride in 2.5% phosphoric acid) in a 96-well plate and incubated at room temperature for 10 min in the dark. Absorbance was read at 540 nm using a microplate reader (BioTek). Nitrite concentrations were calculated using a sodium nitrite standard curve and expressed as µM/mg protein.
2.5. Quantification of Proinflammatory Cytokines by ELISA
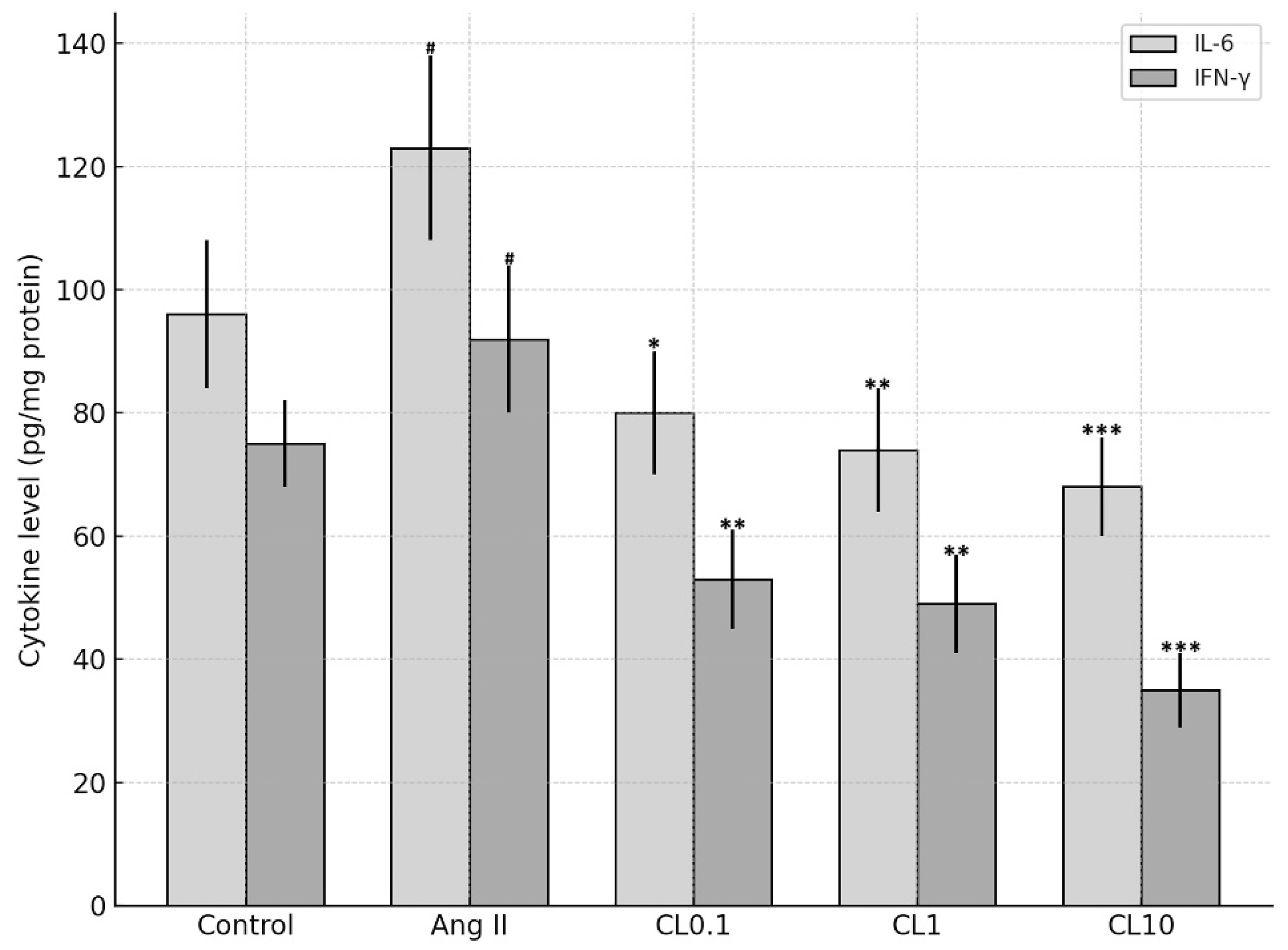
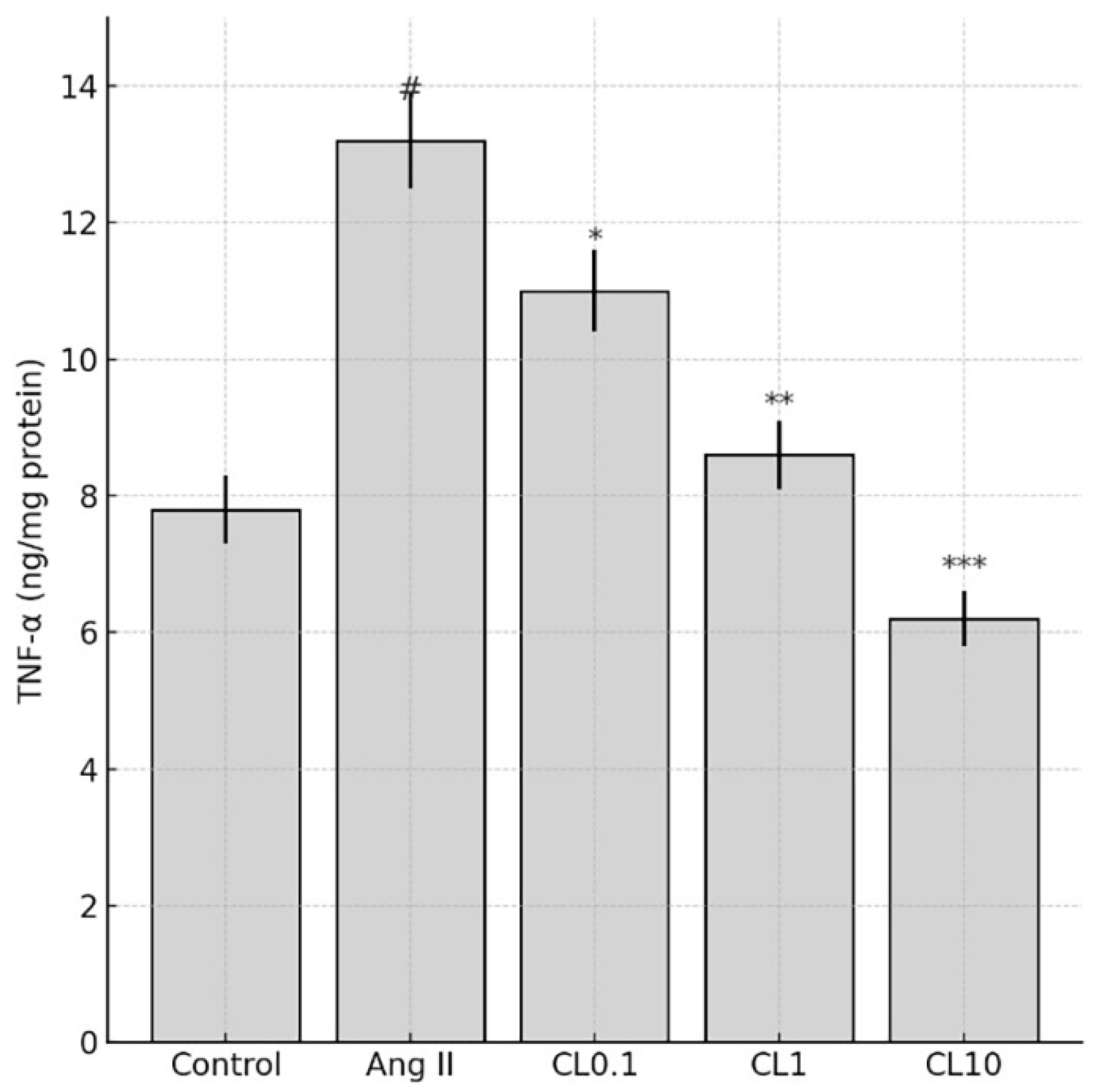
Levels of the inflammatory cytokines TNF-α, IL-6, and IFN-γ were quantified in HBMEC lysates using commercially available sandwich ELISA kits (Invitrogen, Carlsbad, CA, USA; Cat. #13-7341-81 for TNF-α, #EH2IL6 for IL-6, and #14-7311-81 for IFN-γ), following the manufacturer’s protocols. Briefly, 96-well plates pre-coated with specific capture antibodies were incubated with prepared cell lysates. After washing, wells were incubated with biotin-conjugated detection antibodies, followed by streptavidin-HRP. The enzymatic reaction was developed using a chromogenic substrate, and absorbance was measured at 540 nm using a microplate reader. Cytokine concentrations were normalized to the total protein content of each sample and expressed as ng/mg or pg/mg of protein.
2.6. Gene Expression Analysis (RT-qPCR)
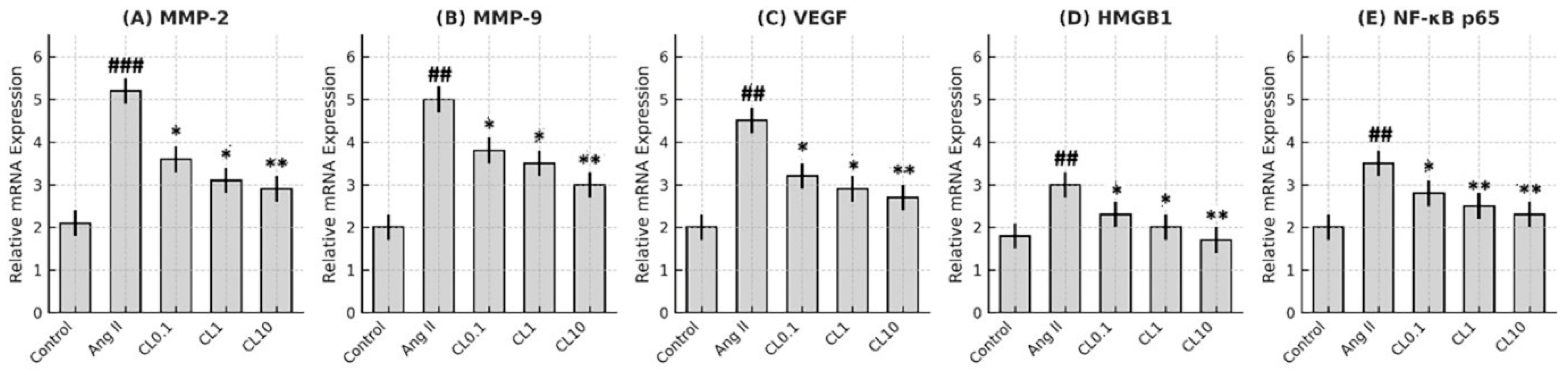
Total RNA was extracted from treated HBMECs using TRIzol Reagent (Invitrogen), and complementary DNA (cDNA) was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific), following the manufacturer’s protocols. Quantitative real-time PCR (RT-qPCR) was performed using the QuantStudio 3 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with SYBR Green Master Mix. The expression levels of VEGF, MMP-2, MMP-9, HMGB1, and NF-κB p65 were assessed using specific primer sequences listed in
Table 1. GAPDH was used as the internal reference gene. Relative gene expression was calculated using the 2
−ΔΔCt method. Primer efficiency was calculated using standard curves generated from serial cDNA dilutions, and all primer sets showed efficiencies between 90 and 110%. Additionally, melt curve analysis was performed for each qPCR reaction and yielded a single distinct peak, confirming specific amplification and excluding nonspecific products or primer-dimer artifacts.
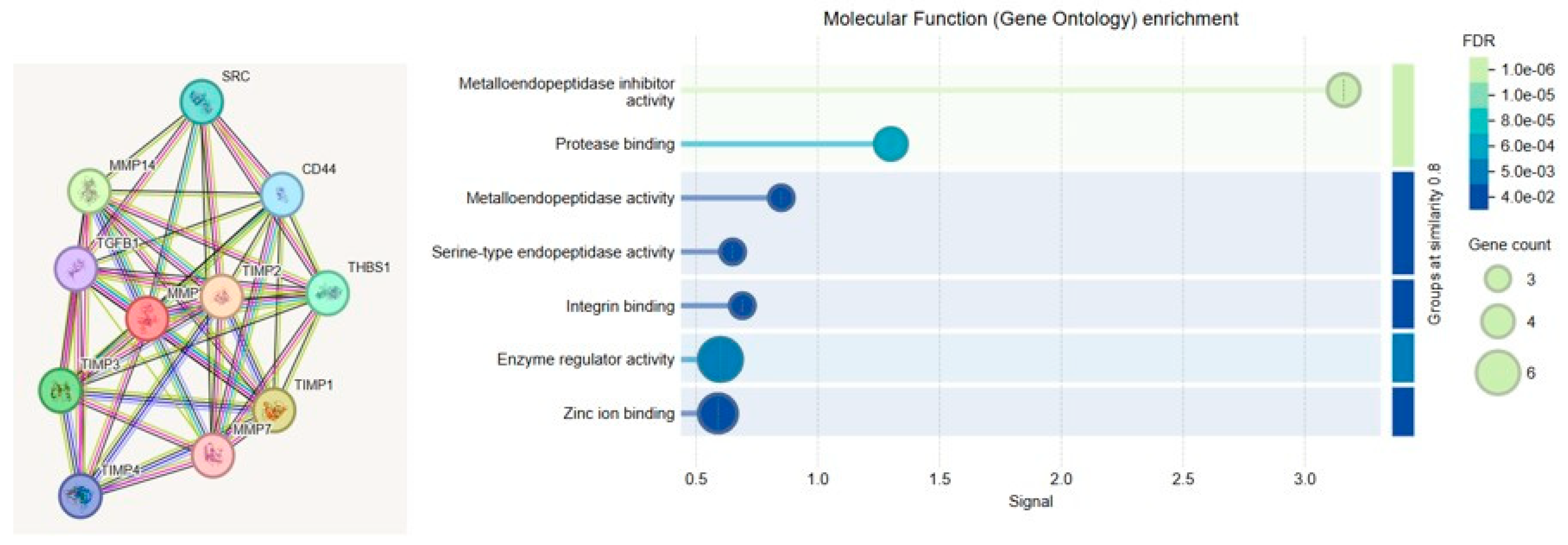
2.7. Bioinformatics/GO Analysis
To elucidate the functional relevance of MMP-2 and MMP-9 in the context of cerebral endothelial dysfunction, Gene Ontology (GO) enrichment analysis was performed. The analysis categorized gene functions into three principal domains: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). MMP-2 and MMP-9 were primarily associated with ECM remodeling, inflammatory signaling, and angiogenesis. Notably, MMP-driven inflammation was linked to upregulation of NF-κB, a central transcription factor in vascular inflammation, while pathological angiogenesis was mediated through VEGF signaling.
GO enrichment analysis was conducted to investigate the biological functions, molecular activities, and subcellular localizations of the genes associated with endothelial dysfunction. The analysis was performed using STRING and ShinyGO (v0.76), and results were classified into three main GO categories: BP, MF, and CC.
2.8. Statistical Analysis
All experiments were conducted with a minimum of three independent biological replicates. Data are expressed as mean ± standard deviation (SD). Statistical comparisons between multiple groups were performed using one-way analysis of variance (ANOVA), followed by Tukey’s post hoc test for pairwise comparisons. Prior to ANOVA, assumptions of normality and homogeneity of variances were evaluated using the Shapiro–Wilk and Levene’s tests, respectively. Differences were considered statistically significant at p < 0.05. All analyses were performed using SPSS software version 20.0 (IBM Corp., Armonk, NY, USA).
4. Discussion
This study demonstrates that CL confers significant protection against Ang II-induced endothelial dysfunction in an in vitro CA model, through its anti-inflammatory and antioxidant properties. Exposure to Ang II markedly reduced HBMEC viability and induced pronounced oxidative stress, as evidenced by increased intracellular ROS and decreased NO levels. These changes were accompanied by elevated expression of proinflammatory cytokines (TNF-α, IL-6, IFN-γ) and genes involved in vascular remodeling and inflammation (MMP-2, MMP-9, VEGF, HMGB1, NF-κB p65). Importantly, CL pre-treatment reversed these pathological alterations in a concentration-dependent manner. Nitric oxide plays a pivotal role in maintaining endothelial integrity by promoting vasodilation, preventing platelet aggregation, and reducing leukocyte adhesion to the vascular wall [
12]. In addition, NO exhibits strong antioxidant and anti-apoptotic properties, making it a key regulator of endothelial cell survival under conditions of oxidative and inflammatory stress [
13]. In the present study, NO levels were significantly reduced following Ang II exposure, reflecting the impairment of endothelial function. CL treatment restored NO concentrations in a dose-dependent manner, suggesting that part of its protective effect involves the preservation of NO bioavailability. Although we did not directly evaluate the expression of endothelial NO synthase (eNOS), which is the primary enzyme responsible for NO synthesis in endothelial cells, the observed increase in NO levels may indirectly reflect eNOS activity. These findings support the conclusion that CL contributes to endothelial protection not only by attenuating oxidative and inflammatory damage but also by enhancing NO-mediated signaling pathways.
The observed increase in cell viability, reduction in apoptosis, normalization of NO/ROS balance, and downregulation of inflammatory and remodeling-associated genes suggest that CL can mitigate endothelial injury through multifaceted mechanisms. These findings support the hypothesis that CL targets core pathological features of aneurysm development—including endothelial cell loss, ECM degradation, and chronic inflammation—thereby providing a potential pharmacological strategy for preventing aneurysmal progression. Our findings are consistent with previous reports indicating that Ang II induces oxidative stress and elevates proinflammatory cytokine production in both endothelial and vascular smooth muscle cells. For example, Dandona et al. demonstrated that Ang II suppresses endothelial NO synthase (eNOS) activity and enhances IL-6 and TNF-α production via ROS-dependent pathways in human vascular cells [
14]. Benicky et al. further showed that Ang II upregulates TNF-α and IL-6 expression both centrally and peripherally, contributing to vascular inflammation [
15]. In our model, treatment with 1 µM Ang II decreased NO levels by approximately 50% and increased TNF-α, IL-6, and IFN-γ levels by 2–3 fold, which falls within the same range as these established studies. Thus, our in vitro model reliably recapitulates key inflammatory and oxidative effects of Ang II, reinforcing its relevance for studying endothelial dysfunction in vitro endothelial injury model simulating cerebral aneurysm-related pathophysiology pathogenesis.
Ang II is a vasoactive peptide widely recognized as a key initiator of vascular inflammation and a critical contributor to CA development. Its pathogenic role is primarily mediated through the generation of ROS, endothelial dysfunction, elevated proinflammatory cytokine production, and transcriptional regulation of vascular remodeling genes [
9,
16]. Consistent with these findings, our study demonstrated that Ang II treatment led to a marked decrease in cell viability and a significant upregulation of inflammatory mediators, including ROS, IL-6, TNF-α, and IFN-γ. CL, an α2-adrenergic receptor agonist, is traditionally known for its central sympatholytic effects but has also been shown to exert potent antioxidant and anti-inflammatory actions in peripheral vascular systems [
17]. Our data support this dual action, showing that CL significantly ameliorated Ang II-induced endothelial dysfunction in a concentration-dependent manner. Notably, treatment with 10 μM CL restored cell viability to near-control levels and substantially reduced both ROS and cytokine levels. Moreover, Ang II-induced overexpression of critical genes involved in aneurysm pathogenesis—such as VEGF, MMP-2, MMP-9, HMGB1, and NF-κB p65—was significantly downregulated following CL treatment. The suppression of VEGF and matrix metalloproteinases is particularly relevant, as these factors contribute to ECM degradation and vascular wall destabilization [
18,
19]. Additionally, the downregulation of NF-κB p65 indicates that CL may inhibit a central transcriptional regulator of inflammation [
20]. Previous studies have also suggested that CL may reduce cAMP levels via a G-protein-coupled mechanism, thereby attenuating MMP expression in Vascular Smooth Muscle Cells (VSMCs) [
21]. This, in conjunction with reduced sympathetic outflow and improved vascular tone, may collectively decrease hemodynamic stress—a known contributor to aneurysm progression [
22].
CL has been reported to exert diverse physiological effects through multiple mechanisms of action. While some studies have suggested that CL may exacerbate endothelial dysfunction under certain conditions [
23], the current study demonstrated that low- concentration Ang II + CL 0.1 µM (CL0.1) was both safe and protective in the context of cerebral endothelial injury. Reactive oxygen species, commonly referred to as free radicals, are typically regulated by endogenous antioxidant defense systems, including superoxide dismutase (SOD), catalase, and glutathione peroxidase. However, an imbalance between ROS production and antioxidant capacity leads to oxidative stress. Mitochondria are the primary intracellular source of ROS, and excessive ROS accumulation can result in damage at the cellular, tissue, and organ levels. Moreover, ROS can inhibit glutamate reuptake and activate NMDA (N-methyl-D-aspartate) receptors, triggering additional superoxide generation, calcium influx, and NO production. CL is also believed to possess antioxidant properties, which may contribute to its protective role in vascular pathologies by attenuating oxidative stress and preserving endothelial function [
24]. Previous studies have demonstrated CL’s antioxidant capacity per se. In particular, Gourdin et al. showed that CL significantly reduced oxidative burst and improved endothelial-dependent vasodilation following ischemia–reperfusion injury in humans, supporting its direct role in modulating oxidative stress [
7].
Histological evaluations of cerebral aneurysms have consistently demonstrated features of both acute and chronic inflammation, as well as medial wall degeneration [
25]. Numerous immunological factors have been implicated in the formation and progression of cerebral aneurysms, along with the underlying mechanisms driving these processes. Excessive hemodynamic stress disrupts endothelial cell function and promotes the activation of inflammatory cells within the vascular media [
26]. Key molecular mediators such as NF-κB and Ang II further amplify these responses, contributing to phenotypic changes in VSMCs, which acquire proinflammatory and matrix-remodeling properties [
27]. This phenotypic switch initiates apoptotic pathways and facilitates ECM degradation. Matrix metalloproteinases, particularly those secreted by macrophages and VSMCs, play a central role in ECM breakdown and pathological vascular remodeling. Additionally, cytokines such as IL-6, TNF-α, and NO are key regulators of vascular inflammation and immune responses [
28]. Elevated levels of these cytokines and associated chemokines have been detected in the bloodstream of patients with cerebral aneurysms [
29]. The present study’s findings are notable in that CL treatment led to significant reductions not only in MMP expression but also in TNF-α and IL-6 levels, supporting its dual role in modulating both immune activation and the inflammatory cascade involved in aneurysm pathophysiology.
These results are consistent with previous studies that have demonstrated the protective effects of CL in models of systemic inflammation and vascular injury [
30]. However, it is important to acknowledge that the present study was conducted using an in vitro model of cerebral endothelial dysfunction. Therefore, further in vivo investigations using relevant animal models are necessary to validate these findings and to comprehensively evaluate the translational potential of CL as a therapeutic agent for CA prevention. CL, an alpha-2 adrenergic agonist, has demonstrated potential in modulating endothelial function and inflammation. In a study by Taguchi et al., co-treatment with CL and a GRK2 inhibitor in diabetic mice prevented rebound hypertension and endothelial dysfunction after withdrawal. This effect was associated with increased Akt/eNOS signaling and NO production, suggesting a protective role of CL in vascular health [
31].
Ang II is known to exert proinflammatory effects on the vascular system. It increases the expression of adhesion molecules, cytokines, and chemokines, leading to endothelial dysfunction. Ang II also promotes oxidative stress by generating ROS, further contributing to vascular inflammation and damage [
14,
15,
32,
33,
34]. Oxidative stress plays a significant role in the pathogenesis of cerebral aneurysms. It induces endothelial injury and smooth muscle cell phenotypic switching, leading to vessel wall remodeling and potential rupture. Laaksamo et al. reported that oxidative stress is associated with cell death and wall degradation in intracranial aneurysms [
35], highlighting its importance in aneurysm progression [
36,
37,
38]. Furthermore, recent studies have identified oxidative stress-related biomarkers in intracranial aneurysms. Zhang et al. utilized machine learning algorithms to identify four key genes associated with oxidative stress in intracranial aneurysms, providing insights into potential diagnostic markers and therapeutic targets [
39]. These findings collectively suggest that CL may attenuate Ang II-induced endothelial dysfunction through anti-inflammatory and antioxidant pathways, offering a potential therapeutic approach in conditions like CA pathogenesis.
Despite the robust molecular and cellular findings presented in this study, several limitations should be acknowledged. In addition, while the current study focused on CL’s protective role under Ang II-induced injury, a CL-only group (without Ang II) was not included. This limits the ability to determine whether the observed effects are specific to stress conditions or also occur under baseline physiology. Future studies should include such groups to distinguish between baseline pharmacological activity and stimulus-specific modulation. First, the experimental model employed was based entirely on in vitro assays using HBMECs to simulate cerebral endothelial dysfunction. While this model effectively mimics early-stage endothelial injury and allows for precise mechanistic exploration, it does not fully capture the complex hemodynamic, immunological, and neurovascular interactions present in in vivo CA development. Second, the effects of CL were evaluated over a relatively short treatment period (24 h), and longer-term outcomes such as endothelial repair, matrix stabilization, or gene expression persistence were not assessed. Additionally, while three concentrations of CL were tested, pharmacokinetic data and physiological relevance to systemic administration in animal or clinical settings were not explored. Interestingly, TNF-α levels in the CL10 group were observed to be slightly lower than those in the control group. This phenomenon may be attributed to the strong anti-inflammatory potential of high-dose CL, which has been shown to suppress not only stimulus-induced cytokine production but also baseline (unstimulated) expression levels via inhibition of NF-κB and MAPK pathways. CL-mediated downregulation of intracellular cAMP has also been linked to reduced activation of transcriptional regulators involved in basal cytokine expression. These mechanisms are particularly evident at supraphysiological concentrations, such as 10 µM, which may explain the slight reduction in TNF-α below control levels observed in our study [
8]. The anti-inflammatory and antioxidant effects observed—though statistically significant—were measured at the transcriptional and biochemical levels, and thus functional outcomes such as endothelial barrier restoration, vascular tone modulation, or aneurysmal wall stabilization were not directly evaluated.
One limitation of our study is the use of a pretreatment protocol with CL, which may not fully represent the clinical context where treatment is typically initiated after pathology onset. Future in vitro and in vivo studies should include posttreatment protocols to evaluate the therapeutic efficacy of CL under more physiologically relevant conditions. Additionally, future studies should confirm the presence of alpha two adrenergic receptors in HBMECs under our experimental conditions and assess whether CL’s protective effects can be reversed by specific alpha two AR antagonists such as yohimbine. These experiments would provide mechanistic insight into receptor specific actions of CL. Future studies should incorporate in vivo aneurysm models such as elastase induced or genetically modified rodent models to validate the protective effects of CL under physiological and pathological conditions. These models would also allow assessment of aneurysm incidence, rupture rate, and histopathological wall integrity following treatment. Moreover, integration of hemodynamic modeling and imaging modalities such as MRI angiography and Doppler flowmetry could provide valuable insights into the vascular remodeling process in response to CL. Finally, it would be important to examine CL’s interaction with other vasoactive or anti-inflammatory agents as well as its long term safety and efficacy profile in the context of aneurysm prevention. Although gene expression analyses were performed to assess key apoptotic and oxidative stress markers, protein level confirmation such as Western blotting for Caspase 3 or related proteins could not be included due to laboratory constraints. This limitation has now been explicitly acknowledged. We agree that future studies should incorporate immunoblotting or immunohistochemical methods to validate mRNA findings and deepen the mechanistic understanding of CL’s protective effects. Elucidating downstream signaling pathways such as MAPK, PI3K Akt, or JAK STAT and confirming protein level expression changes via immunoblotting or immunohistochemistry will also enhance the mechanistic clarity of its therapeutic action.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}