

Sugars to Acids via Thioesters: A Computational Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

2. Computational Methods
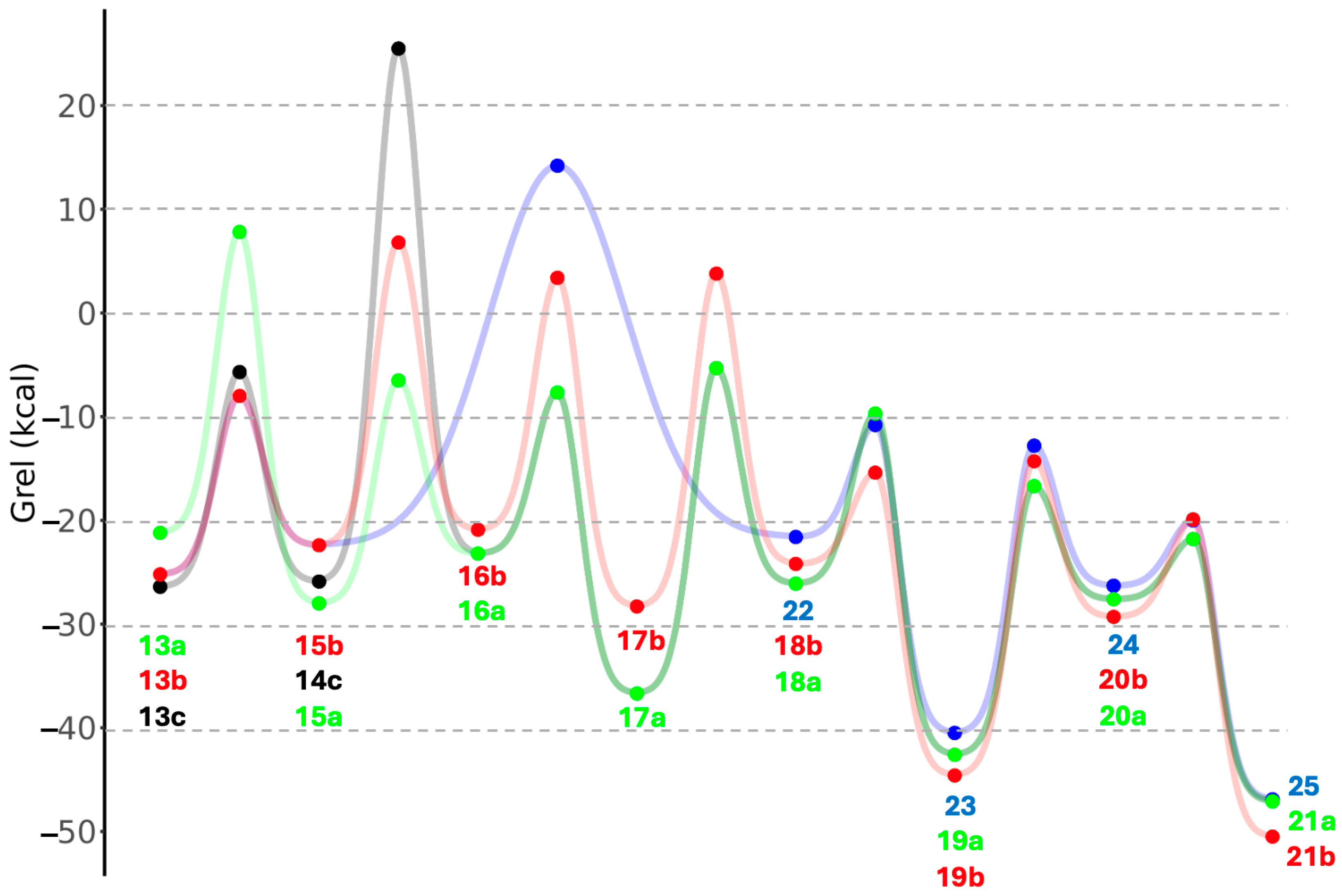
3. Results and Discussion
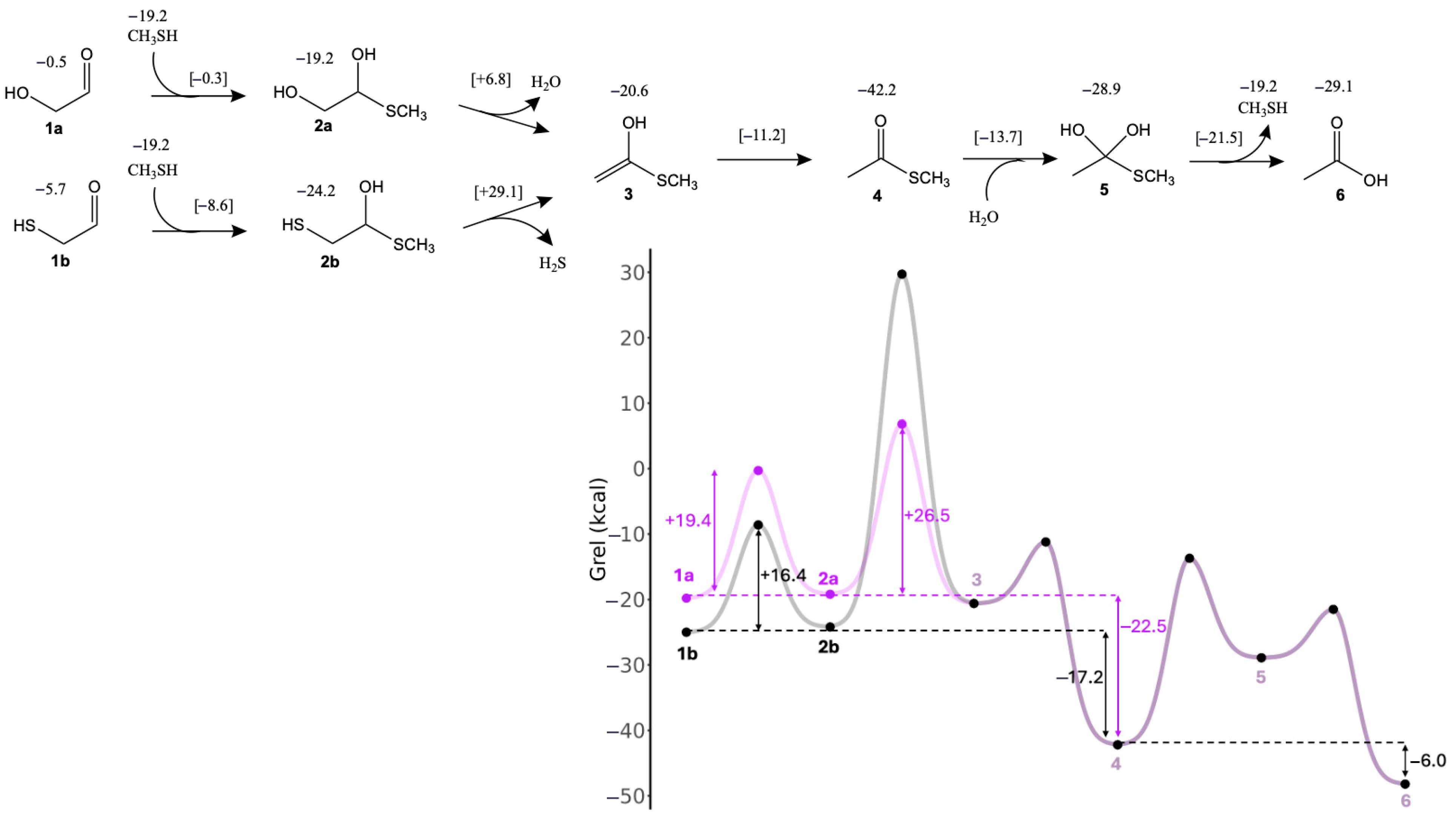
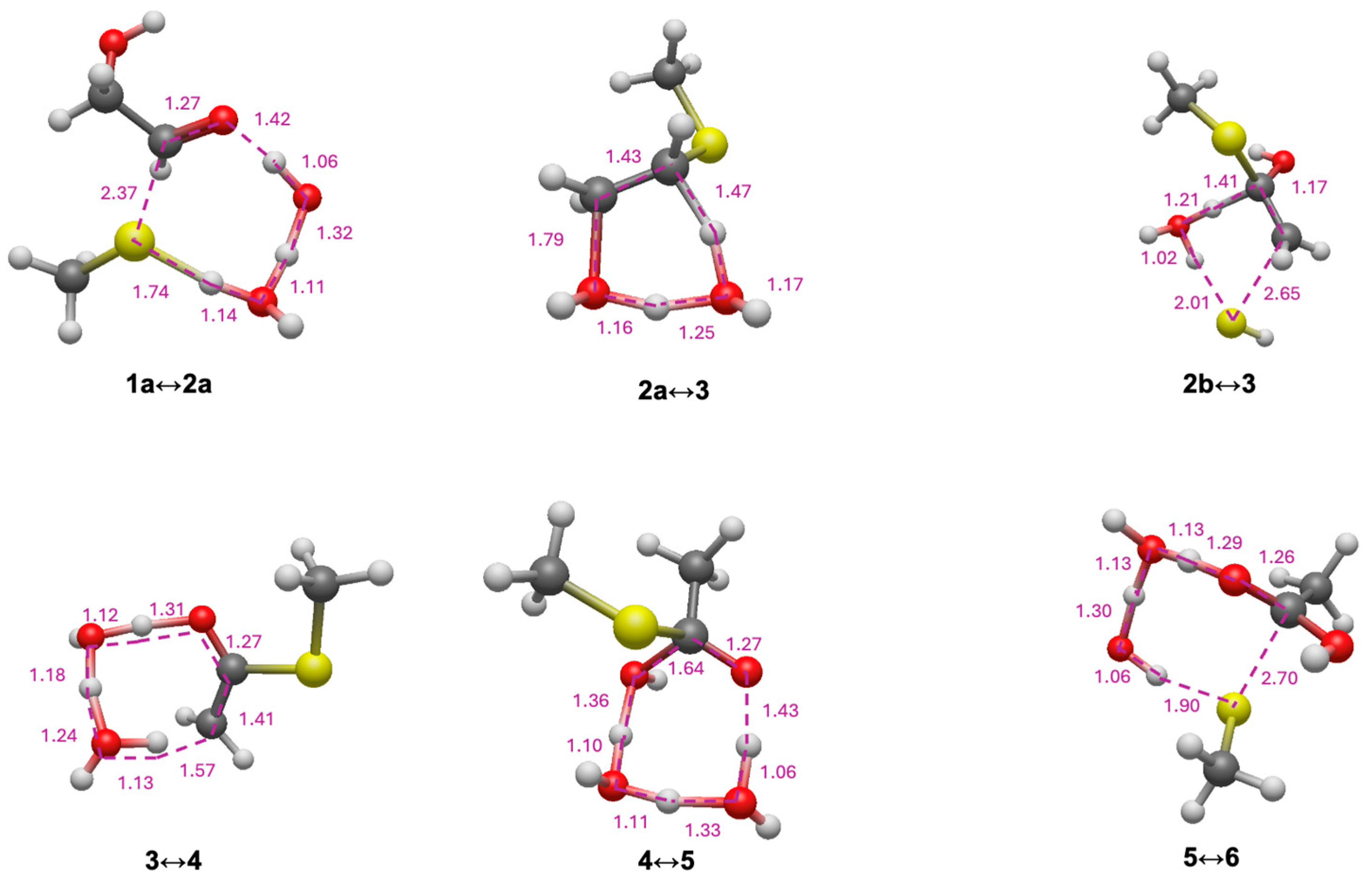
3.1. Glycolaldehyde and Mercaptoaldehyde
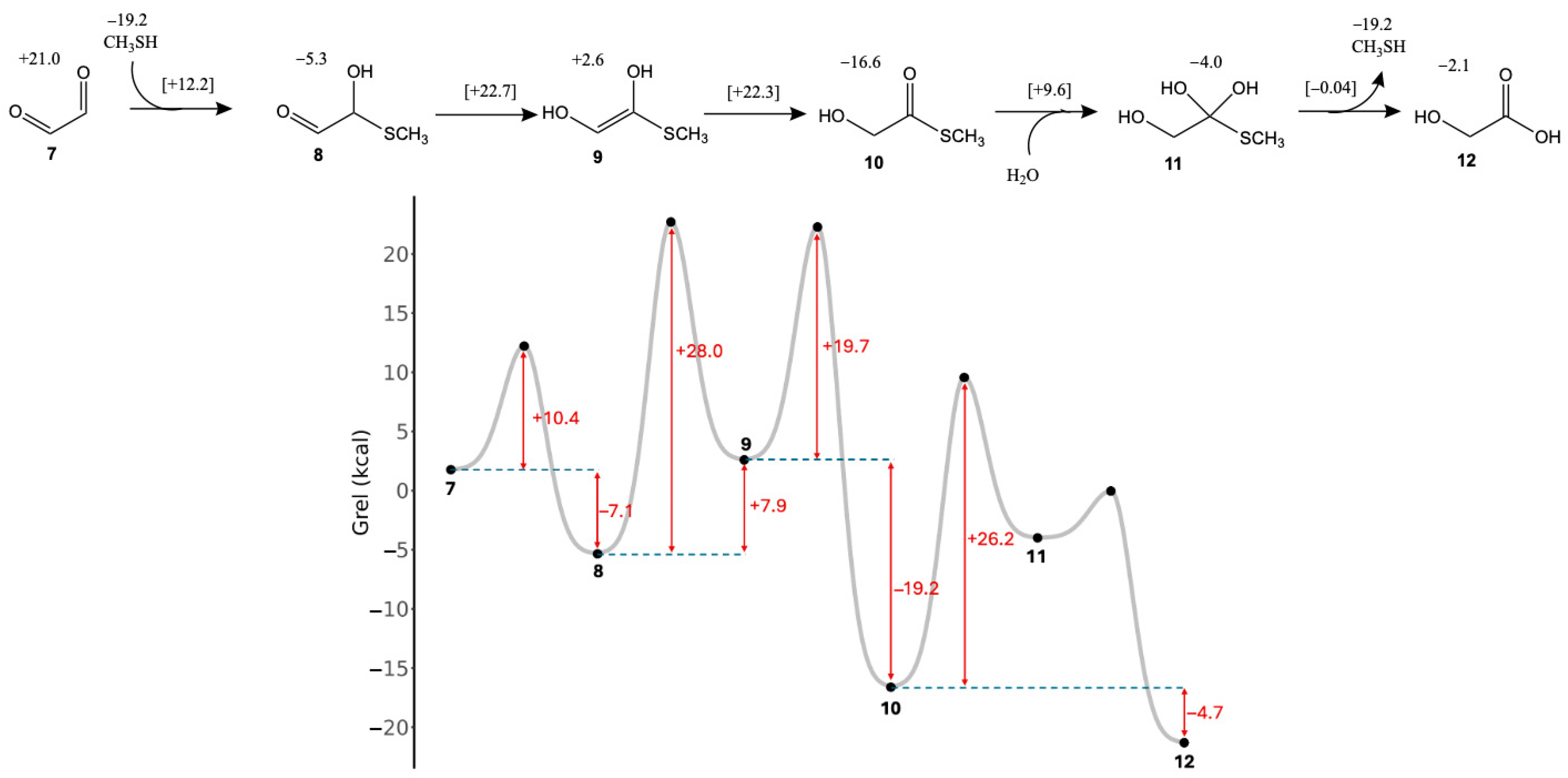
3.2. Glyoxal
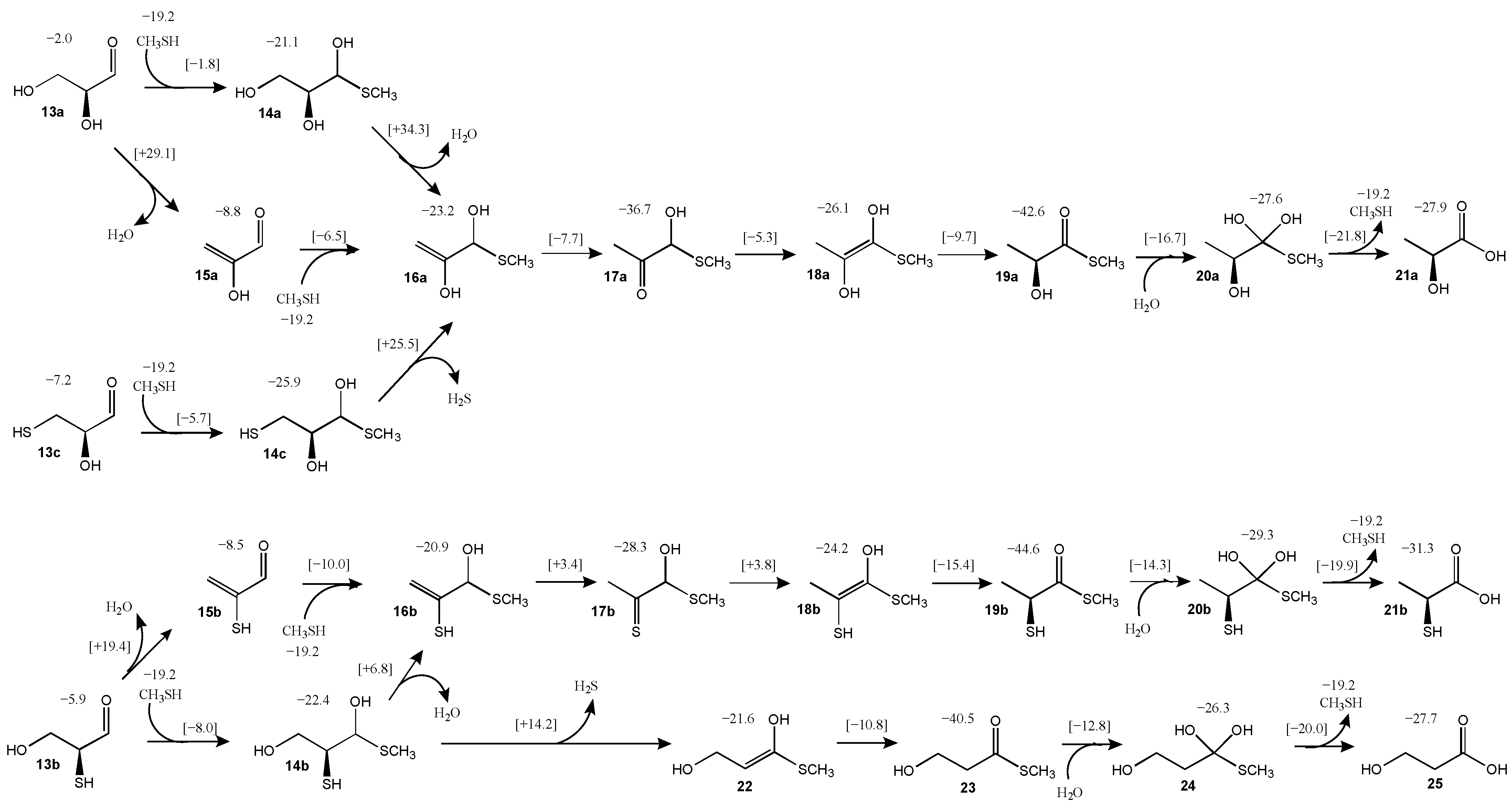
3.3. Glyceraldehyde and Its Sulfur Analogs
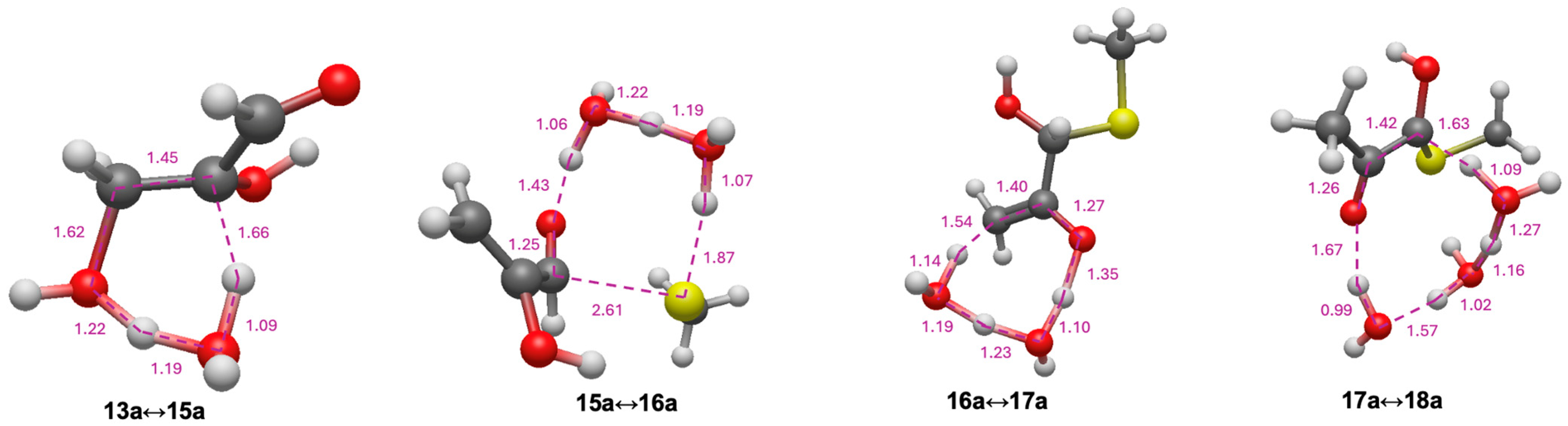
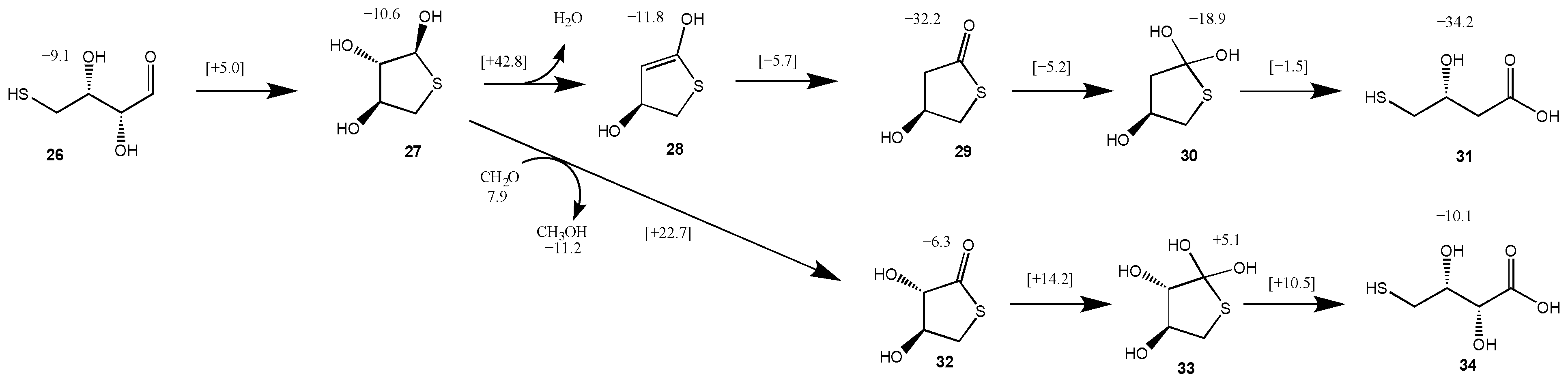
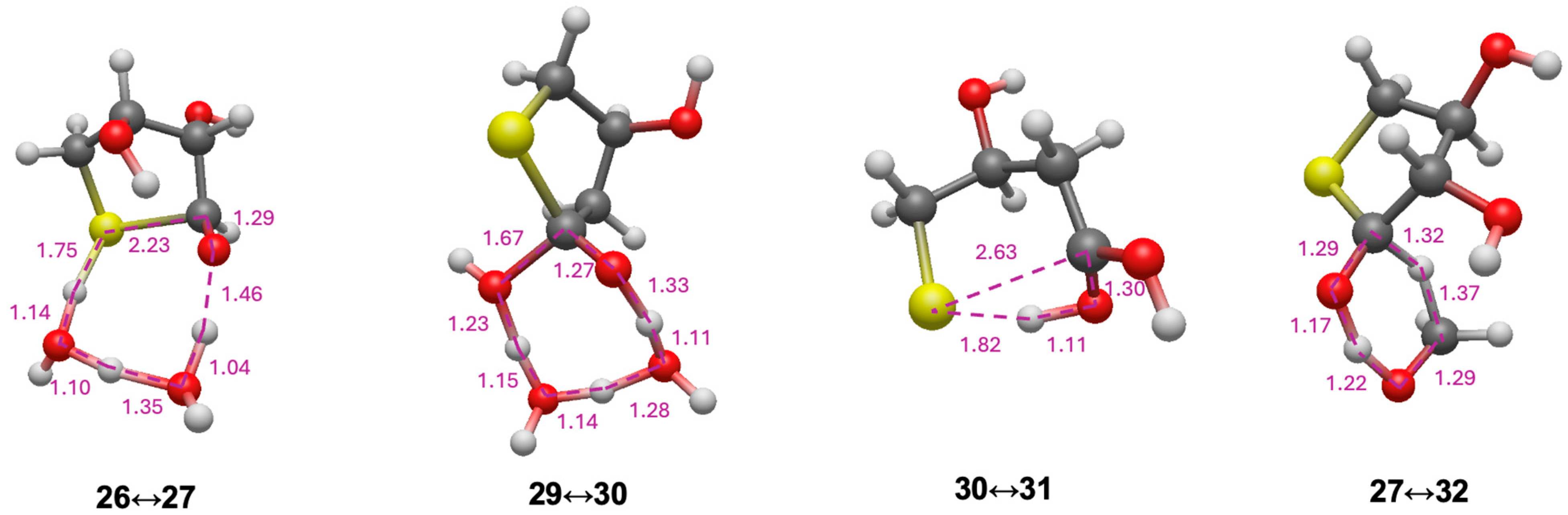
3.4. Intramolecular Disproportionation with a C4 Sugar
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braakman, R.; Smith, E. The compositional and evolutionary logic of metabolism. Phys. Biol. 2013, 10, 11001. [Google Scholar] [CrossRef]
- Xavier, J.C.; Hordijk, W.; Kauffman, S.; Steel, M.; Martin, W.F. Autocatalytic chemical networks at the origin of metabolism. Proc. Biol. Sci. 2020, 287, 20192377. [Google Scholar] [CrossRef]
- Omran, A.; Menor-Salvan, C.; Springsteen, G.; Pasek, M. The Messy Alkaline Formose Reaction and Its Link to Metabolism. Life 2020, 10, 125. [Google Scholar] [CrossRef]
- Breslow, R. On the Mechanism of the Formose Reaction. Tetrahedron Lett. 1959, 1, 22–26. [Google Scholar] [CrossRef]
- Appayee, C.; Breslow, R. Deuterium studies reveal a new mechanism for the formose reaction involving hydride shifts. J. Am. Chem. Soc. 2014, 136, 3720–3723. [Google Scholar] [CrossRef]
- Ricardo, A.; Frye, F.; Carrigan, M.A.; Tipton, J.D.; Powell, D.H.; Benner, S.A. 2-Hydroxymethylboronate as a reagent to detect carbohydrates: Application to the analysis of the formose reaction. J. Org. Chem. 2006, 71, 9503–9505. [Google Scholar] [CrossRef]
- Robinson, W.E.; Daines, E.; van Duppen, P.; de Jong, T.; Huck, W.T.S. Environmental conditions drive self-organization of reaction pathways in a prebiotic reaction network. Nat. Chem. 2022, 14, 623–631. [Google Scholar] [CrossRef]
- van Duppen, P.; Daines, E.; Robinson, W.E.; Huck, W.T.S. Dynamic Environmental Conditions Affect the Composition of a Model Prebiotic Reaction Network. J. Am. Chem. Soc. 2023, 145, 7559–7568. [Google Scholar] [CrossRef]
- Briš, A.; Baltussen, M.G.; Tripodi, G.L.; Huck, W.T.; Franceschi, P.; Roithová, J. Direct Analysis of Complex Reaction Mixtures: Formose Reaction. Angew. Chem. Int. Ed. 2024, 63, e202316621. [Google Scholar] [CrossRef]
- de Duve, C. The Beginnings of Life on Earth. Am. Sci. 1995, 83, 428–437. [Google Scholar]
- Gulick, A. Phosphorus as a factor in the origin of life. Am. Sci. 1955, 43, 479–489. [Google Scholar]
- Keefe, A.D.; Miller, S.L. Are polyphosphates or phosphate esters prebiotic reagents? J. Mol. Evol. 1995, 41, 693–702. [Google Scholar] [CrossRef]
- Brady, M.P.; Tostevin, R.; Tosca, N.J. Marine phosphate availability and the chemical origins of life on Earth. Nat. Commun. 2022, 13, 5162. [Google Scholar] [CrossRef]
- Gull, M.; Feng, T.; Cruz, H.A.; Krishnamurthy, R.; Pasek, M.A. Prebiotic Chemistry of Phosphite: Mild Thermal Routes to Form Condensed-P Energy Currency Molecules Leading Up to the Formation of Organophosphorus Compounds. Life 2023, 13, 920. [Google Scholar] [CrossRef]
- Gull, M.; Feng, T.; Smith, B.; Calcul, L.; Pasek, M.A. Prebiotic Syntheses of Organophosphorus Compounds from Reduced Source of Phosphorus in Non-Aqueous Solvents. Life 2023, 13, 2134. [Google Scholar] [CrossRef]
- Goldford, J.E.; Hartman, H.; Marsland, R., 3rd; Segre, D. Environmental boundary conditions for the origin of life converge to an organo-sulfur metabolism. Nat. Ecol. Evol. 2019, 3, 1715–1724. [Google Scholar] [CrossRef]
- Goldford, J.E.; Hartman, H.; Smith, T.F.; Segre, D. Remnants of an Ancient Metabolism without Phosphate. Cell 2017, 168, 1126–1134 e1129. [Google Scholar] [CrossRef]
- Wachtershauser, G. Before enzymes and templates: Theory of surface metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [CrossRef]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef]
- Youssef-Saliba, S.; Vallee, Y. Sulfur Amino Acids: From Prebiotic Chemistry to Biology and Vice Versa. Synthesis 2021, 53, 2798–2808. [Google Scholar] [CrossRef]
- Cho, C.J.; An, T.; Lai, Y.C.; Vazquez-Salazar, A.; Fracassi, A.; Brea, R.J.; Chen, I.A.; Devaraj, N.K. Protocells by spontaneous reaction of cysteine with short-chain thioesters. Nat. Chem. 2025, 17, 148–155. [Google Scholar] [CrossRef]
- Kua, J.; Miller, N.A. Preliminary Free Energy Map of Prebiotic Compounds Formed from CO2, H2 and H2S. Life 2022, 12, 1763. [Google Scholar] [CrossRef]
- Kua, J.; Peña, M.T.; Cotter, S.N.; Leca, J. Sulfur Analogs of the Core Formose Cycle: A Free Energy Map. Life 2025, 15, 1. [Google Scholar] [CrossRef]
- Weber, A.L. Prebiotic formation of ‘energy-rich’ thioesters from glyceraldehyde and N-acetylcysteine. Orig. Life Evol. Biosph. 1984, 15, 17–27. [Google Scholar] [CrossRef]
- Weber, A.L. Nonenzymatic formation of “energy-rich” lactoyl and glyceroyl thioesters from glyceraldehyde and a thiol. J. Mol. Evol. 1984, 20, 157–166. [Google Scholar] [CrossRef]
- Weber, A.L. Formation of pyrophosphate, tripolyphosphate, and phosphorylimidazole with the thioester, n, s-diacetyl-cysteamine, as the condensing agent. J. Mol. Evol. 1981, 18, 24–29. [Google Scholar] [CrossRef]
- Weber, A.L. Formation of pyrophosphate on hydroxyapatite with thioesters as condensing agents. Biosystems 1982, 15, 183–189. [Google Scholar] [CrossRef]
- El Qami, A.; Hilari, J.I.; Blandin, V.; Gayraud, O.; Milet, A.; Vallee, Y. Prebiotic formation of thioesters via cyclic anhydrides as a key step in the emergence of metabolism. Sci. Rep. 2025, 15, 7039. [Google Scholar] [CrossRef]
- Leqraa, N.; Nicolet, Y.; Milet, A.; Vallee, Y. A way to thioacetate esters compatible with non-oxidative prebiotic conditions. Sci. Rep. 2020, 10, 14488. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Bouza, M.; Fernandez, F.M.; Leman, L.J.; Williams, L.D.; Hud, N.V.; Guzman-Martinez, A. Thioesters provide a plausible prebiotic path to proto-peptides. Nat. Commun. 2022, 13, 2569. [Google Scholar] [CrossRef]
- Kua, J.; Hanley, S.W.; De Haan, D.O. Thermodynamics and Kinetics of Glyoxal Dimer Formation: A Computational Study. J. Phys. Chem. A 2008, 112, 66–72. [Google Scholar] [CrossRef]
- Kua, J.; Avila, J.E.; Lee, C.G.; Smith, W.D. Mapping the Kinetic and Thermodynamic Landscape of Formaldehyde Oligomerization under Neutral Conditions. J. Phys. Chem. A 2013, 117, 12658–12667. [Google Scholar] [CrossRef]
- Kua, J.; Galloway, M.M.; Millage, K.D.; Avila, J.E.; De Haan, D.O. Glycolaldehyde Monomer and Oligomer Equilibria in Aqueous Solution: Comparing Computational Chemistry and NMR Data. J. Phys. Chem. A 2013, 117, 2997–3008. [Google Scholar] [CrossRef]
- Kua, J.; Hernandez, A.L.; Velasquez, D.N. Thermodynamics of Potential CHO Metabolites in a Reducing Environment. Life 2021, 11, 1025. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J. Comput. Chem. 1999, 20, 730–748. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Warshel, A.; Florian, J. Computer simulations of enzyme catalysis: Finding out what has been optimized by evolution. Proc. Natl. Acad. Sci. USA 1998, 95, 5950–5955. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Bailey, W.F. Chiral diamines 4: A computational study of the enantioselective deprotonation of Boc-pyrrolidine with an alkyllithium in the presence of a chiral diamine. J. Am. Chem. Soc. 2001, 123, 8231–8238. [Google Scholar] [CrossRef]
- Nielsen, R.J.; Keith, J.M.; Stoltz, B.M.; Goddard, W.A., 3rd. A computational model relating structure and reactivity in enantioselective oxidations of secondary alcohols by (-)-sparteine-Pd(II) complexes. J. Am. Chem. Soc. 2004, 126, 7967–7974. [Google Scholar] [CrossRef]
- Deubel, D.V.; Lau, J.K. In silico evolution of substrate selectivity: Comparison of organometallic ruthenium complexes with the anticancer drug cisplatin. Chem. Commun. 2006, 23, 2451–2453. [Google Scholar] [CrossRef]
- Wertz, D.H. Relationship between the gas-phase entropies of molecules and their entropies of solvation in water and 1-octanol. J. Am. Chem. Soc. 1980, 102, 5316–5322. [Google Scholar] [CrossRef]
- Abraham, M.H. Relationship between solution entropies and gas phase entropies of nonelectrolytes. J. Am. Chem. Soc. 1981, 103, 6742–6744. [Google Scholar] [CrossRef]
- Krizner, H.E.; De Haan, D.O.; Kua, J. Thermodynamics and Kinetics of Methylglyoxal Dimer Formation: A Computational Study. J. Phys. Chem. A 2009, 113, 6994–7001. [Google Scholar] [CrossRef]
- Kua, J.; Miller, A.S.; Wallace, C.E.; Loli, H. Role of Acid in the Co-oligomerization of Formaldehyde and Pyrrole. ACS Omega 2019, 4, 22251–22259. [Google Scholar] [CrossRef]
- Kua, J.; Tripoli, L.P. Exploring the Core Formose Cycle: Catalysis and Competition. Life 2024, 14, 933. [Google Scholar] [CrossRef]
- eQuilbrator. Available online: http://equilibrator.weizmann.ac.il (accessed on 15 April 2025).
- Yi, R.; Kern, R.; Pollet, P.; Lin, H.; Krishnamurthy, R.; Liotta, C.L. Erythrose and Threose: Carbonyl Migrations, Epimerizations, Aldol, and Oxidative Fragmentation Reactions under Plausible Prebiotic Conditions. Chemistry 2023, 29, e202202816. [Google Scholar] [CrossRef]
- Weber, A.L. Chemical constraints governing the origin of metabolism: The thermodynamic landscape of carbon group transformations under mild aqueous conditions. Orig. Life Evol. Biosph. 2002, 32, 333–357. [Google Scholar] [CrossRef]
- Krishnamurthy, R.; Liotta, C.L. The potential of glyoxylate as a prebiotic source molecule and a reactant in protometabolic pathways-The glyoxylose reaction. Chem 2023, 9, 784–797. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kua, J.; Karin, J.D. Sugars to Acids via Thioesters: A Computational Study. Life 2025, 15, 1189. https://doi.org/10.3390/life15081189
Kua J, Karin JD. Sugars to Acids via Thioesters: A Computational Study. Life. 2025; 15(8):1189. https://doi.org/10.3390/life15081189
Chicago/Turabian StyleKua, Jeremy, and Jonathan D. Karin. 2025. "Sugars to Acids via Thioesters: A Computational Study" Life 15, no. 8: 1189. https://doi.org/10.3390/life15081189
APA StyleKua, J., & Karin, J. D. (2025). Sugars to Acids via Thioesters: A Computational Study. Life, 15(8), 1189. https://doi.org/10.3390/life15081189