
Integrating Soil Physicochemical Properties and Microbial Functional Prediction to Assess Land-Use Impacts in a Cold-Region Wetland Ecosystem


Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description
2.2. Sample Collection
2.3. Analysis of Soil Physicochemical Properties
2.4. DNA Extraction and High-Throughput 16S rRNA Gene Paired-End Sequencing
2.5. Sequencing Data Processing and Analysis
2.6. Statistical Analysis
3. Results
3.1. Soil Physico-Chemical Properties
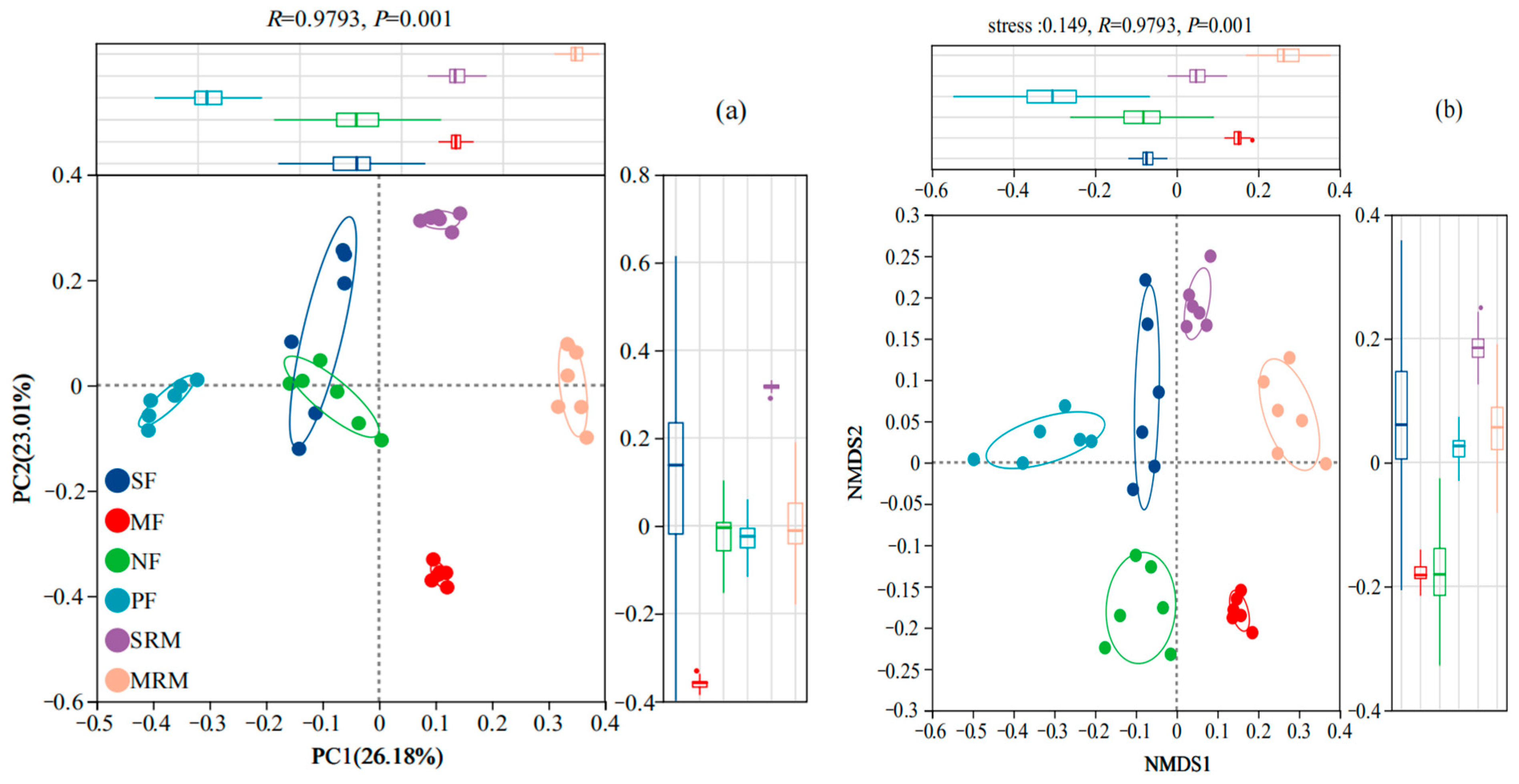
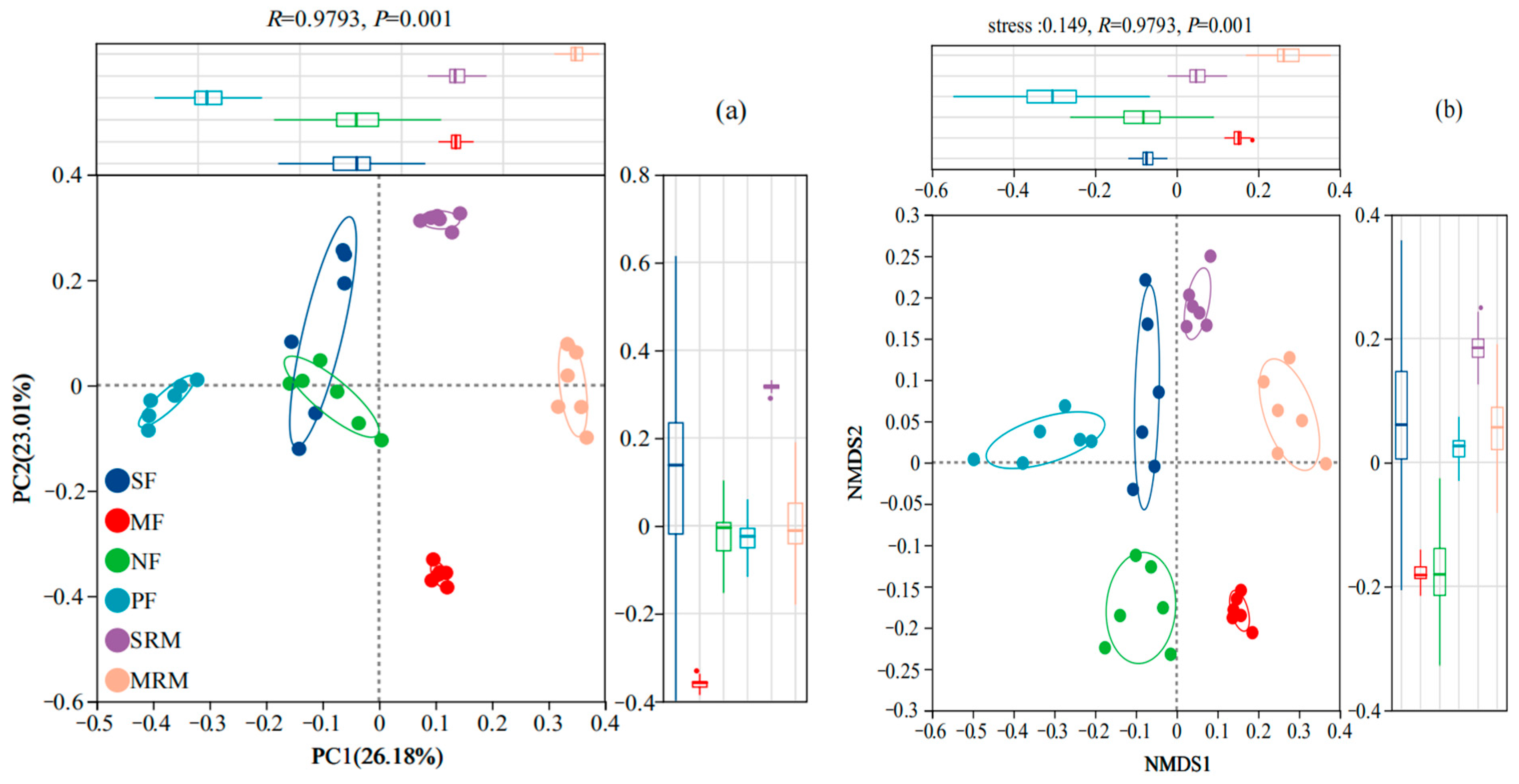
3.2. Microbial Alpha and Beta Diversity
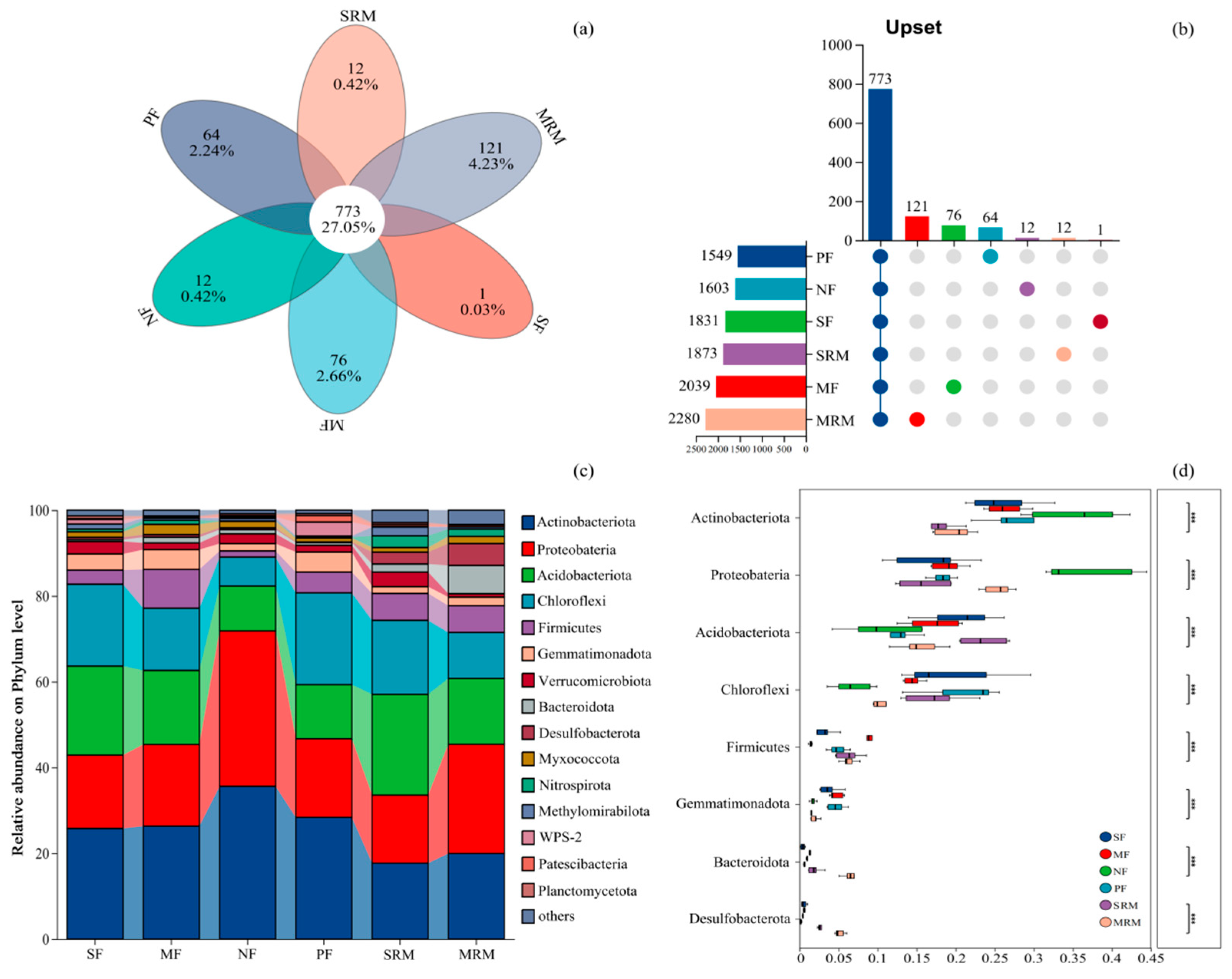
3.3. Species Venn Diagram and Soil Bacterial Structure Analysis
3.4. Correlation Analysis of Environmental Factors
3.5. Phenotypic Prediction Analysis
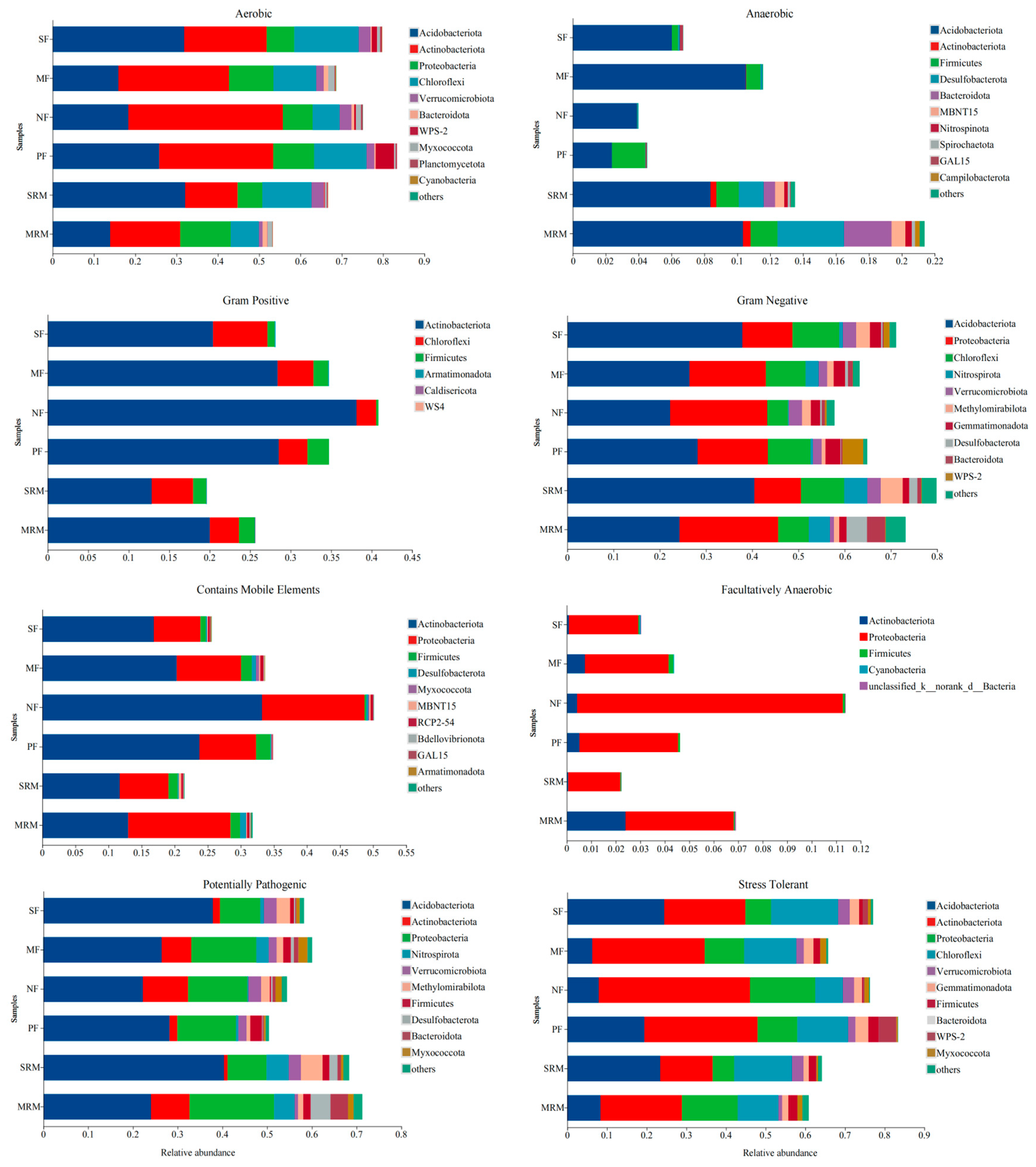
3.6. BugBase Phenotype Prediction
4. Discussion
4.1. Relationship Between Soil Physicochemical Properties and Diversity
4.2. Environmental Factors Influencing Soil Microbial Community Structure
4.3. Functional Prediction Analysis of Soil Bacterial Communities
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, D.; Chi, Q.; Sui, X.; Zhang, M.M.; Jia, H.B.; Sun, G.Y. Metabolic diversity and seasonal variation of soil microbial communities in natural forested wetlands. J. For. Res. 2021, 32, 2619–2631. [Google Scholar] [CrossRef]
- Hu, S.J.; Niu, Z.G.; Chen, Y.F.; Li, L.F.; Zhang, H.Y. Global wetlands: Potential distribution, wetland loss, and status. Sci. Total Environ. 2017, 586, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.J.; Freeman, R.; Spooner, F.; Newbold, T. Vertebrate population trends are influenced by interactions between land use, climatic position, habitat loss and climate change. Glob. Change Biol. 2022, 28, 797–815. [Google Scholar] [CrossRef]
- Pal, S.; Sarkar, R.; Saha, T.K. Exploring the forms of wetland modifications and investigating the causes in lower Atreyee river floodplain area. Ecol. Inform. 2022, 67, 101494. [Google Scholar] [CrossRef]
- Chen, H.; Meng, F.; Yu, Z.; Tan, Y. Spatial–temporal characteristics and influencing factors of farmland expansion in different agricultural regions of Heilongjiang Province, China. Land. Use Policy 2022, 115, 106007. [Google Scholar] [CrossRef]
- Xiong, Y.; Mo, S.H.; Wu, H.P.; Qu, X.Y.; Liu, Y.Y.; Zhou, L. Influence of human activities and climate change on wetland landscape pattern—A review. Sci. Total Environ. 2023, 879, 163112. [Google Scholar] [CrossRef]
- Kreplin, H.N.; Santos Ferreira, C.S.; Destouni, G.; Keesstra, S.D.; Salvati, L.; Kalantari, Z. Arctic wetland system dynamics under climate warming. Wiley Interdiscip. Rev. Water 2021, 8, e1526. [Google Scholar] [CrossRef]
- Bragazza, L.; Parisod, J.; Buttler, A.; Bardgett, R.D. Biogeochemical plant–soil microbe feedback in response to climate warming in peatlands. Nat. Clim. Chang. 2013, 3, 273–277. [Google Scholar] [CrossRef]
- Li, M.; Zhang, K.; Yan, Z.; Liu, L.; Kang, E.; Kang, X. Soil Water Content Shapes Microbial Community Along Gradients of Wetland Degradation on the Tibetan Plateau. Front. Microbiol. 2022, 13, 824267. [Google Scholar] [CrossRef]
- Colette, M.; Guentas, L.; Patrona, L.D.; Ansquer, D.; Callac, N. Dynamic of active microbial diversity in rhizosphere sediments of halophytes used for bioremediation of earthen shrimp ponds. Environ. Microbiome 2023, 18, 58. [Google Scholar] [CrossRef]
- Meng, D.Y.; Cheng, H.X.; Shao, Y.; Luo, M.; Xu, D.D.; Liu, Z.M.; Ma, L.L. Progress on the effect of nitrogen on transformation of soil organic carbon. Processes 2022, 10, 2425. [Google Scholar] [CrossRef]
- Bian, H.F.; Zheng, S.; Liu, Y.; Xu, L.; Chen, Z.; He, N.P. Changes to soil organic matter decomposition rate and its temperature sensitivity along water table gradients in cold-temperate forest swamps. Catena 2020, 194, 104684. [Google Scholar] [CrossRef]
- Ji, Z.X.; Pei, T.T.; Chen, Y.; Wu, H.W.; Hou, Q.Q.; Shi, F.Z.; Xie, B.P.; Zhang, J.X. The driving factors of grassland water use efficiency along degradation gradients on the Qinghai-Tibet Plateau, China. Glob. Ecol. Conserv. 2022, 35, e02090. [Google Scholar] [CrossRef]
- Arunrat, N.; Sereenonchai, S.; Kongsurakan, P.; Iwai, C.B.; Yuttitham, M.; Hatano, R. Post-fire recovery of soil organic carbon, soil total nitrogen, soil nutrients, and soil erodibility in rotational shifting cultivation in Northern Thailand. Front. Environ. Sci. 2023, 11, 1117427. [Google Scholar] [CrossRef]
- Zhang, T.; Song, B.; Han, G.X.; Zhao, H.L.; Hu, Q.L.; Zhao, Y.; Liu, H.J. Effects of coastal wetland reclamation on soil organic carbon, total nitrogen, and total phosphorus in China: A meta-analysis. Land. Degrad. Dev. 2023, 34, 3340–3349. [Google Scholar] [CrossRef]
- Schwalm, M.; Zeitz, J. Concentrations of dissolved organic carbon in peat soils as influenced by land use and site characteristics—A lysimeter study. Catena 2015, 127, 72–79. [Google Scholar] [CrossRef]
- Mustafa, G.; Hussain, S.; Liu, Y.H.; Ali, I.; Liu, J.Y.; Bano, H. Microbiology of wetlands and the carbon cycle in coastal wetland mediated by microorganisms. Sci. Total Environ. 2024, 954, 175734. [Google Scholar] [CrossRef]
- Yousaf, A.; Khalid, N.; Aqeel, M.; Noman, A.; Naeem, N.; Sarfraz, W.; Ejaz, U.; Qaiser, Z.; Khalid, A. Nitrogen Dynamics in Wetland Systems and Its Impact on Biodiversity. Nitrogen 2021, 2, 196–217. [Google Scholar] [CrossRef]
- Pedrinho, A.; Mendes, L.W.; de Araujo Pereira, A.P.; Araujo, A.S.F.; Vaishnav, A.; Karpouzas, D.G.; Singh, B.K. Soil microbial diversity plays an important role in resisting and restoring degraded ecosystems. Plant Soil 2024, 500, 325–349. [Google Scholar] [CrossRef]
- Graham, E.B.; Knelman, J.E. Implications of soil microbial community assembly for ecosystem restoration: Patterns, process, and potential. Microb. Ecol. 2023, 85, 809–819. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Furman, A. Soil redox dynamics under dynamic hydrologic regimes-A review. Sci. Total Environ. 2021, 763, 143026. [Google Scholar] [CrossRef] [PubMed]
- Daunoras, J.; Kačergius, A.; Gudiukaitė, R. Role of soil microbiota enzymes in soil health and activity changes depending on climate change and the type of soil ecosystem. Biology 2024, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.F.; Liu, Y.S.; Wang, H.C.; Wang, C.L.; Xia, M.; Wang, N.; Xiao, D.R.; Wang, H. Plant litter decomposition in wetlands is closely associated with phyllospheric fungi as revealed by microbial community dynamics and co-occurrence network. Sci. Total Environ. 2021, 753, 142194. [Google Scholar] [CrossRef]
- Hartmann, M.; Six, J. Soil structure and microbiome functions in agroecosystems. Nat. Rev. Earth Environ. 2023, 4, 4–18. [Google Scholar] [CrossRef]
- Juottonen, H.; Kieman, M.; Fritze, H.; Hamberg, L.; Laine, A.M.; Merilä, P.; Peltoniemi, K.; Putkinen, A.; Tuittila, E.S. Integrating decomposers, methane-cycling microbes and ecosystem carbon fluxes along a peatland successional gradient in a land uplift region. Ecosystems 2022, 25, 1249–1264. [Google Scholar] [CrossRef]
- Ding, J.N.; Yu, S.P. Stochastic processes dominate the assembly of soil bacterial communities under land use change in cold-region wetlands. Life 2024, 14, 1407. [Google Scholar] [CrossRef]
- Abbott, K.M.; Quirk, T.; Fultz, L.M. Soil microbial community development across a 32-year coastal wetland restoration time series and the relative importance of environmental factors. Sci. Total Environ. 2022, 821, 153359. [Google Scholar] [CrossRef] [PubMed]
- Ghosh Roy, S.; Wimpee, C.F.; McGuire, S.A.; Ehlinger, T.J. Responses of bacterial taxonomical diversity indicators to pollutant loadings in experimental wetland microcosms. Water 2022, 14, 251. [Google Scholar] [CrossRef]
- Pérez-Hernández, J.; Gavilán, R.G. Impacts of land-use changes on vegetation and ecosystem functioning: Old-field secondary succession. Plants 2021, 10, 990. [Google Scholar] [CrossRef]
- Sveen, T.R.; Hannula, S.E.; Bahram, M. Microbial regulation of feedbacks to ecosystem change. Trends Microbiol. 2024, 32, 68–78. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, H.; Huang, L.L.; Kong, Z.Y.; Wu, L. Recovery of soil organic carbon storage driven by microbial communities during long-term natural restoration in wetland ecosystems. Ecol. Eng. 2024, 199, 107170. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Osanai, Y.; Anderson, I.C.; Bange, M.P.; Tissue, D.T.; Singh, B.K. Flooding and prolonged drought have differential legacy impacts on soil nitrogen cycling, microbial communities and plant productivity. Plant Soil 2018, 431, 371–387. [Google Scholar] [CrossRef]
- Talbot, C.J.; Bennett, E.M.; Cassell, K.; Hanes, D.M.; Minor, E.C.; Paerl, H.; Raymond, P.A.; Vargas, R.; Vidon, P.G.; Wollheim, W.; et al. The impact of flooding on aquatic ecosystem services. Biogeochemistry 2018, 141, 439–461. [Google Scholar] [CrossRef] [PubMed]
- Han, D.X.; Gao, C.Y.; Liu, H.X.; Li, Y.H.; Cong, J.X.; Yu, X.F.; Wang, G.P. Anthropogenic and climatic-driven peatland degradation during the past 150 years in the Greater Khingan Mountains, NE China. Land. Degrad. Dev. 2021, 32, 4845–4857. [Google Scholar] [CrossRef]
- Ding, J.N. Effect of cultivation and natural restoration on soil microbial functional structure in cold region wetlands. Appl. Ecol. Environ. Res. 2023, 21, 1471–1484. [Google Scholar] [CrossRef]
- Ding, J.N.; Yu, S.P. Impacts of land use on soil nitrogen-cycling microbial communities: Insights from community structure, functional gene abundance, and network complexity. Life 2025, 15, 466. [Google Scholar] [CrossRef] [PubMed]
- Raymond, P.A.; Saiers, J.E.; Sobczak, W.V. Hydrological and biogeochemical controls on watershed dissolved organic matter transport: Pulse-shunt concept. Ecology 2016, 97, 5–16. [Google Scholar] [CrossRef]
- Kuo, J.; Liu, D.; Lin, C.H. Functional prediction of microbial communities in sediment microbial fuel cells. Bioengineering 2023, 10, 199. [Google Scholar] [CrossRef]
- Song, C.X.; He, H.S.; Liu, K.; Du, H.B.; Krohn, J. Impact of historical pattern of human activities and natural environment on wetland in Heilongjiang River Basin. Front. Environ. Sci. Eng. 2023, 17, 151. [Google Scholar] [CrossRef]
- Xie, S.Y.; Yan, D.D.; Li, J.T.; Liu, Y.; Sheng, Y.F.; Luan, Z.Q. GEE-based spatial-temporal dynamics in a Ramsar wetland, Honghe National Nature Reserve, Northeast China from 1985 to 2021. Land. 2022, 11, 2137. [Google Scholar] [CrossRef]
- Bahadori, M.; Tofighi, H. Investigation of soil organic carbon recovery by the Walkley-Black method under diverse vegetation systems. Soil Sci. 2017, 182, 101–106. [Google Scholar] [CrossRef]
- Sanchez, D.; Armas, C.; Pueyo, J.J.; Trasar-Cepeda, C.; Hernández, T. Soil total nitrogen and its relation to land use changes. Geoderma 2023, 411, 115699. [Google Scholar]
- Jones, D.L.; Willett, V.B. Experimental evaluation of methods to quantify dissolved organic nitrogen (DON) and dissolved organic carbon (DOC) in soil. Soil Biol. Biochem. 2006, 38, 991–999. [Google Scholar] [CrossRef]
- Ashraf, M.N.; Hu, C.; Wu, L.; Duan, Y.H.; Zhang, W.J.; Aziz, T.; Cai, A.D.; Abrar, M.M.; Xu, M. Soil and microbial biomass stoichiometry regulate soil organic carbon and nitrogen mineralization in rice-wheat rotation subjected to long-term fertilization. J. Soils Sediments 2020, 20, 3103–3113. [Google Scholar] [CrossRef]
- Song, Y.Q.; Wu, D.M.; Dörsch, P.; Yue, L.T.; Deng, L.L.; Liao, C.S.; Sha, Z.M.; Dong, W.X.; Yu, Y.C. Improved method for extracting nitrites in soil. Agronomy 2024, 14, 331. [Google Scholar] [CrossRef]
- Cordero, I.; Snell, H.; Bardgett, R.D. High throughput method for measuring urease activity in soil. Soil Biol. Biochem. 2019, 134, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.P.; Zheng, H.Q.; Hu, F.L. Extraction, partial characterization, and storage stability of β-glucosidase from propolis. J. Food Sci. 2011, 76, C75–C79. [Google Scholar] [CrossRef]
- Khan, M.T.; Ejaz, U.; Sohail, M. Evaluation of Factors Affecting Saccharification of Sugarcane Bagasse Using Cellulase Preparation from a Thermophilic Strain of Brevibacillus sp. Curr. Microbiol. 2020, 77, 2422–2429. [Google Scholar]
- Ding, J.N. Soil nitrogen transformation and functional microbial abundance in an agricultural soil amended with biochar. Rev. Bras. Cienc. Solo 2023, 47, e0220156. [Google Scholar] [CrossRef]
- Corwin, D.L.; Plant, R.E. Applications of apparent soil electrical conductivity in precision agriculture. Comput. Electron. Agric. 2005, 46, 1–10. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Weber, C.F. Introducing mothur: Open-source, platform-independent community analysis tools. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.W.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Kennedy, P.G. Fun Guild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Liang, S.C.; Deng, J.J.; Jiang, Y.; Wu, S.; Zhou, Y.; Zhu, W. Functional distribution of bacterial community under different land use patterns based on FaProTax function prediction. Pol. J. Environ. Stud. 2020, 29, 1245–1261. [Google Scholar] [CrossRef]
- Chen, X.; Luo, M.; Liu, Y.X.; Tan, J.; Zhang, C.W.; Tan, F.F.; Huang, J.F. Linking carbon-degrading enzyme activity to microbial carbon-use trophic strategy under salinization in a subtropical tidal wetland. Appl. Soil. Ecol. 2022, 174, 104421. [Google Scholar] [CrossRef]
- Govednik, A.; Potočnik, Ž.; Eler, K.; Suhadolc, M. Combined effects of long-term tillage and fertilisation regimes on soil organic carbon, microbial biomass, and abundance of the total microbial communities and N-functional guilds. Appl. Soil. Ecol. 2023, 188, 104876. [Google Scholar] [CrossRef]
- Fox, A.; Widmer, F.; Lüscher, A. Soil microbial community structures are shaped by agricultural systems revealing little temporal variation. Environ. Res. 2022, 214, 113915. [Google Scholar] [CrossRef]
- Gao, X.L.; Lv, S.H.; Diao, Z.Y.; Wang, D.W.; Li, D.K.; Zheng, Z.R. Responses of vegetation, soil, and microbes and carbon and nitrogen pools to semiarid grassland land-use patterns in Duolun, Inner Mongolia, China. Sustainability 2023, 15, 3434. [Google Scholar] [CrossRef]
- Callaway, J.C.; Sullivan, G.; Zedler, J.B. Species-rich plantings increase biomass and nitrogen accumulation in a wetland restoration experiment. Ecol. Appl. 2003, 13, 1626–1639. [Google Scholar] [CrossRef]
- Naylor, D.; McClure, R.; Jansson, J. Trends in microbial community composition and function by soil depth. Microorganisms 2022, 10, 540. [Google Scholar] [CrossRef]
- He, Z.F.; Xu, X.B.; Muhamethan, A.; Eisanjan, N.; Mahmutjan, M.; Chen, X.L.; Chen, W.L.; Wang, Y.J.; Ye, M.; Wang, Y.H.; et al. Seasonal dynamics of ammonia-oxidizing archaea (AOA) and their contribution to nitrification in wheat rhizospheres. Plant Soil 2025, 1–15. [Google Scholar] [CrossRef]
- Singh, S.K.; Wu, X.X.; Shao, C.Y.; Zhang, H.M. Microbial enhancement of plant nutrient acquisition. Stress Biol. 2022, 2, 3. [Google Scholar] [CrossRef]
- Liao, L.R.; Wang, X.T.; Wang, J.; Liu, G.B.; Zhang, C. Nitrogen fertilization increases fungal diversity and abundance of saprotrophs while reducing nitrogen fixation potential in a semiarid grassland. Plant Soil 2021, 465, 515–532. [Google Scholar] [CrossRef]
- Luo, L.; Meng, H.; Gu, J.D. Microbial extracellular enzymes in biogeochemical cycling of ecosystems. J. Environ. Manag. 2017, 197, 539–549. [Google Scholar] [CrossRef]
- Looby, C.I.; Martin, P.H. Diversity and function of soil microbes on montane gradients: The state of knowledge in a changing world. FEMS Microbiol. Ecol. 2020, 96, fiaa122. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, M.; Zhu, W.H.; Han, L.S.; Qin, L. Soil bacterial communities with an indicative function response to nutrients in wetlands of Northeastern China that have undergone natural restoration. Ecol. Indic. 2019, 101, 562–571. [Google Scholar] [CrossRef]
- Wu, B.; Zhou, L.H.; Liu, S.S.; Liu, F.F.; Saleem, M.; Han, X.G.; Shu, L.F.; Yu, X.L.; Hu, R.W.; He, Z.L.; et al. Biogeography of soil protistan consumer and parasite is contrasting and linked to microbial nutrient mineralization in forest soils at a wide-scale. Soil Biol. Biochem. 2022, 165, 108513. [Google Scholar] [CrossRef]
- Gutknecht, J.L.; Goodman, R.M.; Balser, T.C. Linking soil process and microbial ecology in freshwater wetland ecosystems. Plant. Soil. 2006, 289, 17–34. [Google Scholar] [CrossRef]
- Spieles, D.J. Wetland construction, restoration, and integration: A comparative review. Land 2022, 11, 554. [Google Scholar] [CrossRef]
- Peralta, R.M.; Ahn, C.; Gillevet, P.M. Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands. Sci. Total Environ. 2013, 443, 725–732. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.M.; Wei, G.H. Core microbiota drive functional stability of soil microbiome in reforestation ecosystems. Glob. Change Biol. 2022, 28, 1038–1047. [Google Scholar] [CrossRef]
- Hu, L.N.; Li, Q.; Yan, J.H.; Liu, C.; Zhong, J.X. Vegetation restoration facilitates belowground microbial network complexity and recalcitrant soil organic carbon storage in southwest China karst region. Sci. Total Environ. 2022, 820, 153137. [Google Scholar] [CrossRef]
- Li, T.; Wang, S.C.; Liu, C.E.; Yu, Y.D.; Zong, M.M.; Duan, C.Q. Soil microbial communities’ contributions to soil ecosystem multifunctionality in the natural restoration of abandoned metal mines. J. Environ. Manag. 2024, 353, 120244. [Google Scholar] [CrossRef]
- Bergkessel, M.; Delavaine, L. Diversity in starvation survival strategies and outcomes among heterotrophic proteobacteria. Microb. Physiol. 2021, 31, 146–162. [Google Scholar] [CrossRef]
- Han, X.L.; Luo, Q.; Chen, Y.H.; Xuan, Y.J.; Wu, L.; Qiu, W.H.; Wu, X.G.; Chen, Y.L.; Guo, J.P. Nitrogen Enrichment Alters Plant Root, Soil Microbial Structure, Diversity, and Function in Mountain Forests of North China. Forests 2025, 16, 459. [Google Scholar] [CrossRef]
- Ma, Z.W.; Zhang, M.X.; Xiao, R.; Cui, Y.; Yu, F.H. Changes in soil microbial biomass and community composition in coastal wetlands affected by restoration projects in a Chinese delta. Geoderma 2017, 289, 124–134. [Google Scholar] [CrossRef]
- Mondaca, P.; Celis-Diez, J.L.; Díaz-Siefer, P.; Olmos-Moya, N.; Montero-Silva, F.; Molina, S.; Fontúrbel, F.E.; Aponte, H.; Mandakovic, D.; Bastidas, B.; et al. Effects of sustainable agricultural practices on soil microbial diversity, composition, and functions. Agric. Ecosyst. Environ. 2024, 370, 109053. [Google Scholar] [CrossRef]
- Imhoff, J.F. New dimensions in microbial ecology—Functional genes in studies to unravel the biodiversity and role of functional microbial groups in the environment. Microorganisms 2016, 4, 19. [Google Scholar] [CrossRef]
- Liu, X.D.; Huo, H.R.; Zhang, Y.H.; Yang, H.W.; Li, S.M.; Meng, L.B. Promotion of maize straw degradation rate by altering microbial community structure through the addition of soybean straw. Plant Soil 2024, 1–21. [Google Scholar] [CrossRef]
- Li, X.; Cui, Y.; Ma, D.; Song, D.; Liu, L. Vertical distribution of bacterial community diversity in the Greater Khingan Mountain permafrost region. Ecol. Evol. 2022, 17, e9106. [Google Scholar] [CrossRef]
- Chen, X.; Wang, X.; Song, Y.; Chi, Y. A review of studies on the effects of anthropogenic disturbances on plant–soil–microorganism interactions in grassland ecosystems: Based on grazing and tourism perspectives. Agronomy 2024, 12, 2890. [Google Scholar] [CrossRef]
- Cui, H.; Ou, Y.; Wang, L.X.; Liang, A.Z.; Yan, B.X.; Li, Y.X. Dynamic changes in microbial communities and nutrient stoichiometry associated with soil aggregate structure in restored wetlands. Catena 2021, 197, 104984. [Google Scholar] [CrossRef]
- Palmer, M.; Ruhi, A. Linkages between flow regime, biota, and ecosystem processes: Implications for river restoration. Science 2019, 365, eaaw2087. [Google Scholar] [CrossRef]
- Bissett, A.; Richardson, A.E.; Baker, G.; Thrall, P.H. Long-term land use effects on soil microbial community structure and function. Appl. Soil. Ecol. 2011, 51, 66–78. [Google Scholar] [CrossRef]
- Li, M.C.; Zhou, W.X.; Sun, M.Y.; Shi, W.C.; Lun, J.Q.; Zhou, B.; Hou, L.J.; Gao, Z. Decoupling soil community structure, functional composition, and nitrogen metabolic activity driven by salinity in coastal wetlands. Soil. Biol. Biochem. 2024, 198, 109547. [Google Scholar] [CrossRef]
- Liu, B.Y.; Dai, Y.S.; Cheng, X.; He, X.; Bei, Q.C.; Wang, Y.F.; Zhou, Y.L.; Zhu, B.; Zhang, K.P.; Tian, X.Q.; et al. Straw mulch improves soil carbon and nitrogen cycle by mediating microbial community structure and function in the maize field. Front. Microbiol. 2023, 14, 1217966. [Google Scholar] [CrossRef]
- Hossain, Z.; Komatsu, S. Potentiality of soybean proteomics in untying the mechanism of flood and drought stress tolerance. Proteomes 2014, 2, 107–127. [Google Scholar] [CrossRef]
- Rui, J.; Hu, J.; Wang, F.; Zhao, Y.; Li, C. Altitudinal niches of symbiotic, associative and free-living diazotrophs driven by soil moisture and temperature in the alpine meadow on the Tibetan Plateau. Environ. Res. 2022, 211, 113033. [Google Scholar] [CrossRef]
- Cai, S.Y.; Zhao, X. Responses of bacterial communities and nitrogen-cycling microbiomes to the conversion from cereals to legumes in the rice-based system. Appl. Soil. Ecol. 2024, 195, 105241. [Google Scholar] [CrossRef]
- Pan, H.W.; Qin, Y.; Wang, Y.T.; Liu, S.G.; Yu, B.; Song, Y.P.; Wang, X.M.; Zhu, G.B. Dissimilatory nitrate/nitrite reduction to ammonium (DNRA) pathway dominates nitrate reduction processes in rhizosphere and non-rhizosphere of four fertilized farmland soil. Environ. Res. 2020, 186, 109612. [Google Scholar] [CrossRef]
- Takemura, A.F.; Chien, D.M.; Polz, M.F. Associations and dynamics of Vibrionaceae in the environment, from the genus to the population level. Front. Microbiol. 2014, 5, 38. [Google Scholar] [CrossRef]
- Medvedeff, C.A.; Inglett, K.S.; Inglett, P.W. Patterns and controls of anaerobic soil respiration and methanogenesis following extreme restoration of calcareous subtropical wetlands. Geoderma 2015, 245, 74–82. [Google Scholar] [CrossRef]
- Yi, X.Y.; Ning, C.; Feng, S.L.; Gao, H.Q.; Zhao, J.L.; Liao, J.Y.; Peng, Y.H.; Zhao, S.Q.; Liu, S.G. Urbanization-induced environmental changes strongly affect wetland soil bacterial community composition and diversity. Environ. Res. Lett. 2022, 17, 014027. [Google Scholar] [CrossRef]
- Goh, Y.X.; Anupoju, S.M.B.; Nguyen, A.; Zhang, H.; Ponder, M.; Krometis, L.A.; Pruden, A.; Liao, J. Evidence of horizontal gene transfer and environmental selection impacting antibiotic resistance evolution in soil-dwelling Listeria. Nat. Commun. 2024, 15, 10034. [Google Scholar] [CrossRef] [PubMed]
- Belykh, E.; Maystrenko, T.; Velegzhaninov, I.; Tavleeva, M.; Rasova, E.; Rybak, A. Taxonomic Diversity and Functional Traits of Soil Bacterial Communities under Radioactive Contamination: A Review. Microorganisms 2024, 12, 733. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | SF | MF | NF | PF | SRM | MRM |
|---|---|---|---|---|---|---|
| pH | 6.43 ± 0.07 a | 6.71 ± 0.11 a | 6.72 ± 0.33 a | 6.41 ± 0.11 a | 6.33 ± 0.30 a | 6.42 ± 0.08 a |
| SWC (%) | 26.75 ± 2.11 b | 26.55 ± 4.25 b | 30.07 ± 1.69 ab | 32.79 ± 1.81 ab | 48.01 ± 2.95 a | 47.57 ± 2.77 a |
| SOC (mg·kg−1) | 32.07 ± 1.75 a | 19.75 ± 1.77 b | 30.12 ± 5.41 a | 25.52 ± 6.40 ab | 25.32 ± 7.80 ab | 39.39 ± 6.64 a |
| TN (g·kg−1) | 3.46 ± 0.46 a | 1.85 ± 0.84 b | 3.54 ± 0.19 a | 3.31 ± 0.34 a | 2.54 ± 0.85 a | 4.88 ± 1.42 a |
| DOC (mg·kg−1) | 204.98 ± 8.23 c | 233.95 ± 8.44 b | 367.30 ± 25.41 a | 353.71 ± 17.47 a | 247.64 ± 11.49 b | 234.72 ± 10.19 b |
| MBC (mg·kg−1) | 1437.38 ± 149.91 b | 866.19 ± 136.53 c | 1723.13 ± 138.01 a | 1655.94 ± 164.87 a | 1333.40 ± 122.41 b | 1380.75 ± 112.92 b |
| MBN (mg·kg−1) | 93.68 ± 6.56 c | 66.93 ± 5.31 d | 230.08 ± 26.51 a | 130.35 ± 16.37 b | 122.41 ± 20.54 b | 129.24 ± 22.41 b |
| NH4+-N (mg·kg−1) | 9.71 ± 1.34 d | 11.26 ± 3.09 d | 77.01 ± 5.11 a | 43.92 ± 2.80 b | 24.92 ± 1.47 c | 24.08 ± 1.47 c |
| NO3−-N (mg·kg−1) | 5.51 ± 0.79 c | 3.77 ± 0.81 c | 24.79 ± 3.75 a | 13.68 ± 2.77 b | 14.46 ± 2.23 b | 11.99 ± 1.50 b |
| Urease (IU·g−1) | 6333.17 ± 164.73 c | 6578.33 ± 368.80 c | 11,840.17 ± 879.52 a | 5583 ± 652.96 c | 7663.67 ± 498.84 b | 5881.83 ± 499.07 c |
| β-Glucosidase (IU·g−1) | 40,336.83 ± 985.85 a | 25,559 ± 1035.71 c | 26,542.50 ± 1591.47 c | 30,732.83 ± 794.36 b | 26,515 ± 1598.66 c | 39,304 ± 1320.78 a |
| Cellulose (IU·g−1) | 158.33 ± 21.85 b | 68 ± 6.68 c | 230.08 ± 26.51 a | 130.35 ± 16.37 b | 122.41 ± 20.54 b | 129.24 ± 22.41 b |
| Soil Sample | Sobs | Shannon | Ace | Chao 1 |
|---|---|---|---|---|
| SF | 1181.35 ± 109.18 d | 5.69 ± 0.18 b | 1364.93 ± 128.39 c | 1381.93 ± 135.99 b |
| MF | 1590.53 ± 50.78 b | 6.32 ± 0.07 a | 1765.72 ± 40.44 a | 1812.32 ± 35.18 a |
| NF | 1156.34 ± 91.26 d | 5.26 ± 0.29 b | 1334.45 ± 70.51 c | 1348.37± 77.21 b |
| PF | 1086.32 ± 141.33 d | 5.65 ± 0.21 b | 1228.32 ± 165.85 c | 1256.52 ± 161.71 b |
| SRM | 1365.82 ± 73.49 c | 5.88 ± 0.05 b | 1559.94 ± 88.59 b | 1595.66 ± 105.79 b |
| MRM | 1721.89 ± 56.71 a | 6.43 ± 0.11 a | 1765.77 ± 40.44 a | 1956.14 ± 105.79 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, J.; Yu, S. Integrating Soil Physicochemical Properties and Microbial Functional Prediction to Assess Land-Use Impacts in a Cold-Region Wetland Ecosystem. Life 2025, 15, 972. https://doi.org/10.3390/life15060972
Ding J, Yu S. Integrating Soil Physicochemical Properties and Microbial Functional Prediction to Assess Land-Use Impacts in a Cold-Region Wetland Ecosystem. Life. 2025; 15(6):972. https://doi.org/10.3390/life15060972
Chicago/Turabian StyleDing, Junnan, and Shaopeng Yu. 2025. "Integrating Soil Physicochemical Properties and Microbial Functional Prediction to Assess Land-Use Impacts in a Cold-Region Wetland Ecosystem" Life 15, no. 6: 972. https://doi.org/10.3390/life15060972
APA StyleDing, J., & Yu, S. (2025). Integrating Soil Physicochemical Properties and Microbial Functional Prediction to Assess Land-Use Impacts in a Cold-Region Wetland Ecosystem. Life, 15(6), 972. https://doi.org/10.3390/life15060972