
Seronegative Sicca Syndrome: Diagnostic Considerations and Management Strategies

 , ,
, ,  ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Epidemiology and Patient Demographics
3. Clinical Characteristics and Symptom Spectrum
4. Histopathology and Imaging
5. Immunological Profile and Biomarkers
6. Differential Diagnosis
7. Prognosis and Disease Evolution
8. Therapeutic Strategies and Unmet Needs
9. Discussion
10. Redefining Disease Boundaries
11. Immune Activity Beyond Serology
12. Clinical Implications and Patient Burden
13. Bridging Gaps in Research and Practice
- -
- Embracing the spectrum model of sicca syndromes, with varying degrees of immune involvement;
- -
- Developing new diagnostic categories such as “probable autoimmune exocrinopathy” or “non-criteria sicca syndrome”;
- -
- Incorporating biomarker discovery and advanced immunophenotyping into routine research;
- -
- Ensuring clinical trial access for seronegative patients;
- -
- Validating patient-reported outcomes as core endpoints in future studies.
14. Conclusions
15. Clinical Take-Home Points
- Not all patients with sicca symptoms and immune activation meet criteria for primary Sjögren’s syndrome—seronegative sicca represents a distinct and underdiagnosed entity.
- A subset of seronegative patients may eventually seroconvert or develop systemic features; longitudinal follow-up is essential.
- Histopathologic findings and emerging biomarkers (e.g., BAFF, SP1) can provide critical clues to immune involvement in the absence of anti-SSA/SSB.
- Management should be individualized, balancing symptom relief with careful immune profiling before considering immunosuppression.
- Inclusion of seronegative patients in trials and classification updates is essential to close care gaps and reflect real-world diversity.
Author Contributions
Funding
Conflicts of Interest
References
- Mariette, X.; Criswell, L.A. Primary Sjögren’s syndrome. N. Engl. J. Med. 2018, 378, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: A consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017, 69, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Brito-Zeron, P.; Theander, E.; Baldini, C.; Seror, R.; Retamozo, S.; Quartuccio, L.; Bootsma, H.; Bowman, S.J.; Dörner, T.; Gottenberg, J.-E.; et al. Early diagnosis of primary Sjögren’s syndrome: EULAR-SS task force clinical recommendations. Expert Rev. Clin. Immunol. 2016, 12, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Mariette, X.; Bowman, S.; Baron, G.; Gottenberg, J.E.; Boostma, H.; Theander, E.; Tzioufas, A.; Vitali, C.; Ravaud, P.; et al. Accurate detection of changes in disease activity in primary Sjögren’s syndrome by ESSDAI. Arthritis Care Res. 2010, 62, 551–558. [Google Scholar] [CrossRef]
- Quartuccio, L.; Baldini, C.; Bartoloni, E.; Priori, R.; Carubbi, F.; Corazza, L.; Alunno, A.; Colafrancesco, S.; Luciano, N.; Giacomelli, R. Anti-SSA/SSB-negative Sjögren’s syndrome shows a lower prevalence of lymphoproliferative manifestations, and a lower risk of lymphoma evolution. Autoimmun. Rev. 2015, 14, 1019–1022. [Google Scholar] [CrossRef]
- Maripuri, S.; Grande, J.P.; Osborn, T.G.; Fervenza, F.C.; Matteson, E.L.; Donadio, J.V.; Hogan, M.C. Renal involvement in primary Sjögren’s syndrome: A clinicopathologic study. Clin. J. Am. Soc. Nephrol. 2009, 4, 1423–1431. [Google Scholar] [CrossRef]
- François, H.; Mariette, X. Renal involvement in primary Sjögren syndrome. Nat. Rev. Nephrol. 2016, 12, 82–93. [Google Scholar] [CrossRef]
- Manfrè, V.; Cafaro, G.; Riccucci, I.; Zabotti, A.; Perricone, C.; Bootsma, H.; De Vita, S.; Bartoloni, E. One year in review 2020: Comorbidities, diagnosis and treatment of primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2020, 38 (Suppl. S126), 10–22. [Google Scholar]
- Nocturne, G.; Mariette, X. Advances in understanding the pathogenesis of primary Sjögren’s syndrome. Nat. Rev. Rheumatol. 2013, 9, 544–556. [Google Scholar] [CrossRef]
- Nocturne, G.; Mariette, X. B cells in the pathogenesis of primary Sjögren syndrome. Nat. Rev. Rheumatol. 2018, 14, 133–145. [Google Scholar] [CrossRef]
- Gottenberg, J.-E.; Busson, M.; Cohen-Solal, J.; Lavie, F.; Abbed, K.; Kimberly, R.P.; Sibilia, J.; Mariette, X. Correlation of serum B lymphocyte stimulator and β2 microglobulin with autoantibody secretion and systemic involvement in primary Sjogren’s syndrome. Ann. Rheum. Dis. 2005, 64, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Brito-Zerón, P.; Kostov, B.; Solans, R.; Fraile, G.; Suárez-Cuervo, C.; Casanovas, A.; Rascón, F.J.; Qanneta, R.; Pérez-Alvarez, R.; Ripoll, M.; et al. Systemic activity and mortality in primary Sjögren syndrome: Predicting survival using ESSDAI. Ann. Rheum. Dis. 2016, 75, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Solans, R.; Rosas, J.; Camps, M.T.; Gil, A.; del Pino-Montes, J.; Calvo-Alen, J.; Jiménez-Alonso, J.; Micó, M.L.; Beltrán, J.; et al. Primary Sjögren syndrome in Spain: Clinical and immunologic expression in 1010 patients. Medicine 2008, 87, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Shahane, A. The epidemiology of Sjögren’s syndrome. Clin. Epidemiol. 2014, 6, 247–255. [Google Scholar]
- James, K.; Al-Ali, S.; Tarn, J.; Cockell, S.J.; Gillespie, C.S.; Hindmarsh, V.; Locke, J.; Mitchell, S.; Lendrem, D.; Bowman, S.; et al. A transcriptional signature of fatigue derived from patients with primary Sjögren’s syndrome. PLoS ONE 2015, 10, e0143970. [Google Scholar] [CrossRef]
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.M.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.E.; Chiche, L.; et al. Treatment of primary Sjögren syndrome with rituximab: A randomized trial. Ann. Intern. Med. 2014, 160, 233–242. [Google Scholar] [CrossRef]
- Tu, H.Y.; Yue, S.L.; Mou, L.J. Anti-SSA/SSB-negative primary Sjögren’s syndrome presenting with hypokalemia: A case report. World J. Emerg. Med. 2022, 13, 149–151. [Google Scholar] [CrossRef]
- Acharya, S.; Shrestha, S.; Poddar, E.; Neupane, A.M.; Khadayat, R.M.; Magar, S.R.M.; Lamsal, M.M.; Pathak, R.M. Antimitochondrial antibody-negative primary biliary cirrhosis with secondary Sjogren syndrome: A case report. Ann. Med. Surg. 2023, 85, 5645–5648. [Google Scholar] [CrossRef]
- Gottenberg, J.-E.; Cinquetti, G.; Larroche, C.; Combe, B.; Hachulla, E.; Meyer, O.; Pertuiset, E.; Kaplanski, G.; Chiche, L.; Berthelot, J.-M.; et al. Efficacy of rituximab in systemic manifestations of primary Sjogren’s syndrome: Results in 78 patients of the AutoImmune and Rituximab registry. Ann. Rheum. Dis. 2013, 72, 1026–1031. [Google Scholar] [CrossRef]
- Seror, R.; Bootsma, H.; Saraux, A.; Bowman, S.J.; Theander, E.; Brun, J.G.; Baron, G.; Le Guern, V.; Devauchelle-Pensec, V.; Ramos-Casals, M.; et al. Defining disease activity states in primary Sjögren’s syndrome with ESSDAI and ESSPRI. Ann. Rheum. Dis. 2016, 75, 382–389. [Google Scholar] [CrossRef]
- Seror, R.; Theander, E.; Brun, J.G.; Ramos-Casals, M.; Valim, V.; Dörner, T.; Bootsma, H.; Tzioufas, A.; Solans-Laqué, R.; Mandl, T.; et al. Validation of ESSDAI and ESSPRI. Ann. Rheum. Dis. 2015, 74, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Virone, A.; Ng, W.; Le Guern, V.; Hachulla, E.; Cornec, D.; Daien, C.; Vittecoq, O.; Bienvenu, B.; Marcelli, C.; et al. Rheumatoid factor and disease activity are independent predictors of lymphoma in primary Sjögren’s syndrome. Arthritis Rheumatol. 2016, 68, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Pontarini, E.; Bombardieri, M.; Mariette, X. Lymphomas complicating primary Sjögren’s syndrome: From autoimmunity to lymphoma. Rheumatology 2019, 60, 3513–3521. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]
- Zheng, J.; Haberland, V.; Baird, D.; Walker, V.; Haycock, P.C.; Hurle, M.R.; Gutteridge, A.; Erola, P.; Liu, Y.; Luo, S.; et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 2020, 52, 1122–1131. [Google Scholar] [CrossRef]
- Pertovaara, M.; Korpela, M. Serum β2 microglobulin correlates with the new ESSDAI in patients with Sjögren’s syndrome. Ann. Rheum. Dis. 2011, 70, 2236–2237. [Google Scholar] [CrossRef]
- Stefanski, A.L.; Tomiak, C.; Pleyer, U.; Dietrich, T.; Burmester, G.R.; Dörner, T. The diagnosis and treatment of Sjögren’s syndrome. Dtsch. Arztebl. Int. 2017, 114, 354–361. [Google Scholar] [CrossRef]
- Papageorgiou, A.; Ziogas, D.C.; Mavragani, C.P.; Zintzaras, E.; Tzioufas, A.G.; Moutsopoulos, H.M.; Voulgarelis, M. Predicting the outcome of Sjogren’s syndrome-associated non-Hodgkin’s lymphoma patients. PLoS ONE 2015, 10, e0116189. [Google Scholar] [CrossRef]
- Venables, P.J. Management of patients presenting with Sjogren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2006, 20, 791–807. [Google Scholar] [CrossRef]
- Tobón, G.J.; Saraux, A.; Gottenberg, J.E.; Quartuccio, L.; Fabris, M.; Seror, R.; Devauchelle-Pensec, V.; Morel, J.; Rist, S.; Mariette, X.; et al. Role of Flt3 ligand as a marker of lymphoma in primary Sjögren’s syndrome. Arthritis Rheum. 2013, 65, 3218–3227. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, C.P.; Kapsogeorgou, E.K.; Tzioufas, A.G. Predictive biomarkers in primary Sjögren’s syndrome: Where do we stand? Clin. Exp. Immunol. 2023, 212, 285–295. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, J.; Feng, X.; Xie, L.; Qin, S.; Ma, G.; Zhang, F. Identification of drug targets for Sjögren’s syndrome: Multi-omics Mendelian randomization and colocalization analyses. Front. Immunol. 2024, 15, 1419363. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, A.D.; Kuan, V.; Finan, C.; Kruger, F.A.; Gaulton, A.; Chopade, S.; Sofat, R.; MacAllister, R.J.; Overington, J.P.; Hemingway, H.; et al. Improving the odds of drug development success through human genomics: Modelling study. Sci. Rep. 2019, 9, 18911. [Google Scholar] [CrossRef]
- Musone, S.L.; Taylor, K.E.; Nititham, J.; Chu, C.; Poon, A.; Liao, W.; Lam, E.T.; Ma, A.; Kwok, P.-Y.; Criswell, L.A. Sequencing of TNFAIP3 and association of variants with multiple autoimmune diseases. Genes Immun. 2011, 12, 176–182. [Google Scholar] [CrossRef]
- Zhang, M.; Peng, L.L.; Wang, Y.; Wang, J.S.; Liu, J.; Liu, M.M.; Hu, J.; Song, B.; Yang, H.B. Roles of A20 in autoimmune diseases. Immunol. Res. 2016, 64, 337–344. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Seror, R.; Baron, G.; Camus, M.; Cornec, D.; Perrodeau, E.; Bowman, S.J.; Bombardieri, M.; Bootsma, H.; Gottenberg, J.-E.; Fisher, B.; et al. Development of the STAR (Sjögren’s Tool for Assessing Response) composite responder index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2023, 82, 78–86. [Google Scholar] [CrossRef]
- Arends, S.; de Wolff, L.; van Nimwegen, J.F.; Verstappen, G.M.P.J.; Vehof, J.; Bombardieri, M.; Bowman, S.J.; Pontarini, E.; Baer, A.N.; Nys, M.; et al. Composite of Relevant Endpoints for Sjögren's Syndrome (CRESS): Development and validation of a novel outcome measure. Lancet Rheumatol. 2021, 3, e553–e562. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.A.; Mariette, X.; Papas, A.; Grader-Beck, T.; Bootsma, H.; Ng, W.F.; van Daele, P.L.A.; Finzel, S.; Noaiseh, G.; Elgueta, S.; et al. Safety and Efficacy of Subcutaneous Iscalimab (CFZ533) in Two Distinct Populations of Patients with Sjögren’s Disease (TWINSS): Week 24 Results of a Randomised, Double-Blind, Placebo-Controlled, Phase 2b Dose-Ranging Study. Lancet 2024, 404, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Pringle, S.; Bootsma, H.; Kroese, F.G. Epithelial–immune cell interplay in primary Sjögren syndrome salivary gland pathogenesis. Nat. Rev. Rheumatol. 2021, 17, 333–348. [Google Scholar] [CrossRef]
- Moutsopoulos, H.M.; Chused, T.M.; Mann, D.L.; Klippel, J.H.; Fauci, A.S.; Frank, M.M. Sjögren’s syndrome (sicca syndrome): Current issues. Ann. Intern. Med. 1980, 92 Pt 2, 212–226. [Google Scholar] [CrossRef]
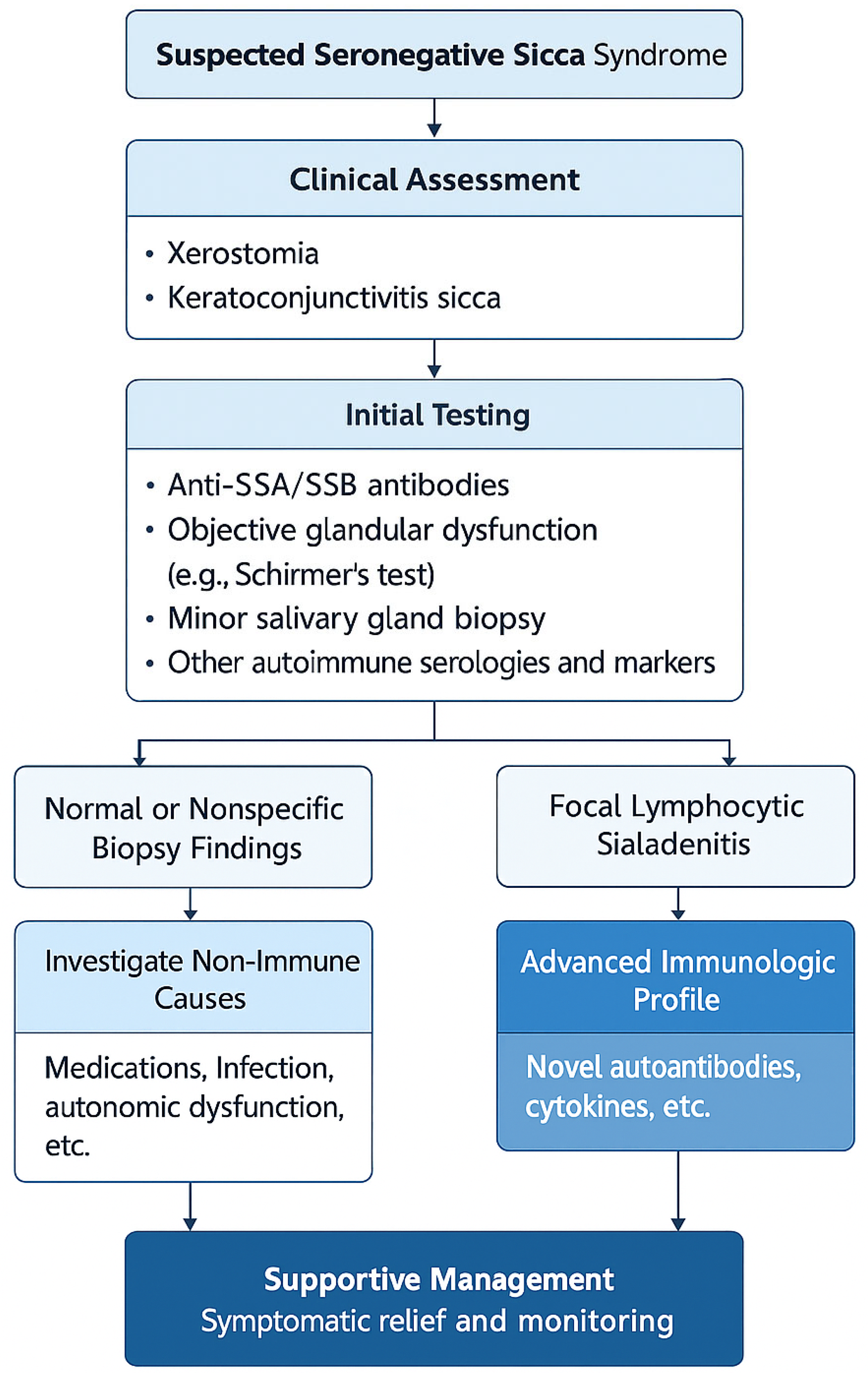

{kind=link}
| Study (Author, Year) | Country | Sample Size | % Female | Mean Age (years) | Anti-SSA/SSB Negative (%) | Systemic Symptoms (%) | Key Notes |
|---|---|---|---|---|---|---|---|
| Quartuccio et al. [5], 2015 | Italy | 133 | 91% | 56.4 | 100% | 24% | Lower lymphoma risk |
| Ramos-Casals et al. [13], 2008 | Spain | 1010 | 94% | 54.2 | ~25% | 32% | Registry data |
| Tu et al. [17], 2022 | China | 1 (case) | Female | 42 | 100% | Hypokalemia | Case report |
| Acharya et al. [18], 2023 | Nepal | 1 (case) | Female | 38 | 100% | Biliary overlap | Rare combo |
| Maripuri et al. [6], 2009 | USA | 25 | 88% | 55.7 | 100% | Renal involvement | Biopsy cohort |
| Feature | Seronegative Patients | Seropositive Patients |
|---|---|---|
| Focus Score ≥ 1 | ~40–60% (variable) | ~80–90% |
| Germinal Centers | Rare (<10%) | Common (~30–40%) |
| Fibrosis | More prevalent | Less common |
| SGUS (hypoechoic areas) | Moderate sensitivity | High sensitivity |
| Biopsy-negative with symptoms | Frequent (~30–40%) | Rare (<10%) |
| Use in classification criteria | Often decisive | Often corroborative |
| Condition | Key Features | Suggested Approach |
|---|---|---|
| Age-related dryness | Older adults, slow onset, no inflammation | Unremarkable labs, normal biopsy |
| Medication-induced sicca | Linked to drug use | Discontinue meds, reassess symptoms |
| Fibromyalgia/CFS | Fatigue, pain, normal labs | Clinical diagnosis, no glandular damage |
| Hepatitis C–related sicca | Positive RF, arthralgia, liver enzymes | HCV serology, liver ultrasound |
| IgG4-related disease | Painless gland swelling | IgG4 levels, biopsy with IgG4 stain |
| Sarcoidosis | Parotid swelling, hilar lymphadenopathy | Chest X-ray, CT, MRI, biopsy |
| Diabetes, hypothyroidism | Metabolic symptoms | Glucose, TSH, antithyroid antibodies |
| Clinical Profile | First-Line Treatment | Optional/Second-Line | Notes |
|---|---|---|---|
| Isolated sicca (glandular only) | Saliva/tear substitutes, pilocarpine | Cevimeline | Focus on symptomatic relief |
| Fatigue, arthralgia (no systemic damage) | Hydroxychloroquine, lifestyle | Cognitive Behavioral Therapy, low-dose antidepressants | Screen for fibromyalgia |
| Biopsy-proven FLS, mild extraglandular | Hydroxychloroquine | Methotrexate or low-dose steroids | Confirm diagnosis before escalation |
| Significant systemic involvement | Not typical | Case-by-case basis, consider MTX/MMF | Rare in true seronegative patients |
| Refractory symptoms, autoantibody-negative | Limited evidence | Experimental biologics (rituximab, BAFF-i) | Consider trial enrollment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basheva-Kraeva, Y.M.; Kraev, K.I.; Uchikov, P.A.; Kraeva, M.I.; Hristov, B.K.; Stoyanova, N.S.; Mitkova-Hristova, V.T.; Ivanov, B.; Karamitev, S.S.; Koleva, N.; et al. Seronegative Sicca Syndrome: Diagnostic Considerations and Management Strategies. Life 2025, 15, 966. https://doi.org/10.3390/life15060966
Basheva-Kraeva YM, Kraev KI, Uchikov PA, Kraeva MI, Hristov BK, Stoyanova NS, Mitkova-Hristova VT, Ivanov B, Karamitev SS, Koleva N, et al. Seronegative Sicca Syndrome: Diagnostic Considerations and Management Strategies. Life. 2025; 15(6):966. https://doi.org/10.3390/life15060966
Chicago/Turabian StyleBasheva-Kraeva, Yordanka M., Krasimir I. Kraev, Petar A. Uchikov, Maria I. Kraeva, Bozhidar K. Hristov, Nina St. Stoyanova, Vesela T. Mitkova-Hristova, Borislav Ivanov, Stanislav S. Karamitev, Nina Koleva, and et al. 2025. "Seronegative Sicca Syndrome: Diagnostic Considerations and Management Strategies" Life 15, no. 6: 966. https://doi.org/10.3390/life15060966
APA StyleBasheva-Kraeva, Y. M., Kraev, K. I., Uchikov, P. A., Kraeva, M. I., Hristov, B. K., Stoyanova, N. S., Mitkova-Hristova, V. T., Ivanov, B., Karamitev, S. S., Koleva, N., Marinkov, A., & Vassilev, V. A. (2025). Seronegative Sicca Syndrome: Diagnostic Considerations and Management Strategies. Life, 15(6), 966. https://doi.org/10.3390/life15060966