

CYTO-SV-ML: A Machine Learning Tool for Cytogenetic Structural Variant Analysis in Somatic Cell Type Using Genome Sequences


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
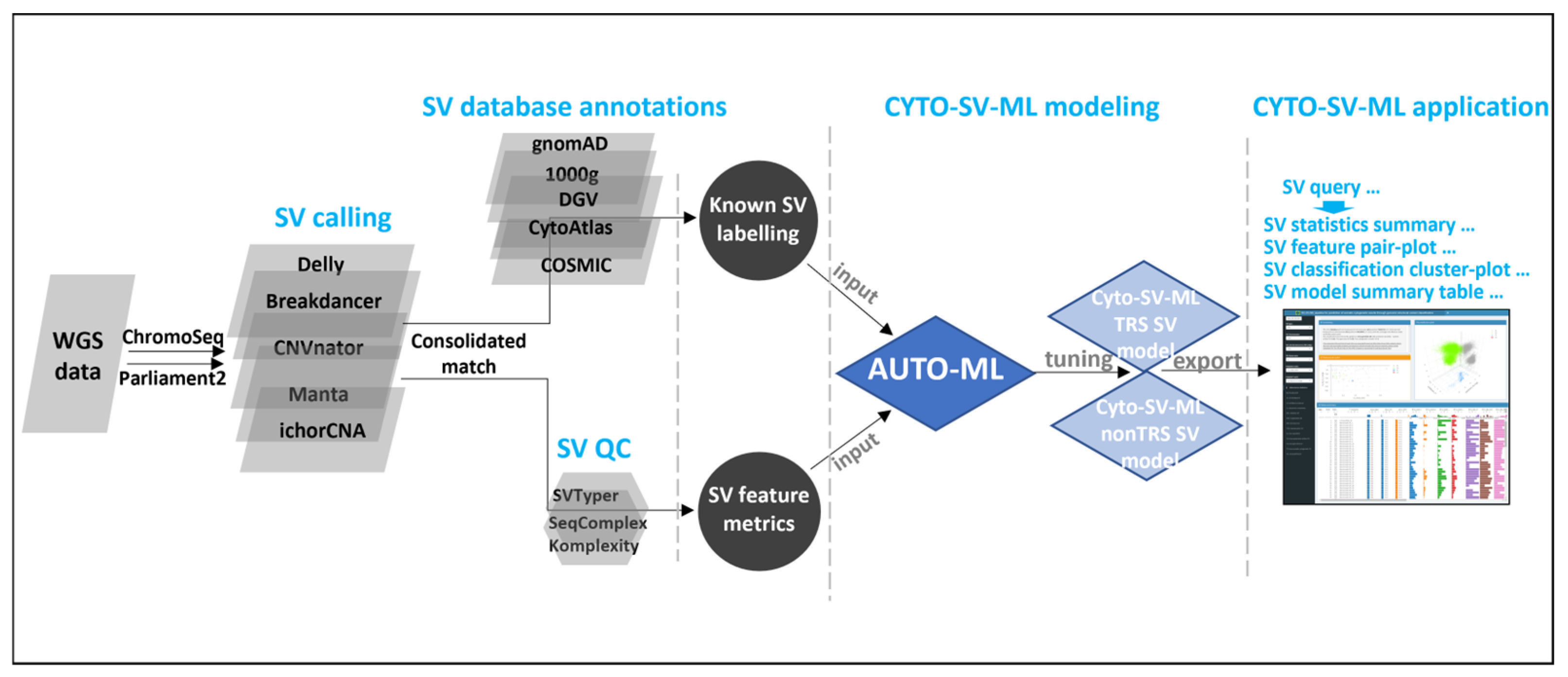
2.1. Implementation
2.1.1. WGS SV Preprocessing
2.1.2. Known SV Labeling
2.1.3. SV Classification Modeling
2.1.4. CYTO-SV-ML Interface Application
2.2. Biological Cohort, DNA Extraction
2.3. Whole Genome Sequencing
3. Results
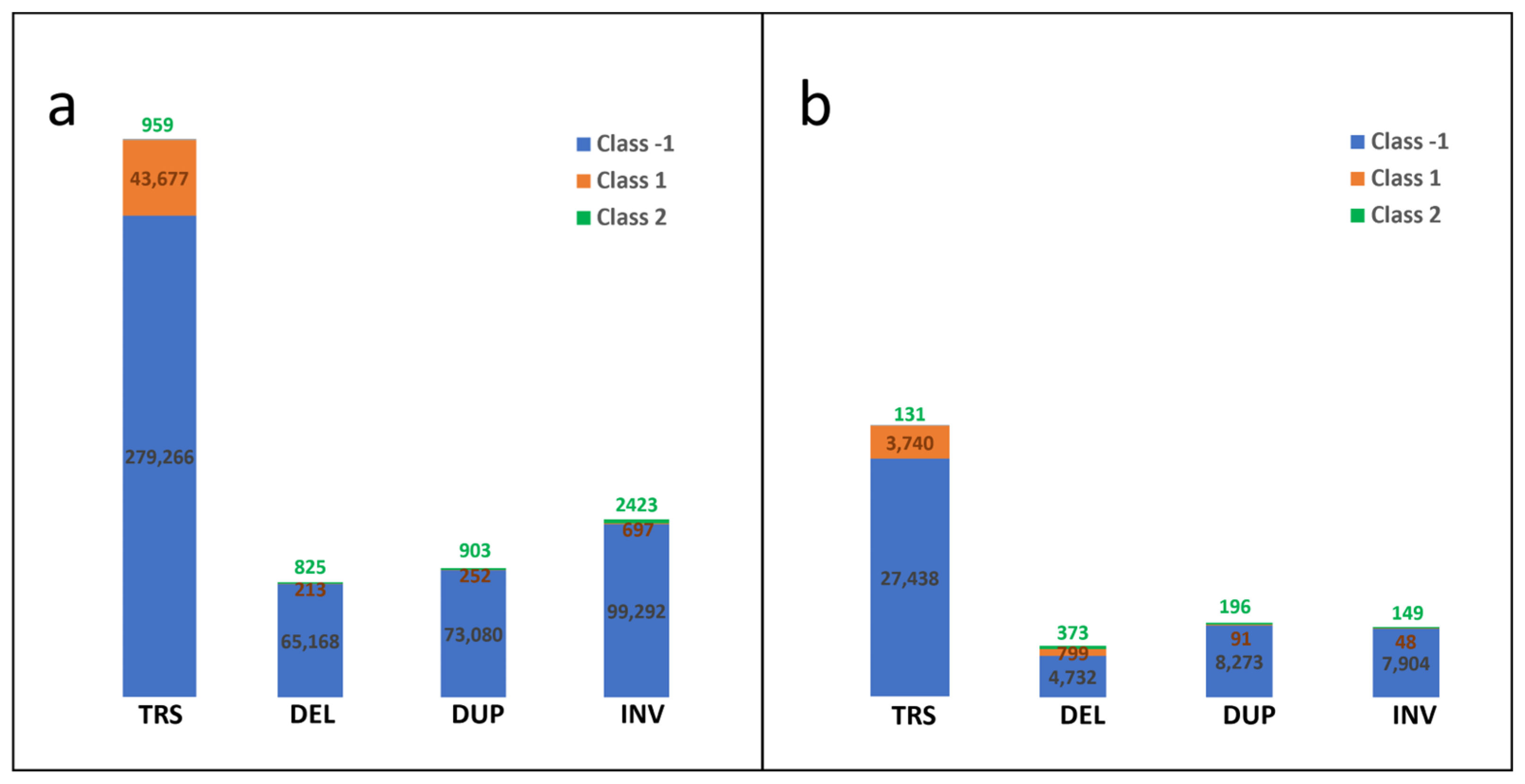
3.1. Preparation of Known SV Data of Cytogenetic Somatic SVs
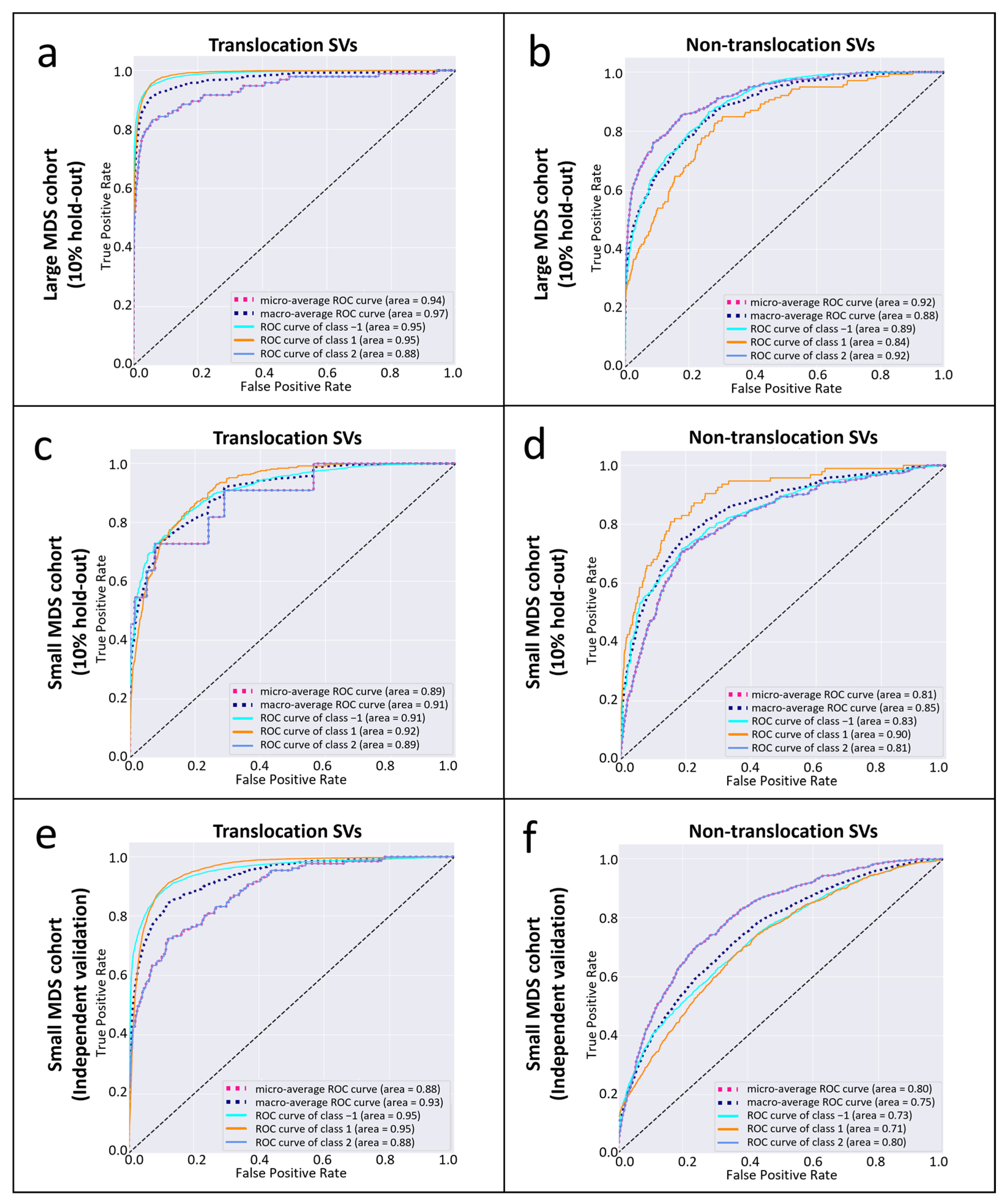
3.2. The Performance of CYTO-SV-ML Pipeline
3.3. Confirmation of SVs from Cytogenetic Records
3.4. The Overview of CYTO-SV-ML Application
3.5. The Runtime of CYTO-SV-ML Pipeline
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| WGS | whole genome sequencing |
| SV | structural variation |
| CNV | copy number variation |
| MDS | myelodysplastic syndromes |
| ML | machine learning |
| AUC | area under the curve |
| FISH | fluorescence in situ hybridization |
| CMA | chromosomal microarray |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
| gnomAD | Genome Aggregation Database |
| TRS | translocation |
| AF | allele frequency |
| PBC | peripheral blood cell |
| SHAP | Shapley Additive Explanations |
References
- Akkari, Y.M.N.; Baughn, L.B.; Dubuc, A.M.; Smith, A.C.; Mallo, M.; Dal Cin, P.; Diez Campelo, M.; Gallego, M.S.; Granada Font, I.; Haase, D.T.; et al. Guiding the global evolution of cytogenetic testing for hematologic malignancies. Blood 2022, 139, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Akkari, Y.; Baughn, L.B.; Kim, A.; Karaca, E.; Raca, G.; Shao, L.; Mikhail, F.M.; ACMG Laboratory Quality Assurance Committee. Section E6.1-6.6 of the American College of Medical Genetics and Genomics (ACMG) Technical Laboratory Standards: Cytogenomic studies of acquired chromosomal abnormalities in neoplastic blood, bone marrow, and lymph nodes. Genet. Med. 2024, 26, 101054. [Google Scholar] [CrossRef] [PubMed]
- Schwabkey, Z.I.; Al Ali, N.; Chan, O.; Sallman, D.A.; Padron, E.; Kuykendall, A.T.; Talati, C.; Sweet, K.; Lancet, J.E.; Komrokji, R.S. Fluorescence in Situ Hybridization (FISH) Utility for Risk Score Assessment in Patients With MDS With Normal Metaphase Karyotype. Clin. Lymphoma Myeloma Leuk. 2021, 21, e52–e56. [Google Scholar] [CrossRef]
- O’Malley, D.P.; Giudice, C.; Chang, A.S.; Chang, D.; Barry, T.S.; Hibbard, M.K.; Chen, R.; Chen, S.T. Comparison of array comparative genomic hybridization (aCGH) to FISH and cytogenetics in prognostic evaluation of chronic lymphocytic leukemia. Int. J. Lab. Hematol. 2011, 33, 238–244. [Google Scholar] [CrossRef]
- Mikhail, F.M.; Heerema, N.A.; Rao, K.W.; Burnside, R.D.; Cherry, A.M.; Cooley, L.D. Section E6.1-6.4 of the ACMG technical standards and guidelines: Chromosome studies of neoplastic blood and bone marrow-acquired chromosomal abnormalities. Genet. Med. 2016, 18, 635–642. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Duncavage, E.J.; Schroeder, M.C.; O’Laughlin, M.; Wilson, R.; MacMillan, S.; Bohannon, A.; Kruchowski, S.; Garza, J.; Du, F.; Hughes, A.E.O.; et al. Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers. N. Engl. J. Med. 2021, 384, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Mack, E.K.M.; Marquardt, A.; Langer, D.; Ross, P.; Ultsch, A.; Kiehl, M.G.; Mack, H.I.D.; Haferlach, T.; Neubauer, A.; Brendel, C. Comprehensive genetic diagnosis of acute myeloid leukemia by next-generation sequencing. Haematologica 2019, 104, 277–287. [Google Scholar] [CrossRef]
- Mareschal, S.; Palau, A.; Lindberg, J.; Ruminy, P.; Nilsson, C.; Bengtzen, S.; Engvall, M.; Eriksson, A.; Neddermeyer, A.; Marchand, V.; et al. Challenging conventional karyotyping by next-generation karyotyping in 281 intensively treated patients with AML. Blood Adv. 2021, 5, 1003–1016. [Google Scholar] [CrossRef]
- Kayser, S.; Hills, R.K.; Langova, R.; Kramer, M.; Guijarro, F.; Sustkova, Z.; Estey, E.H.; Shaw, C.M.; Racil, Z.; Mayer, J.; et al. Characteristics and outcome of patients with acute myeloid leukaemia and t(8;16)(p11;p13): Results from an International Collaborative Study. Br. J. Haematol. 2021, 192, 832–842. [Google Scholar] [CrossRef]
- Uguen, K.; Jubin, C.; Duffourd, Y.; Bardel, C.; Malan, V.; Dupont, J.M.; El Khattabi, L.; Chatron, N.; Vitobello, A.; Rollat-Farnier, P.A.; et al. Genome sequencing in cytogenetics: Comparison of short-read and linked-read approaches for germline structural variant detection and characterization. Mol. Genet. Genom. Med. 2020, 8, e1114. [Google Scholar] [CrossRef] [PubMed]
- Lindstrand, A.; Eisfeldt, J.; Pettersson, M.; Carvalho, C.M.B.; Kvarnung, M.; Grigelioniene, G.; Anderlid, B.M.; Bjerin, O.; Gustavsson, P.; Hammarsjo, A.; et al. From cytogenetics to cytogenomics: Whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med. 2019, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Asadi Fakhr, Z.; Mehrzad, V.; Izaditabar, A.; Salehi, M. Evaluation of the utility of peripheral blood vs bone marrow in karyotype and fluorescence in situ hybridization for myelodysplastic syndrome diagnosis. J. Clin. Lab. Anal. 2018, 32, e22586. [Google Scholar] [CrossRef]
- Coleman, J.F.; Theil, K.S.; Tubbs, R.R.; Cook, J.R. Diagnostic yield of bone marrow and peripheral blood FISH panel testing in clinically suspected myelodysplastic syndromes and/or acute myeloid leukemia: A prospective analysis of 433 cases. Am. J. Clin. Pathol. 2011, 135, 915–920. [Google Scholar] [CrossRef]
- Huret, J.L.; Ahmad, M.; Arsaban, M.; Bernheim, A.; Cigna, J.; Desangles, F.; Guignard, J.C.; Jacquemot-Perbal, M.C.; Labarussias, M.; Leberre, V.; et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013, 41, D920–D924. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Popic, V.; Rohlicek, C.; Cunial, F.; Hajirasouliha, I.; Meleshko, D.; Garimella, K.; Maheshwari, A. Cue: A deep-learning framework for structural variant discovery and genotyping. Nat. Methods 2023, 20, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wang, S.; Audano, P.A.; Meng, D.; Flores, J.I.; Kosters, W.; Yang, X.; Jia, P.; Marschall, T.; Beck, C.R.; et al. SVision: A deep learning approach to resolve complex structural variants. Nat. Methods 2022, 19, 1230–1233. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef]
- Fan, X.; Abbott, T.E.; Larson, D.; Chen, K. BreakDancer: Identification of Genomic Structural Variation from Paired-End Read Mapping. Curr. Protoc. Bioinform. 2014, 45, 15.6.1–15.6.11. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Kallberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Zhang, T.; Auer, P.; Dong, J.; Cutler, C.; Dezern, A.E.; Gadalla, S.M.; Deeg, H.J.; Nazha, A.; Carlson, K.S.; Spellman, S.; et al. Whole-genome sequencing identifies novel predictors for hematopoietic cell transplant outcomes for patients with myelodysplastic syndrome: A CIBMTR study. J. Hematol. Oncol. 2023, 16, 37. [Google Scholar] [CrossRef]
- Wang, W.; Auer, P.; Zhang, T.; Spellman, S.; Carlson, K.S.; Nazha, A.; Bolon, Y.T.; Saber, W. Impact of Epigenomic Hypermethylation at TP53 on Allogeneic Hematopoietic Cell Transplantation Outcomes for Myelodysplastic Syndromes. Transplant. Cell Ther. 2021, 27, 659.e1–659.e6. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; De Haro, L.P.; Wray, J.; Hromas, R. Mechanisms of leukemia translocations. Curr. Opin. Hematol. 2008, 15, 338–345. [Google Scholar] [CrossRef]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef]
- Haferlach, T.; Hutter, S.; Meggendorfer, M. Genome Sequencing in Myeloid Cancers. N. Engl. J. Med. 2021, 384, e106. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Auer, P.; Spellman, S.R.; Dong, J.; Saber, W.; Bolon, Y.-T. CYTO-SV-ML: A Machine Learning Tool for Cytogenetic Structural Variant Analysis in Somatic Cell Type Using Genome Sequences. Life 2025, 15, 929. https://doi.org/10.3390/life15060929
Zhang T, Auer P, Spellman SR, Dong J, Saber W, Bolon Y-T. CYTO-SV-ML: A Machine Learning Tool for Cytogenetic Structural Variant Analysis in Somatic Cell Type Using Genome Sequences. Life. 2025; 15(6):929. https://doi.org/10.3390/life15060929
Chicago/Turabian StyleZhang, Tao, Paul Auer, Stephen R. Spellman, Jing Dong, Wael Saber, and Yung-Tsi Bolon. 2025. "CYTO-SV-ML: A Machine Learning Tool for Cytogenetic Structural Variant Analysis in Somatic Cell Type Using Genome Sequences" Life 15, no. 6: 929. https://doi.org/10.3390/life15060929
APA StyleZhang, T., Auer, P., Spellman, S. R., Dong, J., Saber, W., & Bolon, Y.-T. (2025). CYTO-SV-ML: A Machine Learning Tool for Cytogenetic Structural Variant Analysis in Somatic Cell Type Using Genome Sequences. Life, 15(6), 929. https://doi.org/10.3390/life15060929