

Bicuspid Aortic Valve and Sudden Cardiac Death

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
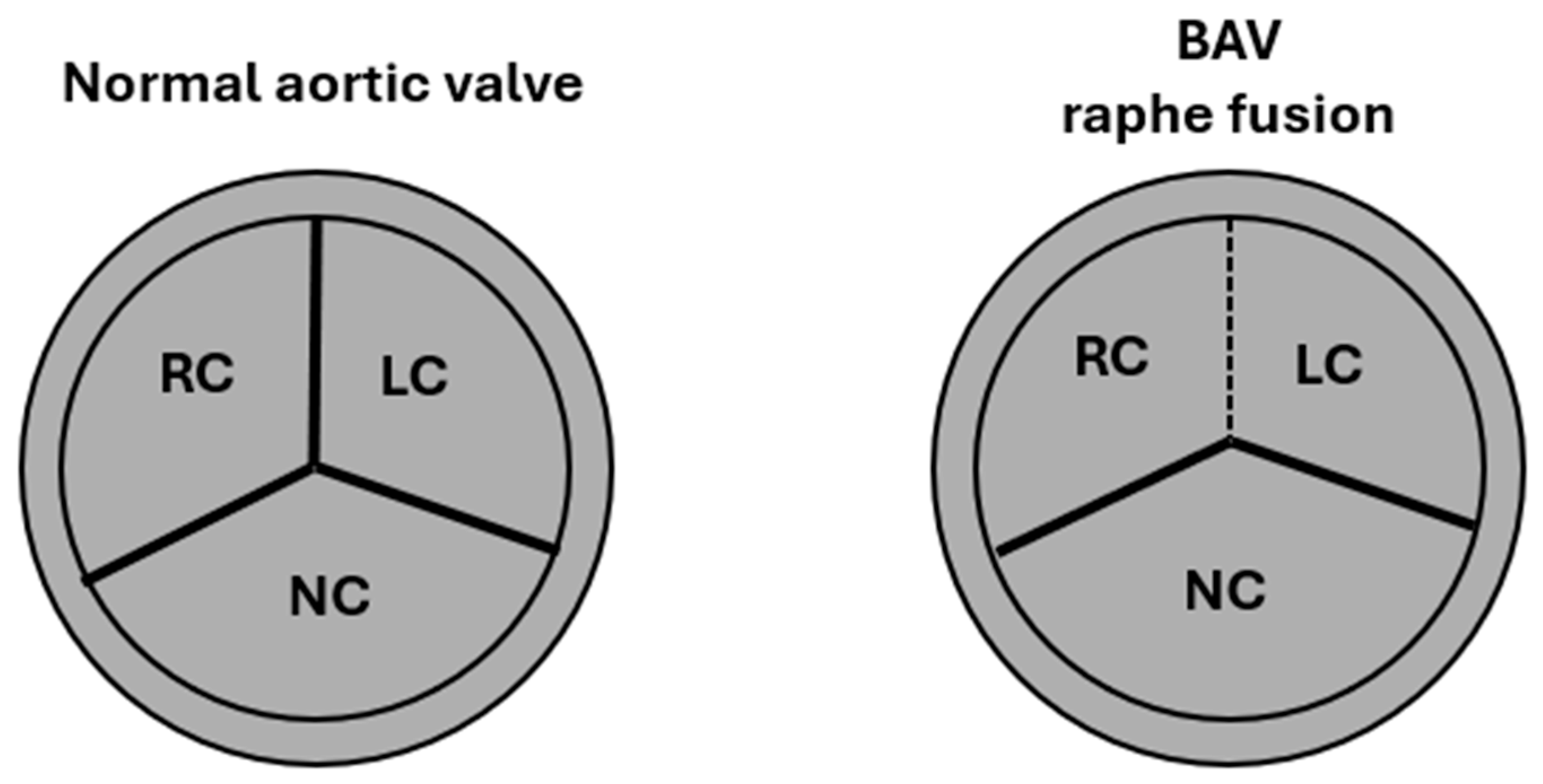
2. Embryology, Anatomy, and Histology
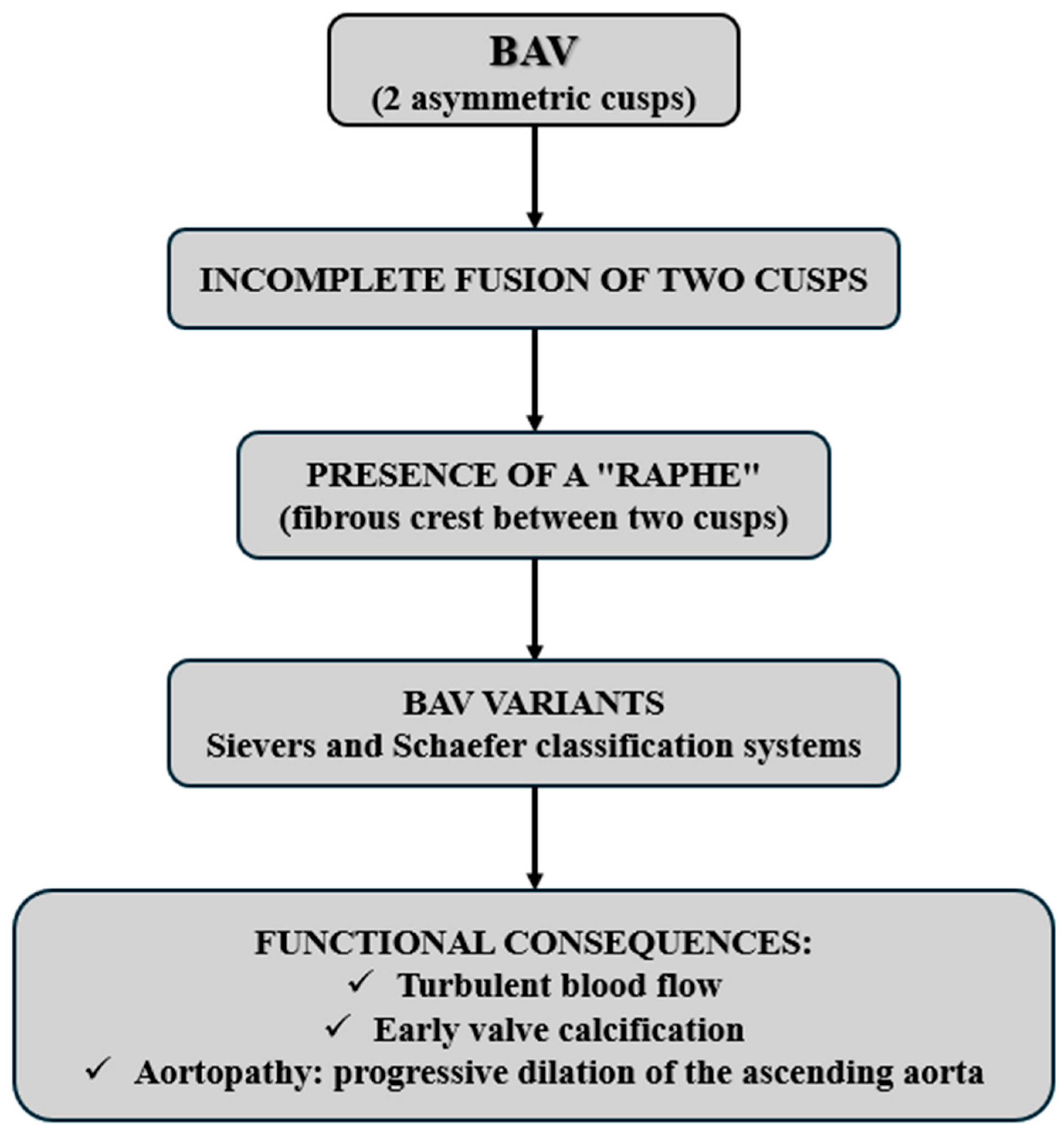
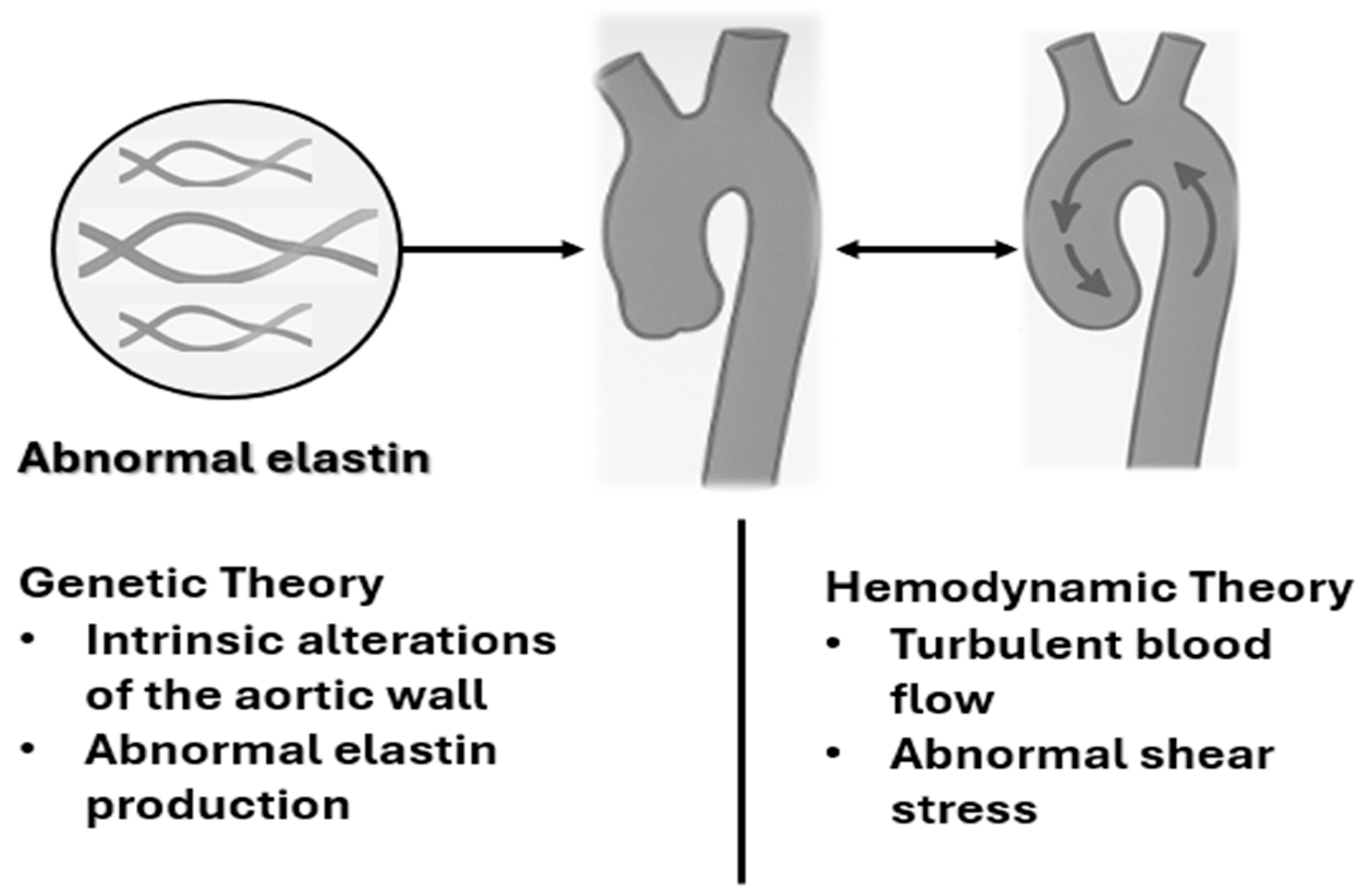
3. Pathogenic Variants and Pathogenesis
4. Valvular Classification and Clinical Implications
5. Sudden Cardiac Death, Other Cardiovascular Complications, and Preventive Strategies
5.1. Haemodynamic Consequences of BAV
- AS, characterised by narrowing of the valve orifice, is a common complication in BAV. This condition results in an increased pressure gradient across the valve, placing excessive strain on the left ventricle, which must work harder to overcome the resistance imposed by the stenotic valve. Consequently, compensatory left ventricular hypertrophy develops, potentially leading to systolic dysfunction and a heightened risk of life-threatening ventricular arrhythmias [52,53,54].
5.2. Major Cardiovascular Complications Associated with BAV
5.3. Prevention and Management of SCD in BAV
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Mubarik, A.; Sharma, S.; Law, M.A. Bicuspid Aortic Valve. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2023. [Google Scholar]
- Kalra, A.; Das, R.; Alkhalil, M.; Dykun, I.; Candreva, A.; Jarral, O.; Rehman, S.M.; Majmundar, M.; Patel, K.N.; Rodes-Cabau, J.; et al. Bicuspid Aortic Valve Disease: Classifications, Treatments, and Emerging Transcatheter Paradigms. Struct. Heart 2023, 8, 100227. [Google Scholar] [CrossRef] [PubMed]
- Chatrath, N.; Westaby, J.; Finocchiaro, G.; Sharma, S.; Esteban, M.T.; Papadakis, M.; Sheppard, M.N. The role of the bicuspid aortic valve in sudden cardiac death-findings at cardiac autopsy. Cardiovasc. Pathol. 2023, 65, 107527. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Rizzo, S.; Basso, C. Bicuspid aortic valve: The most frequent and not so benign congenital heart disease. Cardiovasc. Pathol. 2024, 70, 107604. [Google Scholar] [CrossRef]
- Sillesen, A.S.; Vøgg, O.; Pihl, C.; Raja, A.A.; Sundberg, K.; Vedel, C.; Zingenberg, H.; Jørgensen, F.S.; Vejlstrup, N.; Iversen, K.; et al. Prevalence of Bicuspid Aortic Valve and Associated Aortopathy in Newborns in Copenhagen, Denmark. J. Am. Med. Assoc. 2021, 325, 561–567. [Google Scholar] [CrossRef]
- Bravo-Jaimes, K.; Prakash, S.K. Genetics in bicuspid aortic valve disease: Where are we? Prog. Cardiovasc. Dis. 2020, 63, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, E.M.; Preventza, O.; Hamilton Black, J., 3rd; Augoustides, J.G.; Beck, A.W.; Bolen, M.A.; Braverman, A.C.; Bray, B.E.; Brown-Zimmerman, M.M.; Chen, E.P.; et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2022, 146, e334–e482. [Google Scholar] [CrossRef]
- Roleder, T.; Hawranek, M.; Gąsior, T.; Cieśla, D.; Zembala, M.; Wojakowski, W.; Gąsior, M.; Gąsior, Z. Trends in diagnosis and treatment of aortic stenosis in the years 2006–2016 according to the SILCARD registry. Pol. Arch. Intern. Med. 2018, 128, 739–745. [Google Scholar]
- Aschauer, J.; Zilberszac, R.; Gleiss, A.; Colizzi, C.; Binder, T.; Bruno, P.; Laufer, G.; Massetti, M.; Gabriel, H.; Rosenhek, R. Long-term outcome of bicuspid aortic valve disease. Eur. Heart J. Cardiovasc. Imaging 2024, 25, 425–435. [Google Scholar] [CrossRef]
- Kusner, J.J.; Brown, J.Y.; Gleason, T.G.; Edelman, E.R. The Natural History of Bicuspid Aortic Valve Disease. Struct. Heart J. Heart Team 2022, 7, 100119. [Google Scholar] [CrossRef]
- Masri, A.; Svensson, L.G.; Griffin, B.P.; Desai, M.Y. Contemporary natural history of bicuspid aortic valve disease: A systematic review. Heart 2017, 103, 1323–1330. [Google Scholar] [CrossRef]
- Wirrig, E.E.; Yutzey, K.E. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arter. Thromb. Vasc. Biol. 2014, 34, 737–741. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Koenig, S.N.; Bosse, K.; Majumdar, U.; Bonachea, E.M.; Radtke, F.; Garg, V. Endothelial Notch1 Is Required for Proper Development of the Semilunar Valves and Cardiac Outflow Tract. J. Am. Heart Assoc. 2016, 5, e003075. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelialmesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Soto-Navarrete, M.T.; López-Unzu, M.Á.; Durán, A.C.; Fernández, B. Embryonic development of bicuspid aortic valves. Prog. Cardiovasc. Dis. 2020, 63, 407–418. [Google Scholar] [CrossRef]
- Peterson, J.C.; Chughtai, M.; Wisse, L.J.; Groot, A.C.G.-D.; Feng, Q.; Goumans, M.-J.T.H.; VanMunsteren, J.C.; Jongbloed, M.R.M.; DeRuiter, M.C. Bicuspid aortic valve formation: Nos3 mutation leads to abnormal lineage patterning of neural crest cells and the second heart field. Dis. Model. Mech. 2018, 11, dmm034637. [Google Scholar] [CrossRef] [PubMed]
- Beusch, C.M.; Simonson, O.E.; Wedin, J.O.; Sabatier, P.; Felldin, U.; Kadekar, S.; Österholm, C.; Végvári, Á.; Zubarev, R.A.; Fromell, K.; et al. Analysis of local extracellular matrix identifies different aetiologies behind bicuspid and tricuspid aortic valve degeneration and suggests therapies. Cell. Mol. Life Sci. 2023, 80, 268. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.J.; Song, J.K. Bicuspid aortic valve: Evolving knowledge and new questions. Heart 2022, 109, 10–17. [Google Scholar] [CrossRef]
- Jahanyar, J.; Tsai, P.I.; Arabkhani, B.; Aphram, G.; Mastrobuoni, S.; El Khoury, G.; de Kerchove, L. Functional and pathomorphological anatomy of the aortic valve and root for aortic valve sparing surgery in tricuspid and bicuspid aortic valves. Ann. Cardiothorac. Surg. 2023, 12, 179–193. [Google Scholar] [CrossRef]
- Liu, J.D.; Luo, X.D.; Zhou, Z.P.; Gong, R.; Wu, Y.Q. Annular and supra-annular structure assessments for transcatheter aortic valve replacement in patients with bicuspid aortic stenosis. Rev. Cardiovasc. Med. 2021, 22, 1157–1166. [Google Scholar] [CrossRef]
- Freeze, S.L.; Landis, B.J.; Ware, S.M.; Helm, B.M. Bicuspid Aortic Valve: A Review with Recommendations for Genetic Counseling. J. Genet. Couns. 2016, 25, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Kostina, A.; Shishkova, A.; Ignatieva, E.; Irtyuga, O.; Bogdanova, M.; Levchuk, K.; Golovkin, A.; Zhiduleva, E.; Uspenskiy, V.; Moiseeva, O.; et al. Different Notch signaling in cells from calcified bicuspid and tricuspid aortic valves. J. Mol. Cell. Cardiol. 2018, 114, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Sticchi, E.; De Cario, R.; Magi, A.; Giglio, S.; Provenzano, A.; Nistri, S.; Pepe, G.; Giusti, B. Bicuspid Aortic Valve: Role of Multiple Gene Variants in Influencing the Clinical Phenotype. Biomed Res. Int. 2018, 2018, 8386123. [Google Scholar] [CrossRef]
- Foffa, I.; Ait Alì, L.; Panesi, P.; Mariani, M.; Festa, P.; Botto, N.; Andreassi, M.G. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med. Genet. 2013, 14, 44. [Google Scholar] [CrossRef]
- Bonachea, E.M.; Zender, G.; White, P.; Corsmeier, D.; Newsom, D.; Fitzgerald-Butt, S.; Garg, V.; McBride, K.L. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Med. Genom. 2014, 7, 56. [Google Scholar] [CrossRef]
- Preuss, C.; Capredon, M.; Wünnemann, F.; Chetaille, P.; Prince, A.; Godard, B.; Leclerc, S.; Sobreira, N.; Ling, H.; Awadalla, P.; et al. Family Based Whole Exome Sequencing Reveals the Multifaceted Role of Notch Signalling in Congenital Heart Disease. PLoS Genet. 2016, 12, e1006335. [Google Scholar] [CrossRef] [PubMed]
- Mozzini, C.; Girelli, D.; Cominacini, L.; Soresi, M. An Exploratory Look at Bicuspid Aortic Valve (Bav) Aortopathy: Focus on Molecular and Cellular Mechanisms. Curr. Probl. Cardiol. 2021, 46, 100425. [Google Scholar] [CrossRef]
- Laforest, B.; Nemer, M. GATA5 interacts with GATA4 and GATA6 in outflow tract development. Dev. Biol. 2011, 358, 368–378. [Google Scholar] [CrossRef]
- Bonachea, E.M.; Chang, S.-W.; Zender, G.; LaHaye, S.; Fitzgerald-Butt, S.; McBride, K.L.; Garg, V. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr. Res. 2014, 76, 211–216. [Google Scholar] [CrossRef]
- Xu, Y.-J.; Di, R.-M.; Qiao, Q.; Li, X.-M.; Huang, R.-T.; Xue, S.; Liu, X.-Y.; Wang, J.; Yang, Y.-Q. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene 2018, 663, 115–120. [Google Scholar] [CrossRef]
- Zheng, R.; Zhu, P.; Gu, J.; Ni, B.; Sun, H.; He, K.; Bian, J.; Shao, Y.; Du, J. Transcription factor Sp2 promotes TGFB-mediated interstitial cell osteogenic differentiation in bicuspid aortic valves through a SMAD-dependent pathway. Exp. Cell Res. 2022, 411, 112972. [Google Scholar] [CrossRef] [PubMed]
- Giusti, B.; Sticchi, E.; De Cario, R.; Magi, A.; Nistri, S.; Pepe, G. Genetic Bases of Bicuspid Aortic Valve: The Contribution of Traditional and High-Throughput Sequencing Approaches on Research and Diagnosis. Front. Physiol. 2017, 8, 612. [Google Scholar] [CrossRef] [PubMed]
- Girdauskas, E.; Geist, L.; Disha, K.; Kazakbaev, I.; Groß, T.; Schulz, S.; Ungelenk, M.; Kuntze, T.; Reichenspurner, H.; Kurth, I. Genetic abnormalities in bicuspid aortic valve root phenotype: Preliminary results. Eur. J. Cardiol. Surg. 2017, 52, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Palomares, J.F.; Dux-Santoy, L.; Guala, A.; Galian-Gay, L.; Evangelista, A. Mechanisms of Aortic Dilation in Patients with Bicuspid Aortic Valve: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2023, 82, 448–464. [Google Scholar] [CrossRef]
- Płońska-Gościniak, E.; Tomkiewicz-Pająk, L.; Komar, M.; Kukulski, T.; Kosmala, W.; Kasprzak, J.D.; Mizia-Stec, K.; Hryniewiecki, T.; Kustrzycka-Kratochwil, D.; Niklewski, T.; et al. Bicuspid Aortic Valves (BAV) Registry (RE-BAV): Clinical and echocardiographic characteristics of patients with BAV according to novel classification of bicuspid aortic valves. Kardiol. Pol. 2025, 83, 455–464. [Google Scholar] [CrossRef]
- Făgărășan, A.; Gurzu, S.; Satala, C.B.; Hagău, A.C. The Importance of Aortic Valve Bicuspid Phenotype in Valvular Evolution in Pediatric Patients: A Case Report and Literature Mini-Review. Int. J. Mol. Sci. 2023, 24, 14027. [Google Scholar] [CrossRef]
- Freiholtz, D.; Bergman, O.; Lång, K.; Poujade, F.-A.; Paloschi, V.; Granath, C.; Lindeman, J.H.N.; Olsson, C.; Franco-Cereceda, A.; Eriksson, P.; et al. Bicuspid aortic valve aortopathy is characterized by embryonic epithelial to mesenchymal transition and endothelial instability. J. Mol. Med. 2023, 101, 801–811. [Google Scholar] [CrossRef]
- Michelena, H.I.; Della Corte, A.; Evangelista, A.; Maleszewski, J.J.; Edwards, W.D.; Roman, M.J.; Devereux, R.B.; Fernández, B.; Asch, F.M.; Barker, A.J.; et al. International consensus statement on nomenclature and classification of the congenital bicuspid aortic valve and its aortopathy, for clinical, surgical, interventional and research purposes. Eur. J. Cardio-Thorac. Surg. 2021, 60, 448–476. [Google Scholar] [CrossRef]
- Sievers, H.H.; Schmidtke, C. A classification system for the bicuspid aortic valve from 304 surgical specimens. J. Thorac. Cardiovasc. Surg. 2007, 133, 1226–1233. [Google Scholar] [CrossRef]
- Schaefer, B.M.; Lewin, M.B.; Stout, K.K.; Gill, E.; Prueitt, A.; Byers, P.H.; Otto, C.M. The bicuspid aortic valve: An integrated phenotypic classification of leaflet morphology and aortic root shape. Heart 2008, 94, 1634–1638. [Google Scholar] [CrossRef]
- Michelena, H.I. Speaking a common language: The international consensus on bicuspid aortic valve nomenclature and classification. Ann. Cardiothorac. Surg. 2022, 11, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, A.; Bancone, C.; Dialetto, G.; Covino, F.E.; Manduca, S.; Montibello, M.V.; De Feo, M.; Buonocore, M.; Nappi, G. The ascending aorta with bicuspid aortic valve: A phenotypic classification with potential prognostic significance. Eur. J. Cardio-Thorac. Surg. 2014, 46, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Michelena, H.I.; Prakash, S.K.; Della Corte, A.; Bissell, M.M.; Anavekar, N.; Mathieu, P. Bicuspid aortic valve: Identifying knowledge gaps and rising to the challenge from the International Bicuspid Aortic Valve Consortium (BAVCon). Circulation 2014, 129, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Sabet, H.Y.; Edwards, W.D.; Tazelaar, H.D.; Daly, R.C. Congenitally bicuspid aortic valves: A surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin. Proc. 1999, 74, 14–26. [Google Scholar] [CrossRef]
- Siu, S.C.; Silversides, C.K. Bicuspid aortic valve disease. J. Am. Coll. Cardiol. 2010, 55, 2789–2800. [Google Scholar] [CrossRef]
- Prakash, S.K.; Bossé, Y.; Muehlschlegel, J.D.; Michelena, H.I.; Limongelli, G.; Della Corte, A. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: Insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J. Am. Coll. Cardiol. 2014, 64, 832–839. [Google Scholar] [CrossRef]
- Salzillo, C.; Sansone, V.; Napolitano, F. Sudden Cardiac Death in the Young: State-of-the-Art Review in Molecular Autopsy. Curr. Issues Mol. Biol. 2024, 46, 3313–3327. [Google Scholar] [CrossRef]
- Salzillo, C.; Marzullo, A. Hereditary Aortopathies as Cause of Sudden Cardiac Death in the Young: State-of-the-Art Review in Molecular Medicine. Diseases 2024, 12, 264. [Google Scholar] [CrossRef]
- Salzillo, C.; La Verde, M.; Imparato, A.; Molitierno, R.; Lucà, S.; Pagliuca, F.; Marzullo, A. Cardiovascular Diseases in Public Health: Chromosomal Abnormalities in Congenital Heart Disease Causing Sudden Cardiac Death in Children. Medicina 2024, 60, 1976. [Google Scholar] [CrossRef]
- Verma, S.; Siu, S.C. Aortic dilatation in patients with bicuspid aortic valve. N. Engl. J. Med. 2014, 370, 1920–1929. [Google Scholar] [CrossRef]
- Pellikka, P.A.; Sarano, M.E.; Nishimura, R.A.; Malouf, J.F.; Bailey, K.R.; Scott, C.G.; Barnes, M.E.; Tajik, A.J. Hemodynamically significant aortic stenosis during prolonged follow-up. Circulation 2005, 111, 3290–3295. [Google Scholar] [CrossRef] [PubMed]
- Jabagi, H.; Levine, D.; Gharibeh, L.; Camillo, C.; Castillero, E.; Ferrari, G.; Takayama, H.; Grau, J.B. Implications of Bicuspid Aortic Valve Disease and Aortic Stenosis/Insufficiency as Risk Factors for Thoracic Aortic Aneurysm. Rev. Cardiovasc. Med. 2023, 24, 178. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.S.; Strange, G.; Playford, D.; Stewart, S.; Celermajer, D.S. Characteristics of Bicuspid Aortic Valve Disease and Stenosis: The National Echo Database of Australia. J. Am. Heart Assoc. 2021, 10, e020785. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-T.; Ye, Z.; Ullah, M.W.; Maleszewski, J.J.; Scott, C.G.; Padang, R.; Pislaru, S.V.; Nkomo, V.T.; Mankad, S.V.; Pellikka, P.A.; et al. Bicuspid aortic valve: Long-term morbidity and mortality. Eur. Heart J. 2023, 44, 4549–4562. [Google Scholar] [CrossRef]
- Cotrufo, M.; Della Corte, A.; De Santo, L.S.; Quarto, C.; De Feo, M.; Romano, G.; Amarelli, C.; Scardone, M.; Di Meglio, F.; Guerra, G.; et al. Different patterns of extracellular matrix protein expression in the convexity and the concavity of the dilated aorta with bicuspid aortic valve: Preliminary results. J. Thorac. Cardiovasc. Surg. 2005, 130, 504–511. [Google Scholar] [CrossRef]
- Ehrlich, T.; de Kerchove, L.; Vojacek, J.; Boodhwani, M.; El-Hamamsy, I.; De Paulis, R.; Lansac, E.; Bavaria, J.E.; El Khoury, G.; Schäfers, H.-J. State-of-the art bicuspid aortic valve repair in 2020. Prog. Cardiovasc. Dis. 2020, 63, 457–464. [Google Scholar] [CrossRef]
- Dunne, E.C.; Lacro, R.V.; Flyer, J.N. Bicuspid aortic valve and its ascending aortopathy. Curr. Opin. Pediatr. 2023, 35, 538–545. [Google Scholar] [CrossRef]
- Nappi, F.; Giacinto, O.; Lusini, M.; Garo, M.; Caponio, C.; Nenna, A.; Nappi, P.; Rousseau, J.; Spadaccio, C.; Chello, M. Patients with Bicuspid Aortopathy and Aortic Dilatation. J. Clin. Med. 2022, 11, 6002. [Google Scholar] [CrossRef]
- Wang, J.; Deng, W.; Lv, Q.; Li, Y.; Liu, T.; Xie, M. Aortic Dilatation in Patients with Bicuspid Aortic Valve. Front. Physiol. 2021, 12, 615175. [Google Scholar] [CrossRef]
- Pelliccia, A.; Fagard, R.; Bjørnstad, H.H.; Anastassakis, A. Recommendations for competitive sports participation in athletes with cardiovascular disease: A consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur. Heart J. 2005, 26, 1422–1445. [Google Scholar]
- Pereira, S.C.; Abrantes, A.L.; António, P.S.; Morais, P.; Sousa, C.; David, C.; Pinto, F.J.; Almeida, A.G.; Caldeira, D. Infective endocarditis risk in patients with bicuspid aortic valve: Systematic review and meta-analysis. Int. J. Cardiol. Heart Vasc. 2023, 47, 101249. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, V.; Reyes, F.B.; Gonzalez, D.; Schwartz, M.; Whiltlow, A.; Alegria, J.R. Endocarditis in Adult Congenital Heart Disease Patients: Prevention, Recognition, and Management. Curr. Cardiol. Rep. 2024, 26, 1031–1045. [Google Scholar] [CrossRef]
- Bohbot, Y.; Denev, S.; Benvenga, R.M.; Philip, M.; Michelena, H.I.; Citro, R.; Habib, G.; Tribouilloy, C. Characteristics and prognosis of isolated aortic valve infective endocarditis in patients with bicuspid aortic valves: A propensity matched study. Front. Cardiovasc. Med. 2023, 10, 1304957. [Google Scholar] [CrossRef]
- Presti, F.L.; Guzzardi, D.G.; Bancone, C.; Fedak, P.W.M.; Della Corte, A. The science of BAV aortopathy. Prog. Cardiovasc. Dis. 2020, 63, 465–474. [Google Scholar] [CrossRef]
- Bulut, H.I.; Rad, A.A.; Syrengela, A.-A.; Ttofi, I.; Djordjevic, J.; Kaur, R.; Keiralla, A.; Krasopoulos, G. A Comprehensive Review of Management Strategies for Bicuspid Aortic Valve (BAV): Exploring Epidemiology, Aetiology, Aortopathy, and Interventions in Light of Recent Guidelines. J. Cardiovasc. Dev. Dis. 2023, 10, 398. [Google Scholar] [CrossRef]
- Shimoda, T.; Yokoyama, Y.; Takagi, H.; Kuno, T.; Fukuhara, S. Treatment strategies and outcomes following acute type A aortic dissection repair in patients with bicuspid and tricuspid aortic valves: A meta-analysis. JTCVS Open 2024, 19, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Généreux, P.; Schwartz, A.; Oldemeyer, J.B.; Pibarot, P.; Cohen, D.J.; Blanke, P.; Lindman, B.R.; Babaliaros, V.; Fearon, W.F.; Daniels, D.V.; et al. Transcatheter Aortic-Valve Replacement for Asymptomatic Severe Aortic Stenosis. N. Engl. J. Med. 2025, 392, 217–227. [Google Scholar] [CrossRef]
- Edwards-TAVR-Receives-FDA-Approval-for-Patients-with-Asymptomatic-Severe-Aortic-Stenosis. 2025. Available online: https://www.edwards.com/newsroom/news/2025-05-01-edwards-tavr-receives-fda-approval-for-patients-wi-0da0638 (accessed on 1 May 2025).
- Vahanian, A.; Beyersdorf, F.; Praz, F. 2021 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 2022, 43, 561–632. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Kim, W.-K.; Dhoble, A.; Pio, S.M.; Babaliaros, V.; Jilaihawi, H.; Pilgrim, T.; De Backer, O.; Bleiziffer, S.; Vincent, F.; et al. Bicuspid Aortic Valve Morphology and Outcomes After Transcatheter Aortic Valve Replacement. J. Am. Coll. Cardiol. 2020, 76, 1018–1030. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Bleiziffer, S.; De Backer, O.; Delgado, V.; Arai, T.; Ziegelmueller, J.; Barbanti, M.; Sharma, R.; Perlman, G.Y.; Khalique, O.K.; et al. Outcomes in Transcatheter Aortic Valve Replacement for Bicuspid Versus Tricuspid Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2017, 69, 2579–2589. [Google Scholar] [CrossRef]
- Halim, S.A.; Edwards, F.H.; Dai, D.; Li, Z. Outcomes of Transcatheter Aortic Valve Replacement in Patients with Bicuspid Aortic Valve Disease: A Report From the Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy Registry. Circulation 2020, 141, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Al-Asad, K.S.; Salazar, A.M.; Radwan, Y.; Wang, E.; Salam, M.F.; Sabanci, R.; Saeed, M.; Halboni, A.; Al-Abcha, A.; Abela, G. Transcatheter Aortic Valve Replacement in Bicuspid Versus Tricuspid Aortic Valve Stenosis: Meta-Analysis and Systemic Review. Am. J. Cardiol. 2023, 203, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Zito, A.; Buono, A.; Scotti, A.; Kim, W.-K.; Fabris, T.; de Biase, C.; Bellamoli, M.; Montarello, N.; Costa, G.; Alfadhel, M.; et al. Incidence, Predictors, and Outcomes of Paravalvular Regurgitation After TAVR in Sievers Type 1 Bicuspid Aortic Valves. JACC Cardiovasc. Interv. 2024, 17, 1652–1663. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Tunica/Layers | Structure | Function |
|---|---|---|
| Fibrous tunica (parietal layer) | Collagen | It is stretched in diastole to maintain flap adhesion |
| Tunica spongiosa (middle layer) | Proteoglycans | Absorbs and distributes forces and cyclic movements of the valve |
| Elastic tunica (axial or ventricular layer) | Elastin | It relaxes in diastole and contracts in systole |
| Histology | Description |
|---|---|
| Fibrosis and calcification | Accumulation of fibrotic tissue and calcium deposits on valve cusps |
| Endothelial thickening | Irregular endothelium with anomalous distribution of interstitial cells |
| Elastic fiber fragmentation | Disintegration of the elastic structure of the aortic wall |
| Smooth muscle cell loss | Reduction of muscle cells in the aortic media, contributing to wall weakness |
| Extracellular matrix alterations | Pathologic remodeling of the extracellular matrix with accumulation of proteoglycans |
| Pathogenic Variants | Function | Role in BAV |
|---|---|---|
| NOTCH1 | Receptor involved in endothelial-mesenchymal transition and heart valve development | Variant most frequently associated with BAV, and loss-of-function mutations accelerate aortic valve calcification |
| ARHGAP31, MAML1, SMARCA4, JARID2, JAG1 | NOTCH pathway genes | Rare variants associated with BAV and aortic coarctation |
| GATA4-6 | Transcription factors with zinc finger domain | Regulate early cardiac gene expression and cardiac cell differentiation, common variants of GATA4 associated with BAV, while rare variants of GATA4, GATA5, and GATA6 have been identified in other studies |
| SMAD4, SMAD6 | TGF-β signalling proteins | Variants identified in BAV |
| ROBO4 | Expressed in endothelial cells | Rare variants identified in BAV |
| ACTA2 | Encodes smooth muscle alpha-actin | Relatively rare mutations in BAV |
| FBN1 | Encodes extra-cellular glycoprotein fibrillin 1 | Rare variants found in BAV and aortic root aneurysms |
| Type | Description | Subtype | Main Features |
|---|---|---|---|
| 0 | No raphe, two completely separated cusps | A-P Lateral | More symmetrical structure and less predisposition to calcifications |
| 1 | A raphe, partial fusion of two cusps | L-R, R-NC, L-NC | The most common variant and often associated with aortic stenosis and dilation of the ascending aorta |
| 2 | Two raphes, extensive fusion | L-R/R-NC R-NC/NC-L NC-L/L-R | Greater alteration of blood flow and associated with more abnormal valves |
| Type | Description | Main Features |
|---|---|---|
| A-P | The cusps are oriented anteroposteriorly | Most common, often associated with aortic stenosis |
| L-L | The cusps are laterally oriented | Less common, it can affect hemodynamics differently |
| Type | Main Phenotypes | Prevalence | Anatomical Features | Clinical Implications |
|---|---|---|---|---|
| Fused BAV | Right–left cusp fusion | 70–80% | Fusion between left and right cusp | Increased risk of aortic stenosis, aortic root dilation, aortic regurgitation (especially in males) |
| Right non-cusp fusion | 20–30% | Fusion between right and non-coronary cusp | Faster progression of aortic stenosis and regurgitation | |
| Left non-cusp fusion | 3–6% | Fusion between left and non-coronary cusp | Rare phenotype, further studies needed | |
| Two-sinus BAV | No specific phenotype | Rare | Presence of only two aortic sinuses | Alterations in hemodynamic flow, potential dilation of the ascending aorta |
| Partial-fusion BAV | No specific phenotype | Undetermined | Incomplete or less evident fusion of the cusps | Slower progression of valvular disease, difficult to diagnose with traditional imaging |
| Imaging Modality | Main Advantages | Limitations | Application in BAV Phenotypes |
|---|---|---|---|
| Transthoracic Echocardiography (TTE) | Non-invasive, widely available, useful for assessing valve function. | Limited accuracy for distal aortic measurements or in patients with poor acoustic window. | First approach for screening, useful for valve follow-up. |
| Transoesophageal Echocardiography (TEE) | Higher resolution than TTE, excellent visualization of the aortic root and valve. | Invasive, not optimal for distal ascending aorta. | Useful for detailed morphological evaluation of BAV. |
| Computed Tomography (CT) | High spatial resolution, excellent for aorta evaluation. | Exposure to radiation and iodinated contrast medium. | Optimal for accurate aortic measurements, especially presurgical. |
| Cardiovascular Magnetic Resonance Imaging (MRI) | No radiation exposure, good for aortic measurements and ventricular function. | Limitations in patients with metallic devices or claustrophobia. | Useful in young patients or for long-term follow-up of aortic dilation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salzillo, C.; Quaranta, A.; Di Lizia, F.; Lombardo, M.; Ciccone, M.M.; Santobuono, V.E.; Macorano, E.; Introna, F.; Solarino, B.; Marzullo, A. Bicuspid Aortic Valve and Sudden Cardiac Death. Life 2025, 15, 868. https://doi.org/10.3390/life15060868
Salzillo C, Quaranta A, Di Lizia F, Lombardo M, Ciccone MM, Santobuono VE, Macorano E, Introna F, Solarino B, Marzullo A. Bicuspid Aortic Valve and Sudden Cardiac Death. Life. 2025; 15(6):868. https://doi.org/10.3390/life15060868
Chicago/Turabian StyleSalzillo, Cecilia, Andrea Quaranta, Fabrizia Di Lizia, Michela Lombardo, Marco Matteo Ciccone, Vincenzo Ezio Santobuono, Enrica Macorano, Francesco Introna, Biagio Solarino, and Andrea Marzullo. 2025. "Bicuspid Aortic Valve and Sudden Cardiac Death" Life 15, no. 6: 868. https://doi.org/10.3390/life15060868
APA StyleSalzillo, C., Quaranta, A., Di Lizia, F., Lombardo, M., Ciccone, M. M., Santobuono, V. E., Macorano, E., Introna, F., Solarino, B., & Marzullo, A. (2025). Bicuspid Aortic Valve and Sudden Cardiac Death. Life, 15(6), 868. https://doi.org/10.3390/life15060868