
Insights into CYP1B1-Related Ocular Diseases Through Genetics and Animal Studies

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
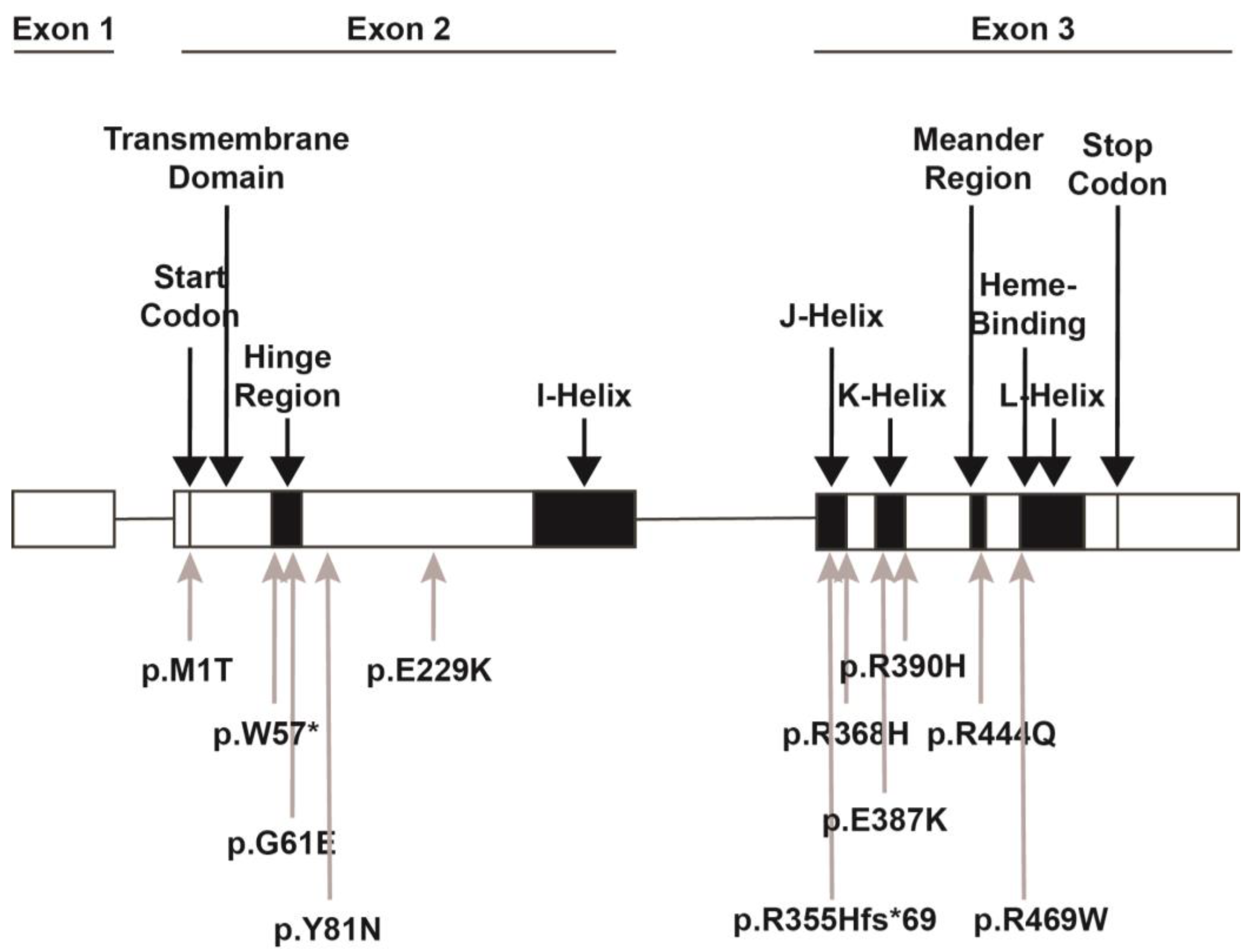
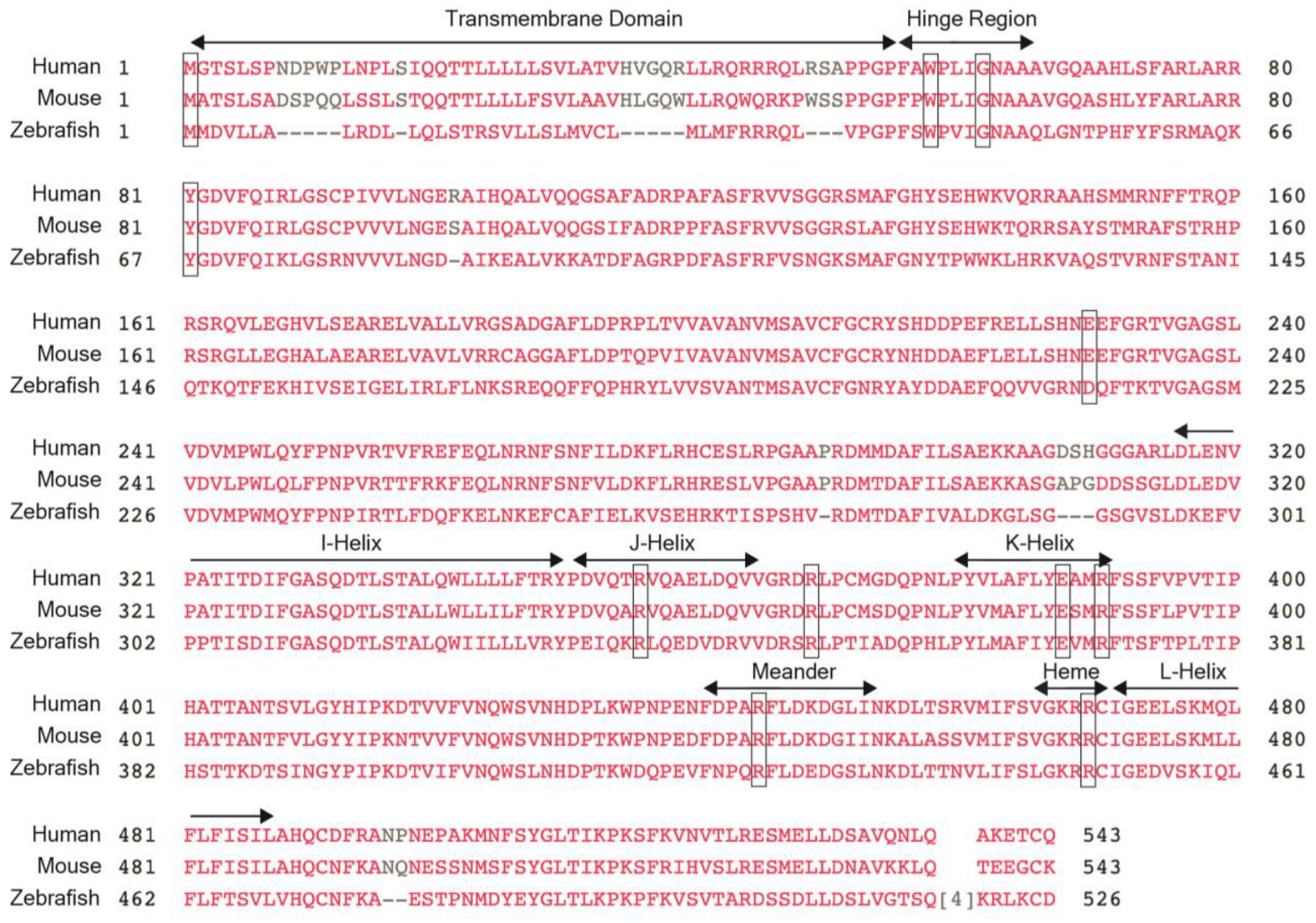
2. CYP1B1 Gene
3. CYP1B1 Genetics in Human Eye Diseases
4. CYP1B1 in Zebrafish Eye Development
5. CYP1B1 in Mouse Eye Development
6. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARS | Axenfeld–Rieger syndrome |
| CCO | Congenital corneal opacity |
| CCS | Conserved core structures |
| CEU | Congenital ectropion uvea |
| DEAB | N,N-diethylaminobenzaldehyde |
| EET | Epoxyeicosatrienoic |
| HETE | Hydroxyeicosatetraenoic |
| JOAG | Juvenile open-angle glaucoma |
| PCG | Primary congenital glaucoma |
| POAG | Primary open-angle glaucoma |
| RA | Retinoic acid |
| VUS | Variant of uncertain significance |
References
- Chavarria-Soley, G.; Michels-Rautenstrauss, K.; Pasutto, F.; Flikier, D.; Flikier, P.; Cirak, S.; Bejjani, B.; Winters, D.L.; Lewis, R.A.; Mardin, C.; et al. Primary congenital glaucoma and Rieger’s anomaly: Extended haplotypes reveal founder effects for eight distinct CYP1B1 mutations. Mol. Vis. 2006, 12, 523–531. [Google Scholar] [PubMed]
- Vasiliou, V.; Gonzalez, F.J. Role of CYP1B1 in glaucoma. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 333–358. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Tyler, R.C.; Weh, E.; Hendee, K.E.; Kariminejad, A.; Abdul-Rahman, O.; Ben-Omran, T.; Manning, M.A.; Yesilyurt, A.; McCarty, C.A.; et al. Analysis of CYP1B1 in pediatric and adult glaucoma and other ocular phenotypes. Mol. Vis. 2016, 22, 1229–1238. [Google Scholar] [PubMed]
- Bolton, E.M.; Bohnsack, B.L. Anterior segment dysgenesis: Part I—Current perspectives on management. Exp. Rev. Ophthalmol. 2024, 19, 173–186. [Google Scholar] [CrossRef]
- Bolton, E.M.; Bohnsack, B.L. Anterior segment dysgenesis: Part II—Genetics and pathogenesis. Exp. Rev. Ophthalmol. 2024, 19, 187–199. [Google Scholar] [CrossRef]
- Reis, L.M.; Seese, S.E.; Costakos, D.; Semina, E.V. Congenital anterior segment ocular disorders: Genotype-phenotype correlations and emerging novel mechanisms. Prog. Retin. Eye Res. 2024, 102, 101288. [Google Scholar] [CrossRef]
- Reis, L.M.; Semina, E.V. Genetics of anterior segment dysgenesis disorders. Curr. Opin. Ophthalmol. 2011, 22, 314–324. [Google Scholar] [CrossRef]
- Michels, K.; Bohnsack, B.L. Ophthalmological manifestations of axenfeld-rieger syndrome: Current perspectives. Clin. Ophthalmol. 2023, 17, 819–828. [Google Scholar] [CrossRef]
- Melki, R.; Colomb, E.; Lefort, N.; Brézin, A.P.; Garchon, H.J. CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J. Med. Genet. 2004, 41, 647–651. [Google Scholar] [CrossRef]
- Patel, H.Y.; Richards, A.J.; De Karolyi, B.; Best, S.J.; Danesh-Meyer, H.V.; Vincent, A.L. Screening glaucoma genes in adult glaucoma suggests a multiallelic contribution of CYP1B1 to open-angle glaucoma phenotypes. Clin. Exp. Ophthalmol. 2012, 40, e208–e217. [Google Scholar] [CrossRef]
- Sarfarazi, M.; Akarsu, A.N.; Hossain, A.; Turacli, M.E.; Aktan, S.G.; Barsoum-Homsy, M.; Chevrette, L.; Sayli, B.S. Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics 1995, 30, 171–177. [Google Scholar] [CrossRef]
- Tang, Y.M.; Wo, Y.Y.; Stewart, J.; Hawkins, A.L.; Griffin, C.A.; Sutter, T.R.; Greenlee, W.F. Isolation and characterization of the human cytochrome P450 CYP1B1 gene. J. Biol. Chem. 1996, 271, 28324–28330. [Google Scholar] [CrossRef]
- Stoilov, I.; Akarsu, A.N.; Sarfarazi, M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 1997, 6, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Sutter, T.R.; Tang, Y.M.; Hayes, C.L.; Wo, Y.Y.; Jabs, E.W.; Li, X.; Yin, H.; Cody, C.W.; Greenlee, W.F. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J. Biol. Chem. 1994, 269, 13092–13099. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Stoilov, I.; Akarsu, A.N.; Alozie, I.; Child, A.; Barsoum-Homsy, M.; Turacli, M.E.; Or, M.; Lewis, R.A.; Ozdemir, N.; Brice, G.; et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am. J. Hum. Genet. 1998, 62, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sorenson, C.M.; Sheibani, N. Cytochrome P450 1B1 and Primary Congenital Glaucoma. J. Ophthalmic Vis. Res. 2015, 10, 60–67. [Google Scholar] [CrossRef]
- Achary, M.S.; Reddy, A.B.; Chakrabarti, S.; Panicker, S.G.; Mandal, A.K.; Ahmed, N.; Balasubramanian, D.; Hasnain, S.E.; Nagarajaram, H.A. Disease-causing mutations in proteins: Structural analysis of the CYP1B1 mutations causing primary congenital glaucoma in humans. Biophys. J. 2006, 91, 4329–4339. [Google Scholar] [CrossRef]
- Jansson, I.; Stoilov, I.; Sarfarazi, M.; Schenkman, J.B. Effect of two mutations of human CYP1B1, G61E and R469W, on stability and endogenous steroid substrate metabolism. Pharmacogenetics 2001, 11, 793–801. [Google Scholar] [CrossRef]
- Choudhary, D.; Jansson, I.; Stoilov, I.; Sarfarazi, M.; Schenkman, J.B. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab. Dispos. 2004, 32, 840–847. [Google Scholar] [CrossRef]
- Chambers, D.; Wilson, L.; Maden, M.; Lumsden, A. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 2007, 134, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Haduch, A.; Bromek, E.; Kuban, W.; Daniel, W.A. The engagement of cytochrome P450 enzymes in tryptophan metabolism. Metabolites 2023, 13, 629. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Li, H.; Zheng, J. Effects of xenobiotics on CYP1 enzyme-mediated biotransformation and bioactivation of estradiol. Drug Metab. Rev. 2023, 55, 1–49. [Google Scholar] [CrossRef]
- Faiq, M.A.; Singh, H.N.; Ali, M.; Dada, R.; Chan, K.C.; Dada, T.; Saluja, D. Functional genomics of primary congenital glaucoma by pathway analysis and functional characterization of CYP1B1 mutations. Vis. Res. 2024, 227, 108534. [Google Scholar] [CrossRef] [PubMed]
- Mokhosoev, I.M.; Astakhov, D.V.; Terentiev, A.A.; Moldogazieva, N.T. Human cytochrome P450 cancer-related metabolic activities and gene polymorphisms: A Review. Cells 2024, 13, 1958. [Google Scholar] [CrossRef]
- Elfaki, I.; Mir, R.; Almutairi, F.M.; Duhier, F.M.A. Cytochrome P450: Polymorphisms and roles in cancer, diabetes and atherosclerosis. Asian Pac. J. Cancer Prev. 2018, 19, 2057–2070. [Google Scholar] [CrossRef]
- Kwon, Y.J.; Shin, S.; Chun, Y.J. Biological roles of cytochrome P450 1A1, 1A2, and 1B1 enzymes. Arch. Pharm. Res. 2021, 44, 63–83. [Google Scholar] [CrossRef]
- Alsubait, A.; Aldossary, W.; Rashid, M.; Algamdi, A.; Alrfaei, B.M. CYP1B1 gene: Implications in glaucoma and cancer. J. Cancer 2020, 11, 4652–4661. [Google Scholar] [CrossRef]
- Shah, M.; Bouhenni, R.; Benmerzouga, I. Geographical variability in CYP1B1 mutations in primary congenital glaucoma. J. Clin. Med. 2022, 11, 2048. [Google Scholar] [CrossRef]
- Ferák, V.; Gencik, A.; Gencikova, A. Population genetic aspects of primary congenital glaucoma. II. Fitness, parental consanguinity, founder effect. Hum. Genet. 1982, 61, 198–200. [Google Scholar] [CrossRef]
- Gencik, A.; Gencikova, A.; Ferák, V. Population genetical aspects of primary congenital glaucoma. I. Incidence, prevalence, gene frequency, and age of onset. Hum. Genet. 1982, 61, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Plásilová, M.; Stoilov, I.; Sarfarazi, M.; Kádasi, L.; Feráková, E.; Ferák, V. Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary congenital glaucoma. J. Med. Genet. 1999, 36, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Sivadorai, P.; Cherninkova, S.; Bouwer, S.; Kamenarova, K.; Angelicheva, D.; Seeman, P.; Hollingsworth, K.; Mihaylova, V.; Oscar, A.; Dimitrova, G.; et al. Genetic heterogeneity and minor CYP1B1 involvement in the molecular basis of primary congenital glaucoma in Gypsies. Clin. Genet. 2008, 74, 82–87. [Google Scholar] [CrossRef]
- Chitsazian, F.; Tusi, B.K.; Elahi, E.; Saroei, H.A.; Sanati, M.H.; Yazdani, S.; Pakravan, M.; Nilforooshan, N.; Eslami, Y.; Mehrjerdi, M.A.; et al. CYP1B1 mutation profile of Iranian primary congenital glaucoma patients and associated haplotypes. J. Mol. Diagn. 2007, 9, 382–393. [Google Scholar] [CrossRef]
- Ou, Z.; Liu, G.; Liu, W.; Deng, Y.; Zheng, L.; Zhang, S.; Feng, G. Bioinformatics analysis of CYP1B1 mutation hotspots in Chinese primary congenital glaucoma patients. Biosci. Rep. 2018, 38, BSR20180056. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Luthra-Guptasarma, M.; Prasher, D.; Dhingra, D.; Singh, N.; Kumar, A.; Sharma, S.P.; Kaur, H.; Snehi, S.; Thattaruthody, F.; et al. CYP1B1 and MYOC variants in neonatal-onset versus infantile-onset primary congenital glaucoma. Br. J. Ophthalmol. 2023, 107, 227–233. [Google Scholar] [CrossRef]
- Su, C.C.; Liu, Y.F.; Li, S.Y.; Yang, J.J.; Yen, Y.C. Mutations in the CYP1B1 gene may contribute to juvenile-onset open-angle glaucoma. Eye 2012, 26, 1369–1377. [Google Scholar] [CrossRef]
- Kaushik, S.; Choudhary, S.; Kaur, A.; Srivastava, P.; Pokharel, B.; Akella, M.; Pandav, S.S. Neonatal-onset congenital ectropion uveae may be caused by a distinct cyp1b1 pathologic variant. Am. J. Ophthalmol. 2022, 239, 54–65. [Google Scholar] [CrossRef]
- Bejjani, B.A.; Lewis, R.A.; Tomey, K.F.; Anderson, K.L.; Dueker, D.K.; Jabak, M.; Astle, W.F.; Otterud, B.; Leppert, M.; Lupski, J.R. Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital glaucoma in Saudi Arabia. Am. J. Hum. Genet. 1998, 62, 325–333. [Google Scholar] [CrossRef]
- Sarfarazi, M.; Stoilov, I.; Schenkman, J.B. Genetics and biochemistry of primary congenital glaucoma. Ophthalmol. Clin. 2003, 16, 543–554. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Ghanekar, Y.; Kaur, K.; Kaur, I.; Mandal, A.K.; Rao, K.N.; Parikh, R.S.; Thomas, R.; Majumder, P.P. A polymorphism in the CYP1B1 promoter is functionally associated with primary congenital glaucoma. Hum. Mol. Genet. 2010, 19, 4083–4090. [Google Scholar] [CrossRef] [PubMed]
- Badeeb, O.M.; Micheal, S.; Koenekoop, R.K.; den Hollander, A.I.; Hedrawi, M.T. CYP1B1 mutations in patients with primary congenital glaucoma from Saudi Arabia. BMC Med. Genet. 2014, 15, 109. [Google Scholar] [CrossRef]
- Banerjee, A.; Chakraborty, S.; Chakraborty, A.; Chakrabarti, S.; Ray, K. Functional and structural analyses of cyp1b1 variants linked to congenital and adult-onset glaucoma to investigate the molecular basis of these diseases. PLoS ONE 2016, 11, e0156252. [Google Scholar] [CrossRef] [PubMed]
- Campos-Mollo, E.; López-Garrido, M.P.; Blanco-Marchite, C.; Garcia-Feijoo, J.; Peralta, J.; Belmonte-Martínez, J.; Ayuso, C.; Escribano, J. CYP1B1 mutations in Spanish patients with primary congenital glaucoma: Phenotypic and functional variability. Mol. Vis. 2009, 15, 417–431. [Google Scholar]
- López-Garrido, M.P.; Blanco-Marchite, C.; Sánchez-Sánchez, F.; López-Sánchez, E.; Chaqués-Alepuz, V.; Campos-Mollo, E.; Salinas-Sánchez, A.S.; Escribano, J. Functional analysis of CYP1B1 mutations and association of heterozygous hypomorphic alleles with primary open-angle glaucoma. Clin. Genet. 2010, 77, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Al-Abdi, L.; Mohamed, J.Y.; Aldahmesh, M.A.; Alkuraya, F.S. Familial juvenile glaucoma with underlying homozygous p.G61E CYP1B1 mutations. J. AAPOS 2011, 15, 198–199. [Google Scholar] [CrossRef]
- Bashir, R.; Tahir, H.; Yousaf, K.; Naz, S.; Naz, S. Homozygous p.G61E mutation in a consanguineous Pakistani family with co-existence of juvenile-onset open angle glaucoma and primary congenital glaucoma. Gene 2015, 570, 295–298. [Google Scholar] [CrossRef]
- Reis, L.M.; Tyler, R.C.; Volkmann Kloss, B.A.; Schilter, K.F.; Levin, A.V.; Lowry, R.B.; Zwijnenburg, P.J.; Stroh, E.; Broeckel, U.; Murray, J.C.; et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur. J. Hum. Genet. 2012, 20, 1224–1233. [Google Scholar] [CrossRef]
- Samant, M.; Chauhan, B.K.; Lathrop, K.L.; Nischal, K.K. Congenital aniridia: Etiology, manifestations and management. Expert Rev. Ophthalmol. 2016, 11, 135–144. [Google Scholar] [CrossRef]
- Chavarria-Soley, G.; Michels-Rautenstrauss, K.; Caliebe, A.; Kautza, M.; Mardin, C.; Rautenstrauss, B. Novel CYP1B1 and known PAX6 mutations in anterior segment dysgenesis (ASD). J. Glaucoma 2006, 15, 499–504. [Google Scholar] [CrossRef]
- Franco, E.; Gagrani, M.; Scanga, H.L.; Areaux, R.G., Jr.; Chu, C.T.; Nischal, K.K. Variable phenotype of congenital corneal opacities in biallelic cyp1b1 pathogenic variants. Cornea 2024, 43, 195–200. [Google Scholar] [CrossRef]
- Tanwar, M.; Dada, T.; Dada, R. Axenfeld-rieger syndrome associated with congenital glaucoma and cytochrome p4501b1 gene mutations. Case Rep. Med. 2010, 2010, 212656. [Google Scholar] [CrossRef] [PubMed]
- Millá, E.; Mañé, B.; Duch, S.; Hernan, I.; Borràs, E.; Planas, E.; Dias Mde, S.; Carballo, M.; Gamundi, M.J. Survey of familial glaucoma shows a high incidence of cytochrome P450, family 1, subfamily B, polypeptide 1 (CYP1B1) mutations in non-consanguineous congenital forms in a Spanish population. Mol. Vis. 2013, 19, 1707–1722. [Google Scholar] [PubMed]
- Kelberman, D.; Islam, L.; Jacques, T.S.; Russell-Eggitt, I.; Bitner-Glindzicz, M.; Khaw, P.T.; Nischal, K.K.; Sowden, J.C. CYP1B1-related anterior segment developmental anomalies novel mutations for infantile glaucoma and von Hippel’s ulcer revisited. Ophthalmology 2011, 118, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Beedanagari, S.R.; Taylor, R.T.; Bui, P.; Wang, F.; Nickerson, D.W.; Hankinson, O. Role of epigenetic mechanisms in differential regulation of the dioxin-inducible human CYP1A1 and CYP1B1 genes. Mol. Pharmacol. 2010, 78, 608–616. [Google Scholar] [CrossRef]
- Mookherjee, S.; Acharya, M.; Banerjee, D.; Bhattacharjee, A.; Ray, K. Molecular basis for involvement of CYP1B1 in MYOC upregulation and its potential implication in glaucoma pathogenesis. PLoS ONE 2012, 7, e45077. [Google Scholar] [CrossRef]
- Reddy, A.B.; Panicker, S.G.; Mandal, A.K.; Hasnain, S.E.; Balasubramanian, D. Identification of R368H as a predominant CYP1B1 allele causing primary congenital glaucoma in Indian patients. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4200–4203. [Google Scholar] [CrossRef]
- Gupta, V.; Somarajan, B.I.; Walia, G.K.; Kaur, J.; Kumar, S.; Gupta, S.; Chaurasia, A.K.; Gupta, D.; Kaushik, A.; Mehta, A.; et al. Role of CYP1B1, p.E229K and p.R368H mutations among 120 families with sporadic juvenile onset open-angle glaucoma. Graefe’s Arch. Clin. Exp. Ophthalmol. 2018, 256, 355–362. [Google Scholar] [CrossRef]
- Bejjani, B.A.; Stockton, D.W.; Lewis, R.A.; Tomey, K.F.; Dueker, D.K.; Jabak, M.; Astle, W.F.; Lupski, J.R. Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Hum. Mol. Genet. 2000, 9, 367–374. [Google Scholar] [CrossRef]
- Pasutto, F.; Chavarria-Soley, G.; Mardin, C.Y.; Michels-Rautenstrauss, K.; Ingelman-Sundberg, M.; Fernández-Martínez, L.; Weber, B.H.; Rautenstrauss, B.; Reis, A. Heterozygous loss-of-function variants in CYP1B1 predispose to primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 249–254. [Google Scholar] [CrossRef]
- López-Garrido, M.P.; Sánchez-Sánchez, F.; López-Martínez, F.; Aroca-Aguilar, J.D.; Blanco-Marchite, C.; Coca-Prados, M.; Escribano, J. Heterozygous CYP1B1 gene mutations in Spanish patients with primary open-angle glaucoma. Mol. Vis. 2006, 12, 748–755. [Google Scholar] [PubMed]
- Acharya, M.; Mukhopadhyay, A.; Bhattacharjee, A.; Thakur, S.K.; Bandyopadhyay, A.K.; Ray, K. Complex genetics of glaucoma: Defects in CYP1B1, and not MYOC, cause pathogenesis in an early-onset POAG patient with double variants at both loci. J. Genet. 2008, 87, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Doshi, M.; Marcus, C.; Bejjani, B.A.; Edward, D.P. Immunolocalization of CYP1B1 in normal, human, fetal and adult eyes. Exp. Eye Res. 2006, 82, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.L.; Bohnsack, B.L. Multi-photon time lapse-imaging to visualize development in real-time: Visualization of migrating neural crest cells in zebrafish embryos. J. Vis. Exp. 2017, 9, E56214. [Google Scholar]
- Soules, K.A.; Link, B.A. Morphogenesis of the anterior segment in the zebrafish eye. BMC Dev. Biol. 2005, 5, 12. [Google Scholar] [CrossRef]
- Gray, M.P.; Smith, R.S.; Soules, K.A.; John, S.W.M.; Link, B.A. The aqueous humor outflow pathway of zebrafish. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1515–1521. [Google Scholar] [CrossRef]
- Scornaienchi, M.L.; Thornton, C.; Willett, K.L.; Wilson, J.Y. Functional differences in the cytochrome P450 1 family enzymes from zebrafish (Danio rerio) using heterologously expressed proteins. Arch. Biochem. Biophys. 2010, 502, 17–22. [Google Scholar] [CrossRef]
- Williams, A.L.; Eason, J.; Chawla, B.; Bohnsack, B.L. Cyp1b1 regulates ocular fissure closure through a retinoic acid-independent pathway. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1084–1097. [Google Scholar] [CrossRef]
- Hocking, J.C.; Famulski, J.K.; Yoon, K.H.; Widen, S.A.; Bernstein, C.S.; Koch, S.; Weiss, O.; Agarwala, S.; Inbal, A.; Lehmann, O.J.; et al. Morphogenetic defects underlie Superior Coloboma, a newly identified closure disorder of the dorsal eye. PLoS Genet. 2018, 14, e1007246. [Google Scholar] [CrossRef]
- Alexandre-Moreno, S.; Bonet-Fernández, J.M.; Atienzar-Aroca, R.; Aroca-Aguilar, J.D.; Escribano, J. Null cyp1b1 activity in zebrafish leads to variable craniofacial defects associated with altered expression of extracellular matrix and lipid metabolism genes. Int. J. Mol. Sci. 2021, 22, 6430. [Google Scholar] [CrossRef]
- Eason, J.; Williams, A.L.; Chawla, B.; Apsey, C.; Bohnsack, B.L. Differences in neural crest sensitivity to ethanol account for the infrequency of anterior segment defects in the eye cmpared with craniofacial anomalies in a zebrafish model of fetal alcohol syndrome. Birth Defects Res. 2017, 109, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Billingsley, G.; Priston, M.; Williams-Lyn, D.; Sutherland, J.; Glaser, T.; Oliver, E.; Walter, M.A.; Heathcote, G.; Levin, A.; et al. Phenotypic heterogeneity of CYP1B1: Mutations in a patient with Peters’ anomaly. J. Med. Genet. 2001, 38, 324–326. [Google Scholar] [CrossRef]
- Rauf, B.; Irum, B.; Kabir, F.; Firasat, S.; Naeem, M.A.; Khan, S.N.; Husnain, T.; Riazuddin, S.; Akram, J.; Riazuddin, S.A. A spectrum of CYP1B1 mutations associated with primary congenital glaucoma in families of Pakistani descent. Hum. Genome Var. 2016, 3, 16021. [Google Scholar] [CrossRef] [PubMed]
- Bejjani, B.A.; Xu, L.; Armstrong, D.; Lupski, J.R.; Reneker, L.W. Expression patterns of cytochrome P4501B1 (Cyp1b1) in FVB/N mouse eyes. Exp. Eye Res. 2002, 75, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, I.; Rezaie, T.; Jansson, I.; Schenkman, J.B.; Sarfarazi, M. Expression of cytochrome P4501b1 (Cyp1b1) during early murine development. Mol. Vis. 2004, 10, 629–636. [Google Scholar]
- Choudhary, D.; Jansson, I.; Rezaul, K.; Han, D.K.; Sarfarazi, M.; Schenkman, J.B. Cyp1b1 protein in the mouse eye during development: An immunohistochemical study. Drug Metab. Dispos. 2007, 35, 987–994. [Google Scholar] [CrossRef]
- Libby, R.T.; Smith, R.S.; Savinova, O.V.; Zabaleta, A.; Martin, J.E.; Gonzalez, F.J.; John, S.W. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science 2003, 299, 1578–1581. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, S.; Sorenson, C.M.; Teixeira, L.; Dubielzig, R.R.; Peters, D.M.; Conway, S.J.; Jefcoate, C.R.; Sheibani, N. Cyp1b1 mediates periostin regulation of trabecular meshwork development by suppression of oxidative stress. Mol. Cell. Biol. 2013, 33, 4225–4240. [Google Scholar] [CrossRef]
- Teixeira, L.B.; Zhao, Y.; Dubielzig, R.R.; Sorenson, C.M.; Sheibani, N. Ultrastructural abnormalities of the trabecular meshwork extracellular matrix in Cyp1b1-deficient mice. Vet. Pathol. 2015, 52, 397–403. [Google Scholar] [CrossRef]
- Falero-Perez, J.; Larsen, M.C.; Teixeira, L.B.C.; Zhang, H.F.; Lindner, V.; Sorenson, C.M.; Jefcoate, C.R.; Sheibani, N. Targeted deletion of Cyp1b1 in pericytes results in attenuation of retinal neovascularization and trabecular meshwork dysgenesis. Trends Dev. Biol. 2019, 12, 1–12. [Google Scholar]
- Lakkappa, N.; Krishnamurthy, P.T.; Hammock, B.D.; Velmurugan, D.; Bharath, M.M. Possible role of epoxyeicosatrienoic acid in prevention of oxidative stress mediated neuroinflammation in Parkinson disorders. Med. Hypotheses 2016, 93, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Y.; Tian, S.; Na, S.; Wei, H.; Wu, Y.; Yang, Y.; Shen, Z.; Ding, J.; Bao, S.; et al. CYP1B1 affects the integrity of the blood-brain barrier and oxidative stress in the striatum: An investigation of manganese-induced neurotoxicity. CNS Neurosci. Ther. 2024, 30, e14633. [Google Scholar] [CrossRef]
- Falero-Perez, J.; Sorenson, C.M.; Sheibani, N. Cyp1b1-deficient retinal astrocytes are more proliferative and migratory and are protected from oxidative stress and inflammation. Am. J. Physiol. Cell Physiol. 2019, 316, C767–C781. [Google Scholar] [CrossRef]
- Falero-Perez, J.; Sorenson, C.M.; Sheibani, N. Retinal astrocytes transcriptome reveals Cyp1b1 regulates the expression of genes involved in cell adhesion and migration. PLoS ONE 2020, 15, e0231752. [Google Scholar] [CrossRef] [PubMed]
- Amirmokhtari, N.; Foresi, B.D.; Dewan, S.S.; Bouhenni, R.A.; Smith, M.A. Absence of cytochrome p450-1b1 increases susceptibility of pressure-induced axonopathy in the murine retinal projection. Front. Cell Dev. Biol. 2021, 9, 636321. [Google Scholar] [CrossRef] [PubMed]
- Szabó, V.; Varsányi, B.; Barboni, M.; Takács, Á.; Knézy, K.; Molnár, M.J.; Nagy, Z.Z.; György, B.; Rivolta, C. Insights into eye genetics and recent advances in ocular gene therapy. Mol. Cell. Probes 2025, 79, 102008. [Google Scholar] [CrossRef]
- Conley, S.M.; Cai, X.; Naash, M.I. Nonviral ocular gene therapy: Assessment and future directions. Curr. Opin. Mol. Ther. 2008, 10, 456–463. [Google Scholar]
- Banou, L.; Sarrafpour, S.; Teng, C.C.; Liu, J. Ocular gene therapy: An overview of viral vectors, immune responses, and future directions. Yale J. Biol. Med. 2024, 97, 491–503. [Google Scholar] [CrossRef]
- Anton, N.; Geamănu, A.; Iancu, R.; Pîrvulescu, R.A.; Popa-Cherecheanu, A.; Barac, R.I.; Bandol, G.; Bogdănici, C.M. A mini-review on gene therapy in glaucoma and future directions. Int. J. Mol. Sci. 2024, 25, 11019. [Google Scholar] [CrossRef]
- Vasudevan, A.; Jozić, A.; Curtis, A.G.; Bodi, E.; Ryals, R.C.; Sahay, G. Lipid nanoparticle-mediated intracameral mRNA delivery facilitates gene expression and editing in the anterior chamber of the eye. J. Control. Release 2025, 379, 1022–1028. [Google Scholar] [CrossRef]
- Amankwa, C.E.; Young, O.; DebNath, B.; Gondi, S.R.; Rangan, R.; Ellis, D.Z.; Zode, G.; Stankowska, D.L.; Acharya, S. Modulation of mitochondrial metabolic parameters and antioxidant enzymes in healthy and glaucomatous trabecular meshwork cells with hybrid small molecule SA-2. Int. J. Mol. Sci. 2023, 24, 11557. [Google Scholar] [CrossRef] [PubMed]
- Iorga, R.E.; Moraru, A.D.; Costin, D.; Munteanu-Dănulescu, R.S.; Brănișteanu, D.C. Current trends in targeting the oxidative stress in glaucoma (Review). Eur. J. Ophthalmol. 2024, 34, 328–337. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolton, E.M.; Drackley, A.; Williams, A.L.; Bohnsack, B.L. Insights into CYP1B1-Related Ocular Diseases Through Genetics and Animal Studies. Life 2025, 15, 395. https://doi.org/10.3390/life15030395
Bolton EM, Drackley A, Williams AL, Bohnsack BL. Insights into CYP1B1-Related Ocular Diseases Through Genetics and Animal Studies. Life. 2025; 15(3):395. https://doi.org/10.3390/life15030395
Chicago/Turabian StyleBolton, Elizabeth M., Andy Drackley, Antionette L. Williams, and Brenda L. Bohnsack. 2025. "Insights into CYP1B1-Related Ocular Diseases Through Genetics and Animal Studies" Life 15, no. 3: 395. https://doi.org/10.3390/life15030395
APA StyleBolton, E. M., Drackley, A., Williams, A. L., & Bohnsack, B. L. (2025). Insights into CYP1B1-Related Ocular Diseases Through Genetics and Animal Studies. Life, 15(3), 395. https://doi.org/10.3390/life15030395