Notch and Hedgehog Signaling Unveiled: Crosstalk, Roles, and Breakthroughs in Cancer Stem Cell Research


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structure and Function of Notch Signaling
3. CSC and Notch Inhibition
4. Notch Signaling and the TME
4.1. Endothelial Cells
4.2. Angiogenesis
4.3. Adipose-Derived Stem Cells (ADSCs)
4.4. Hypoxia
5. Crosstalk with Other Signaling Pathways
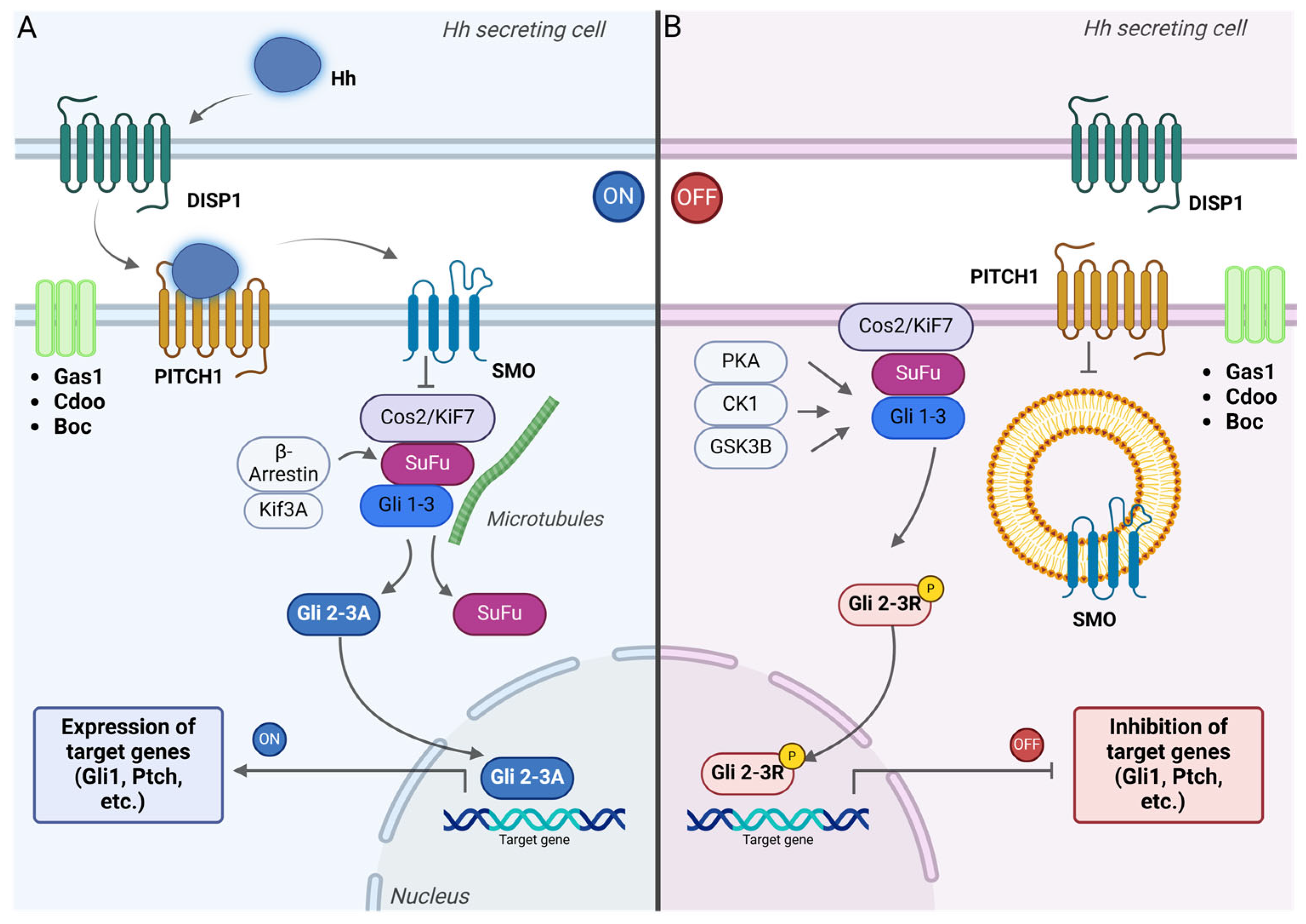
6. Role of Hedgehog Signaling Pathway in CSCs
7. Hedgehog Signaling Pathway and Its Role in Cancer Onset
8. Hedgehog Signaling (Hh) and CSCs
9. Hh and Crosstalk with Other Signaling Pathways
10. Conclusions
Author Contributions
Funding
Data Availability
Acknowledgments
Conflicts of Interest
References
- Penton, A.L.; Leonard, L.D.; Spinner, N.B. Notch signaling in human development and disease. Semin. Cell Dev. Biol. 2012, 23, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, C.T.; Huang, P. Complex crosstalk of Notch and Hedgehog signalling during the development of the central nervous system. Cell. Mol. Life Sci. 2021, 78, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Xu, M.; Yang, J.; Ma, X. The role of Hedgehog and Notch signaling pathway in cancer. Mol. Biomed. 2022, 3, 44. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Jesse, A.M.; Kohn, A.; Gunnell, L.M.; Honjo, T.; Zuscik, M.J.; O’Keefe, R.J.; Hilton, M.J. RBPjkappa-dependent Notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development 2010, 137, 1461–1471. [Google Scholar] [CrossRef]
- Hankenson, K.D.; Gagne, K.; Shaughnessy, M. Extracellular signaling molecules to promote fracture healing and bone regeneration. Adv. Drug Deliv. Rev. 2015, 94, 3–12. [Google Scholar] [CrossRef]
- MacGrogan, D.; Nus, M.; de la Pompa, J.L. Notch signaling in cardiac development and disease. Curr. Top. Dev. Biol. 2010, 92, 333–365. [Google Scholar] [CrossRef]
- Luxán, G.; D’Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes. Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef]
- Lobov, I.B.; Renard, R.A.; Papadopoulos, N.; Gale, N.W.; Thurston, G.; Yancopoulos, G.D.; Wiegand, S.J. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2007, 104, 3219–3224. [Google Scholar] [CrossRef]
- Baeten, J.T.; Lilly, B. Differential Regulation of NOTCH2 and NOTCH3 Contribute to Their Unique Functions in Vascular Smooth Muscle Cells. J. Biol. Chem. 2015, 290, 16226–16237. [Google Scholar] [CrossRef]
- Sparks, E.E.; Huppert, K.A.; Brown, M.A.; Washington, M.K.; Huppert, S.S. Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 2010, 51, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Panikkar, A.; Xu, J.; Antoniou, A.; Raynaud, P.; Lemaigre, F.; Stanger, B.Z. Notch signaling controls liver development by regulating biliary differentiation. Development 2009, 136, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Raynaud, P.; Cordi, S.; Zong, Y.; Tronche, F.; Stanger, B.Z.; Jacquemin, P.; Pierreux, C.E.; Clotman, F.; Lemaigre, F.P. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology 2009, 136, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Espinoza, L.; Dowbaj, A.M.; Kohler, T.N.; Strauss, B.; Sarlidou, O.; Belenguer, G.; Pacini, C.; Martins, N.P.; Dobie, R.; Wilson-Kanamori, J.R.; et al. Dynamic cell contacts between periportal mesenchyme and ductal epithelium act as a rheostat for liver cell proliferation. Cell Stem Cell 2021, 28, 1907–1921.e8. [Google Scholar] [CrossRef]
- Jensen, J.; Pedersen, E.E.; Galante, P.; Hald, J.; Heller, R.S.; Ishibashi, M.; Kageyama, R.; Guillemot, F.; Serup, P.; Madsen, O.D. Control of endodermal endocrine development by Hes-1. Nat. Genet. 2000, 24, 36–44. [Google Scholar] [CrossRef]
- Seymour, P.A.; Collin, C.A.; Egeskov-Madsen, A.R.; Jørgensen, M.C.; Shimojo, H.; Imayoshi, I.; de Lichtenberg, K.H.; Kopan, R.; Kageyama, R.; Serup, P. Jag1 Modulates an Oscillatory Dll1-Notch-Hes1 Signaling Module to Coordinate Growth and Fate of Pancreatic Progenitors. Dev. Cell 2020, 52, 731–747.e8. [Google Scholar] [CrossRef]
- Wendorff, A.A.; Koch, U.; Wunderlich, F.T.; Wirth, S.; Dubey, C.; Brüning, J.C.; MacDonald, H.R.; Radtke, F. Hes1 is a critical but context-dependent mediator of canonical Notch signaling in lymphocyte development and transformation. Immunity 2010, 33, 671–684. [Google Scholar] [CrossRef]
- López-López, S.; Romero de Ávila, M.J.; Hernández de León, N.C.; Ruiz-Marcos, F.; Baladrón, V.; Nueda, M.L.; Laborda, J.; García-Ramírez, J.J.; Monsalve, E.M.; Díaz-Guerra, M.J.M. NOTCH4 Exhibits Anti-Inflammatory Activity in Activated Macrophages by Interfering With Interferon-γ and TLR4 Signaling. Front. Immunol. 2021, 12, 734966. [Google Scholar] [CrossRef]
- Radke, A.L.; Reynolds, L.E.; Melo, R.C.; Dvorak, A.M.; Weller, P.F.; Spencer, L.A. Mature human eosinophils express functional Notch ligands mediating eosinophil autocrine regulation. Blood 2009, 113, 3092–3101. [Google Scholar] [CrossRef]
- Lewis, K.L.; Caton, M.L.; Bogunovic, M.; Greter, M.; Grajkowska, L.T.; Ng, D.; Klinakis, A.; Charo, I.F.; Jung, S.; Gommerman, J.L.; et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 2011, 35, 780–791. [Google Scholar] [CrossRef]
- Butler, J.M.; Nolan, D.J.; Vertes, E.L.; Varnum-Finney, B.; Kobayashi, H.; Hooper, A.T.; Seandel, M.; Shido, K.; White, I.A.; Kobayashi, M.; et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell 2010, 6, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Chen, T.; Wang, Y.; Tian, R.; Zhang, L.; Song, P.; Yang, S.; Zhu, Y.; Guo, X.; Huang, Y.; et al. Mesenchymal stem cells overexpressing Ihh promote bone repair. J. Orthop. Surg. Res. 2014, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Koleva, M.; Kappler, R.; Vogler, M.; Herwig, A.; Fulda, S.; Hahn, H. Pleiotropic effects of sonic hedgehog on muscle satellite cells. Cell Mol. Life Sci. 2005, 62, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Tanaka, N. Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer. J. Dev. Biol. 2017, 5, 12. [Google Scholar] [CrossRef]
- Frey, M.R. Sonic Hedgehog: Powering up Intestinal Regeneration? Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 650–651. [Google Scholar] [CrossRef]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef]
- Dessaud, E.; Ribes, V.; Balaskas, N.; Yang, L.L.; Pierani, A.; Kicheva, A.; Novitch, B.G.; Briscoe, J.; Sasai, N. Dynamic assignment and maintenance of positional identity in the ventral neural tube by the morphogen sonic hedgehog. PLoS Biol. 2010, 8, e1000382. [Google Scholar] [CrossRef]
- Yeo, S.Y.; Chitnis, A.B. Jagged-mediated Notch signaling maintains proliferating neural progenitors and regulates cell diversity in the ventral spinal cord. Proc. Natl. Acad. Sci. USA 2007, 104, 5913–5918. [Google Scholar] [CrossRef]
- Wall, D.S.; Mears, A.J.; McNeill, B.; Mazerolle, C.; Thurig, S.; Wang, Y.; Kageyama, R.; Wallace, V.A. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J. Cell Biol. 2009, 184, 101–112. [Google Scholar] [CrossRef]
- Jacobs, C.T.; Huang, P. Notch signalling maintains Hedgehog responsiveness via a Gli-dependent mechanism during spinal cord patterning in zebrafish. Elife 2019, 8, e49252. [Google Scholar] [CrossRef]
- Ringuette, R.; Atkins, M.; Lagali, P.S.; Bassett, E.A.; Campbell, C.; Mazerolle, C.; Mears, A.J.; Picketts, D.J.; Wallace, V.A. A Notch-Gli2 axis sustains Hedgehog responsiveness of neural progenitors and Müller glia. Dev. Biol. 2016, 411, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Karamboulas, C.; Ailles, L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Lai, A.G. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br. J. Cancer 2019, 121, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J. Nevoid basal-cell carcinoma syndrome. Medicine 1987, 66, 98–113. [Google Scholar] [CrossRef]
- Lombardo, Y.; Faronato, M.; Filipovic, A.; Vircillo, V.; Magnani, L.; Coombes, R.C. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. 2014, 16, R62. [Google Scholar] [CrossRef]
- Arcaroli, J.J.; Tai, W.M.; McWilliams, R.; Bagby, S.; Blatchford, P.J.; Varella-Garcia, M.; Purkey, A.; Quackenbush, K.S.; Song, E.-K.; Pitts, T.M.; et al. A NOTCH1 gene copy number gain is a prognostic indicator of worse survival and a predictive biomarker to a Notch1 targeting antibody in colorectal cancer. Int. J. Cancer 2016, 138, 195–205. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Abou-Antoun, T.J.; Hale, J.S.; Lathia, J.D.; Dombrowski, S.M. Brain Cancer Stem Cells in Adults and Children: Cell Biology and Therapeutic Implications. Neurotherapeutics 2017, 14, 372–384. [Google Scholar] [CrossRef]
- Donnem, T.; Andersen, S.; Al-Shibli, K.; Al-Saad, S.; Busund, L.T.; Bremnes, R.M. Prognostic impact of Notch ligands and receptors in nonsmall cell lung cancer: Coexpression of Notch-1 and vascular endothelial growth factor-A predicts poor survival. Cancer 2010, 116, 5676–5685. [Google Scholar] [CrossRef]
- Asnaghi, L.; Ebrahimi, K.B.; Schreck, K.C.; Bar, E.E.; Coonfield, M.L.; Bell, W.R.; Handa, J.; Merbs, S.L.; Harbour, J.W.; Eberhart, C.G. Notch signaling promotes growth and invasion in uveal melanoma. Clin. Cancer Res. 2012, 18, 654–665. [Google Scholar] [CrossRef]
- García-Peydró, M.; Fuentes, P.; Mosquera, M.; García-León, M.J.; Alcain, J.; Rodríguez, A.; García de Miguel, P.; Menéndez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 axis drives pathogenesis in a T cell acute lymphoblastic leukemia model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.J.; Brandt, W.D.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Bar, E.E.; Eberhart, C.G. Lateral inhibition of Notch signaling in neoplastic cells. Oncotarget 2015, 6, 1666–1677. [Google Scholar] [CrossRef] [PubMed]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Fendler, A.; Bauer, D.; Busch, J.; Jung, K.; Wulf-Goldenberg, A.; Kunz, S.; Song, K.; Myszczyszyn, A.; Elezkurtaj, S.; Erguen, B.; et al. Inhibiting WNT and NOTCH in renal cancer stem cells and the implications for human patients. Nat. Commun. 2020, 11, 929. [Google Scholar] [CrossRef]
- Liu, R.; Yu, Y.; Wang, Q.; Zhao, Q.; Yao, Y.; Sun, M.; Zhuang, J.; Sun, C.; Qi, Y. Interactions between hedgehog signaling pathway and the complex tumor microenvironment in breast cancer: Current knowledge and therapeutic promises. Cell Commun. Signal. 2024, 22, 432. [Google Scholar] [CrossRef]
- Aval, S.F.; Lotfi, H.; Sheervalilou, R.; Zarghami, N. Tuning of major signaling networks (TGF-β, Wnt, Notch and Hedgehog) by miRNAs in human stem cells commitment to different lineages: Possible clinical application. Biomed. Pharmacother. 2017, 91, 849–860. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.-K.; Kittappa, R.; McKay, R.D.G. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef]
- Fleming, R.J. Structural conservation of Notch receptors and ligands. Semin. Cell Dev. Biol. 1998, 9, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Rebay, I.; Fleming, R.J.; Fehon, R.G.; Cherbas, L.; Cherbas, P.; Artavanis-Tsakonas, S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: Implications for Notch as a multifunctional receptor. Cell 1991, 67, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A Ligand-Induced Extracellular Cleavage Regulates γ-Secretase-like Proteolytic Activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Struhl, G.; Greenwald, I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 1999, 398, 522–525. [Google Scholar] [CrossRef]
- Fortini, M.E.; Artavanis-Tsakonas, S. The suppressor of hairless protein participates in notch receptor signaling. Cell 1994, 79, 273–282. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Zhou, S.; Chen, L.; Young, D.B.; Hayward, S.D. CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 23–28. [Google Scholar] [CrossRef]
- Nagel, A.C.; Krejci, A.; Tenin, G.; Bravo-Patiño, A.; Bray, S.; Maier, D.; Preiss, A. Hairless-mediated repression of notch target genes requires the combined activity of Groucho and CtBP corepressors. Mol. Cell Biol. 2005, 25, 10433–10441. [Google Scholar] [CrossRef]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Banerjee, S.; Sarkar, F.H. Exploitation of the Notch signaling pathway as a novel target for cancer therapy. Anticancer. Res. 2008, 28, 3621–3630. [Google Scholar]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006, 66, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, M.; Kawaguchi, T.; Nigro, J.M.; Feuerstein, B.G.; Berger, M.S.; Miele, L.; Pieper, R.O. Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 2007, 106, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Demichelis, F.; Riva, A.; Varambally, S.; Hofer, M.D.; Kutok, J.L.; Kim, R.; Tang, J.; Montie, J.E.; Chinnaiyan, A.M.; et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004, 64, 6854–6857. [Google Scholar] [CrossRef] [PubMed]
- Real, P.J.; Ferrando, A.A. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia 2009, 23, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Maitra, A.; Ghosh, B.; Zechner, U.; Argani, P.; Iacobuzio-Donahue, C.A.; Sriuranpong, V.; Iso, T.; Meszoely, I.M.; Wolfe, M.S.; et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003, 3, 565–576. [Google Scholar] [CrossRef]
- Hopfer, O.; Zwahlen, D.; Fey, M.F.; Aebi, S. The Notch pathway in ovarian carcinomas and adenomas. Br. J. Cancer 2005, 93, 709–718. [Google Scholar] [CrossRef]
- Qiao, L.; Wong, B.C. Role of Notch signaling in colorectal cancer. Carcinogenesis 2009, 30, 1979–1986. [Google Scholar] [CrossRef]
- South, A.P.; Cho, R.J.; Aster, J.C. The double-edged sword of Notch signaling in cancer. Semin. Cell Dev. Biol. 2012, 23, 458–464. [Google Scholar] [CrossRef]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Dotto, G.P. Notch tumor suppressor function. Oncogene 2008, 27, 5115–5123. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, Y.; Wu, K.C.; Liu, J.; Zhao, Y.Q.; Pan, Y.L.; Du, R.; Zheng, G.R.; Xiong, Y.M.; Xu, H.L.; et al. RUNX3 directly interacts with intracellular domain of Notch1 and suppresses Notch signaling in hepatocellular carcinoma cells. Exp. Cell Res. 2010, 316, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V.; Zurrida, S.; Maisonneuve, P.; Viale, G.; Di Fiore, P.P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J. Cell Biol. 2004, 167, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Greve, B.; Kelsch, R.; Müller-Uthoff, H.; Weiss, K.; Kharabi Masouleh, B.; Sibrowski, W.; Kiesel, L.; Buchweitz, O. The adult stem cell marker Musashi-1 modulates endometrial carcinoma cell cycle progression and apoptosis via Notch-1 and p21WAF1/CIP1. Int. J. Cancer 2011, 129, 2042–2049. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Yu, H.; Linnoila, R.I.; Li, L.; Li, D.; Mo, B.; Okano, H.; Penalva, L.O.; Glazer, R.I. Musashi1 as a potential therapeutic target and diagnostic marker for lung cancer. Oncotarget 2013, 4, 739–750. [Google Scholar] [CrossRef]
- Wang, X.Y.; Penalva, L.O.; Yuan, H.; Linnoila, R.I.; Lu, J.; Okano, H.; Glazer, R.I. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol. Cancer 2010, 9, 221. [Google Scholar] [CrossRef]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther. 2013, 6, 1249–1259. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Mol. Asp. Med. 2014, 39, 3–32. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef]
- Chu, Q.; Orr, B.A.; Semenkow, S.; Bar, E.E.; Eberhart, C.G. Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clin. Cancer Res. 2013, 19, 3224–3233. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in cancer onset and progression. Bull. Acad. Natl. Med. 2009, 193, 1969–1978; discussion 1978–1969. [Google Scholar] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Hollier, B.G.; Evans, K.; Mani, S.A. The epithelial-to-mesenchymal transition and cancer stem cells: A coalition against cancer therapies. J. Mammary Gland. Biol. Neoplasia 2009, 14, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.; Reiman, J.M.; Asiedu, M.K.; Behrens, M.D.; Nassar, A.; Kalli, K.R.; Haluska, P.; Ingle, J.N.; Hartmann, L.C.; Manjili, M.H.; et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009, 69, 2887–2895. [Google Scholar] [CrossRef]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Böttinger, E.P. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef]
- Yabuuchi, S.; Pai, S.G.; Campbell, N.R.; de Wilde, R.F.; De Oliveira, E.; Korangath, P.; Streppel, M.M.; Rasheed, Z.A.; Hidalgo, M.; Maitra, A.; et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013, 335, 41–51. [Google Scholar] [CrossRef]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.-J.J.; et al. Chronic Treatment with the γ-Secretase Inhibitor LY-411,575 Inhibits β-Amyloid Peptide Production and Alters Lymphopoiesis and Intestinal Cell Differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef] [PubMed]
- Dandawate, P.; Padhye, S.; Ahmad, A.; Sarkar, F.H. Novel strategies targeting cancer stem cells through phytochemicals and their analogs. Drug Deliv. Transl. Res. 2013, 3, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Das, T.P.; Damodaran, C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br. J. Cancer 2013, 109, 2587–2596. [Google Scholar] [CrossRef]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H.; et al. A high Notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- Albini, A.; Sporn, M.B. The tumour microenvironment as a target for chemoprevention. Nat. Rev. Cancer 2007, 7, 139–147. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, F.; Wang, A.; Zheng, S.; Lu, Y. Dll4-Notch signaling in regulation of tumor angiogenesis. J. Cancer Res. Clin. Oncol. 2014, 140, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, R.E.; Seftor, E.A.; Gruman, L.M.; Lee, L.M.; Nickoloff, B.J.; Miele, L.; Sheriff, D.D.; Schatteman, G.C. Transendothelial function of human metastatic melanoma cells: Role of the microenvironment in cell-fate determination. Cancer Res. 2002, 62, 665–668. [Google Scholar] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Hess, A.R.; Seftor, R.E. Vasculogenic mimicry and tumour-cell plasticity: Lessons from melanoma. Nat. Rev. Cancer 2003, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Yoo, S.H.; Kim, S.H. Adipose-derived stem cells induced EMT-like changes in H358 lung cancer cells. Anticancer. Res. 2013, 33, 4421–4430. [Google Scholar]
- Horrée, N.; van Diest, P.J.; Sie-Go, D.M.; Heintz, A.P. The invasive front in endometrial carcinoma: Higher proliferation and associated derailment of cell cycle regulators. Hum. Pathol. 2007, 38, 1232–1238. [Google Scholar] [CrossRef]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar] [CrossRef]
- Yeung, T.M.; Gandhi, S.C.; Bodmer, W.F. Hypoxia and lineage specification of cell line-derived colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4382–4387. [Google Scholar] [CrossRef]
- Haase, V.H. Oxygen regulates epithelial-to-mesenchymal transition: Insights into molecular mechanisms and relevance to disease. Kidney Int. 2009, 76, 492–499. [Google Scholar] [CrossRef]
- Poon, E.; Harris, A.L.; Ashcroft, M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert. Rev. Mol. Med. 2009, 11, e26. [Google Scholar] [CrossRef]
- Soares, R.; Balogh, G.; Guo, S.; Gärtner, F.; Russo, J.; Schmitt, F. Evidence for the notch signaling pathway on the role of estrogen in angiogenesis. Mol. Endocrinol. 2004, 18, 2333–2343. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef]
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.C.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J. Exp. Med. 2011, 208, 1809–1822. [Google Scholar] [CrossRef]
- Shepherd, C.; Banerjee, L.; Cheung, C.W.; Mansour, M.R.; Jenkinson, S.; Gale, R.E.; Khwaja, A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660. [Google Scholar] [CrossRef]
- Calzavara, E.; Chiaramonte, R.; Cesana, D.; Basile, A.; Sherbet, G.V.; Comi, P. Reciprocal regulation of Notch and PI3K/Akt signalling in T-ALL cells in vitro. J. Cell. Biochem. 2008, 103, 1405–1412. [Google Scholar] [CrossRef]
- Chan, S.M.; Weng, A.P.; Tibshirani, R.; Aster, J.C.; Utz, P.J. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood 2007, 110, 278–286. [Google Scholar] [CrossRef]
- Meurette, O.; Stylianou, S.; Rock, R.; Collu, G.M.; Gilmore, A.P.; Brennan, K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res. 2009, 69, 5015–5022. [Google Scholar] [CrossRef]
- Campbell, K.J.; Perkins, N.D. Regulation of NF-kappaB function. Biochem. Soc. Symp. 2006, 165–180. [Google Scholar] [CrossRef]
- Cheng, P.; Zlobin, A.; Volgina, V.; Gottipati, S.; Osborne, B.; Simel, E.J.; Miele, L.; Gabrilovich, D.I. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J. Immunol. 2001, 167, 4458–4467. [Google Scholar] [CrossRef] [PubMed]
- Oakley, F.; Mann, J.; Ruddell, R.G.; Pickford, J.; Weinmaster, G.; Mann, D.A. Basal expression of IkappaBalpha is controlled by the mammalian transcriptional repressor RBP-J (CBF1) and its activator Notch1. J. Biol. Chem. 2003, 278, 24359–24370. [Google Scholar] [CrossRef] [PubMed]
- Moran, S.T.; Cariappa, A.; Liu, H.; Muir, B.; Sgroi, D.; Boboila, C.; Pillai, S. Synergism between NF-kappa B1/p50 and Notch2 during the development of marginal zone B lymphocytes. J. Immunol. 2007, 179, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef]
- Bash, J.; Zong, W.X.; Banga, S.; Rivera, A.; Ballard, D.W.; Ron, Y.; Gélinas, C. Rel/NF-kappaB can trigger the Notch signaling pathway by inducing the expression of Jagged1, a ligand for Notch receptors. EMBO J. 1999, 18, 2803–2811. [Google Scholar] [CrossRef]
- Guan, E.; Wang, J.; Laborda, J.; Norcross, M.; Baeuerle, P.A.; Hoffman, T. T cell leukemia-associated human Notch/translocation-associated Notch homologue has I kappa B-like activity and physically interacts with nuclear factor-kappa B proteins in T cells. J. Exp. Med. 1996, 183, 2025–2032. [Google Scholar] [CrossRef]
- Shin, H.M.; Minter, L.M.; Cho, O.H.; Gottipati, S.; Fauq, A.H.; Golde, T.E.; Sonenshein, G.E.; Osborne, B.A. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006, 25, 129–138. [Google Scholar] [CrossRef]
- Hanlon, L.; Avila, J.L.; Demarest, R.M.; Troutman, S.; Allen, M.; Ratti, F.; Rustgi, A.K.; Stanger, B.Z.; Radtke, F.; Adsay, V.; et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010, 70, 4280–4286. [Google Scholar] [CrossRef]
- Veenendaal, L.M.; Kranenburg, O.; Smakman, N.; Klomp, A.; Borel Rinkes, I.H.; van Diest, P.J. Differential Notch and TGFβ signaling in primary colorectal tumors and their corresponding metastases. Cell. Oncol. 2008, 30, 1–11. [Google Scholar] [CrossRef]
- Xu, P.; Qiu, M.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H.; Pu, P. The oncogenic roles of Notch1 in astrocytic gliomas in vitro and in vivo. J. Neurooncol. 2010, 97, 41–51. [Google Scholar] [CrossRef]
- Mittal, S.; Subramanyam, D.; Dey, D.; Kumar, R.V.; Rangarajan, A. Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol. Cancer 2009, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Weijzen, S.; Rizzo, P.; Braid, M.; Vaishnav, R.; Jonkheer, S.M.; Zlobin, A.; Osborne, B.A.; Gottipati, S.; Aster, J.C.; Hahn, W.C.; et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat. Med. 2002, 8, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Holland, E.C. Notch signaling enhances nestin expression in gliomas. Neoplasia 2006, 8, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Rodilla, V.; Villanueva, A.; Obrador-Hevia, A.; Robert-Moreno, A.; Fernández-Majada, V.; Grilli, A.; López-Bigas, N.; Bellora, N.; Albà, M.M.; Torres, F.; et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 6315–6320. [Google Scholar] [CrossRef] [PubMed]
- Ungerbäck, J.; Elander, N.; Grünberg, J.; Sigvardsson, M.; Söderkvist, P. The Notch-2 gene is regulated by Wnt signaling in cultured colorectal cancer cells. PLoS ONE 2011, 6, e17957. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Persano, L.; Pistollato, F.; Moro, E.; Frasson, C.; Porazzi, P.; Della Puppa, A.; Bresolin, S.; Battilana, G.; Indraccolo, S.; et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 2013, 4, e500. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. NUMB is a break of WNT-Notch signaling cycle. Int. J. Mol. Med. 2006, 18, 517–521. [Google Scholar] [CrossRef]
- Katoh, M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007, 3, 30–38. [Google Scholar] [CrossRef]
- Zhao, X.; Malhotra, G.K.; Lele, S.M.; Lele, M.S.; West, W.W.; Eudy, J.D.; Band, H.; Band, V. Telomerase-immortalized human mammary stem/progenitor cells with ability to self-renew and differentiate. Proc. Natl. Acad. Sci. USA 2010, 107, 14146–14151. [Google Scholar] [CrossRef]
- Kamakura, S.; Oishi, K.; Yoshimatsu, T.; Nakafuku, M.; Masuyama, N.; Gotoh, Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 2004, 6, 547–554. [Google Scholar] [CrossRef]
- Ingham, P.W.; Placzek, M. Orchestrating ontogenesis: Variations on a theme by sonic hedgehog. Nat. Rev. Genet. 2006, 7, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Spek, C.A.; Peppelenbosch, M.P. Hedgehog: An unusual signal transducer. Bioessays 2004, 26, 387–394. [Google Scholar] [CrossRef]
- Gallet, A. Hedgehog morphogen: From secretion to reception. Trends Cell Biol. 2011, 21, 238–246. [Google Scholar] [CrossRef]
- Danesin, C.; Agius, E.; Escalas, N.; Ai, X.; Emerson, C.; Cochard, P.; Soula, C. Ventral neural progenitors switch toward an oligodendroglial fate in response to increased Sonic hedgehog (Shh) activity: Involvement of Sulfatase 1 in modulating Shh signaling in the ventral spinal cord. J. Neurosci. 2006, 26, 5037–5048. [Google Scholar] [CrossRef]
- Thérond, P.P. Release and transportation of Hedgehog molecules. Curr. Opin. Cell Biol. 2012, 24, 173–180. [Google Scholar] [CrossRef]
- Bischoff, M.; Gradilla, A.-C.; Seijo, I.; Andrés, G.; Rodríguez-Navas, C.; González-Méndez, L.; Guerrero, I. Cytonemes are required for the establishment of a normal Hedgehog morphogen gradient in Drosophila epithelia. Nat. Cell Biol. 2013, 15, 1269–1281. [Google Scholar] [CrossRef]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Jia, J.; Tong, C.; Wang, B.; Luo, L.; Jiang, J. Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 2004, 432, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Struhl, G. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 1998, 391, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, L.; Scott, M.P.; Rohatgi, R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 2009, 187, 365–374. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Walsh, C.T.; McMahon, A.P. Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation. Proc. Natl. Acad. Sci. USA 2009, 106, 2623–2628. [Google Scholar] [CrossRef]
- Shevde, L.A.; Samant, R.S. Nonclassical hedgehog-GLI signaling and its clinical implications. Int. J. Cancer 2014, 135, 1–6. [Google Scholar] [CrossRef]
- Mille, F.; Thibert, C.; Fombonne, J.; Rama, N.; Guix, C.; Hayashi, H.; Corset, V.; Reed, J.C.; Mehlen, P. The Patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat. Cell Biol. 2009, 11, 739–746. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Hegde, G.V.; Peterson, K.J.; Emanuel, K.; Mittal, A.K.; Joshi, A.D.; Dickinson, J.D.; Kollessery, G.J.; Bociek, R.G.; Bierman, P.; Vose, J.M.; et al. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: A potential new therapeutic target. Mol. Cancer Res. 2008, 6, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Seeley, E.S.; Carrière, C.; Goetze, T.; Longnecker, D.S.; Korc, M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009, 69, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef]
- Li, X.; Deng, W.; Nail, C.D.; Bailey, S.K.; Kraus, M.H.; Ruppert, J.M.; Lobo-Ruppert, S.M. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 2006, 25, 609–621. [Google Scholar] [CrossRef]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- He, J.; Sheng, T.; Stelter, A.A.; Li, C.; Zhang, X.; Sinha, M.; Luxon, B.A.; Xie, J. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J. Biol. Chem. 2006, 281, 35598–35602. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7, re8. [Google Scholar] [CrossRef]
- Kurita, S.; Mott, J.L.; Almada, L.L.; Bronk, S.F.; Werneburg, N.W.; Sun, S.Y.; Roberts, L.R.; Fernandez-Zapico, M.E.; Gores, G.J. GLI3-dependent repression of DR4 mediates hedgehog antagonism of TRAIL-induced apoptosis. Oncogene 2010, 29, 4848–4858. [Google Scholar] [CrossRef]
- Athar, M.; Li, C.; Tang, X.; Chi, S.; Zhang, X.; Kim, A.L.; Tyring, S.K.; Kopelovich, L.; Hebert, J.; Epstein, E.H., Jr.; et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004, 64, 7545–7552. [Google Scholar] [CrossRef]
- Carballo, G.B.; Matias, D.; Ribeiro, J.H.; Pessoa, L.S.; Arrais-Neto, A.M.; Spohr, T. Cyclopamine sensitizes glioblastoma cells to temozolomide treatment through Sonic hedgehog pathway. Life Sci. 2020, 257, 118027. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, S.A.; Grinenko, N.F.; Antonova, O.M.; Kurapov, P.B.; Shepeleva, I.I.; Chekhonin, V.P. Relationship between Hedgehog Signaling Pathway and Drug Resistance of Poorly Differentiated Gliomas. Bull. Exp. Biol. Med. 2018, 164, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, R.; Wang, Z.; Chen, S.; Chen, S.; Guo, G.; Liu, Z. Cyclopamine Suppresses Human Esophageal Carcinoma Cell Growth by Inhibiting Glioma-Associated Oncogene Protein-1, a Marker of Human Esophageal Carcinoma Progression. Med. Sci. Monit. 2019, 25, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Fan, S.; Wang, H.; Mao, J.; Shi, Y.; Ibrahim, M.M.; Ma, W.; Yu, X.; Hou, Z.; Wang, B.; et al. Genistein decreases the breast cancer stem-like cell population through Hedgehog pathway. Stem Cell. Res. Ther. 2013, 4, 146. [Google Scholar] [CrossRef]
- Coward, L.; Barnes, N.C.; Setchell, K.D.R.; Barnes, S. Genistein, daidzein, and their.beta.-glycoside conjugates: Antitumor isoflavones in soybean foods from American and Asian diets. J. Agric. Food Chem. 1993, 41, 1961–1967. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Sureda, A.; Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; et al. Genistein and cancer: Current status, challenges, and future directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef]
- Fiaschi, M.; Rozell, B.; Bergström, A.; Toftgård, R. Development of mammary tumors by conditional expression of GLI1. Cancer Res. 2009, 69, 4810–4817. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Yue, W.; Wei, B.; Wang, N.; Li, T.; Guan, L.; Shi, S.; Zeng, Q.; Pei, X.; Chen, L. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS ONE 2011, 6, e17687. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Zacchetti, G.; Ruiz i Altaba, A. Hedgehog pathway activity is required for the lethality and intestinal phenotypes of mice with hyperactive Wnt signaling. Mech. Dev. 2010, 127, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Xu, X.F.; Xu, L.; Niu, P.Q.; Wang, F.; Hu, G.Y.; Wang, X.P.; Guo, C.Y. Cyclopamine blocked the growth of colorectal cancer SW116 cells by modulating some target genes of Gli1 in vitro. Hepatogastroenterology 2011, 58, 1511–1518. [Google Scholar] [CrossRef]
- Chen, X.; Lingala, S.; Khoobyari, S.; Nolta, J.; Zern, M.A.; Wu, J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J. Hepatol. 2011, 55, 838–845. [Google Scholar] [CrossRef]
- Jimeno, A.; Feldmann, G.; Suárez-Gauthier, A.; Rasheed, Z.; Solomon, A.; Zou, G.M.; Rubio-Viqueira, B.; García-García, E.; López-Ríos, F.; Matsui, W.; et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol. Cancer Ther. 2009, 8, 310–314. [Google Scholar] [CrossRef]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef]
- Fu, J.; Rodova, M.; Nanta, R.; Meeker, D.; Van Veldhuizen, P.J.; Srivastava, R.K.; Shankar, S. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro Oncol. 2013, 15, 691–706. [Google Scholar] [CrossRef]
- Nanta, R.; Kumar, D.; Meeker, D.; Rodova, M.; Van Veldhuizen, P.J.; Shankar, S.; Srivastava, R.K. NVP-LDE-225 (Erismodegib) inhibits epithelial-mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rγ null mice by regulating Bmi-1 and microRNA-128. Oncogenesis 2013, 2, e42. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef]
- Read, T.A.; Fogarty, M.P.; Markant, S.L.; McLendon, R.E.; Wei, Z.; Ellison, D.W.; Febbo, P.G.; Wechsler-Reya, R.J. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell 2009, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Sims-Mourtada, J.; Izzo, J.G.; Apisarnthanarax, S.; Wu, T.T.; Malhotra, U.; Luthra, R.; Liao, Z.; Komaki, R.; van der Kogel, A.; Ajani, J.; et al. Hedgehog: An attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin. Cancer Res. 2006, 12, 6565–6572. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef]
- Lauth, M. RAS and Hedgehog—Partners in crime. Front. Biosci. 2011, 16, 2259–2270. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes. Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133+ glioma stem cells to temozolomide therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iluta, S.; Nistor, M.; Buruiana, S.; Dima, D. Notch and Hedgehog Signaling Unveiled: Crosstalk, Roles, and Breakthroughs in Cancer Stem Cell Research. Life 2025, 15, 228. https://doi.org/10.3390/life15020228
Iluta S, Nistor M, Buruiana S, Dima D. Notch and Hedgehog Signaling Unveiled: Crosstalk, Roles, and Breakthroughs in Cancer Stem Cell Research. Life. 2025; 15(2):228. https://doi.org/10.3390/life15020228
Chicago/Turabian StyleIluta, Sabina, Madalina Nistor, Sanda Buruiana, and Delia Dima. 2025. "Notch and Hedgehog Signaling Unveiled: Crosstalk, Roles, and Breakthroughs in Cancer Stem Cell Research" Life 15, no. 2: 228. https://doi.org/10.3390/life15020228
APA StyleIluta, S., Nistor, M., Buruiana, S., & Dima, D. (2025). Notch and Hedgehog Signaling Unveiled: Crosstalk, Roles, and Breakthroughs in Cancer Stem Cell Research. Life, 15(2), 228. https://doi.org/10.3390/life15020228