2.1. Synthesis
Chromatographic purification of compounds was carried out using column chromatography on Acros silica gel (60–200 mesh). The reaction progress and purity of compounds were monitored by TLC on Sorbfil PTLC-AF-A-UF plates. Melting points of the products were determined using a Stanford Research Systems MPA-100 OptiMelt appliance. 1H, 13C, HSQC, and COSY NMR spectra were recorded on a Bruker Avance 400 WB spectrometer (400.13 and 100.62 MHz). Signals of dimethyl sulfoxide-d6 (δH 2.50, δC 39.51) were used as references in the 1H and 13C NMR spectra. Coupling constants (J) are reported in Hz (splitting abbreviations: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad; and combinations thereof). The HPLC/MS experiment was carried out using a TripleTOF 5600 AB Sciex superhigh resolution mass spectrometer (Germany) from the solution in methanol using the turboionic spray (TIS) ionization method with the collision energy with nitrogen molecules of 10 eV.
2.1.1. 2,2,8-trimethyl-4H-[1,3]dioxino[4,5-c]pyridine-5-carboxylic acid (3)
KMnO
4 (4.54 g, 28.7 mmol) was added to a solution of compound
2 (2.00 g, 9.57 mmol) in 50 mL of H
2O and 50 mL of Me
2CO. The reaction mixture was stirred at 50 °C for 24 h. Then, the precipitate was filtered off, and the filtrate was concentrated. The resulting solution was acidified with an aqueous solution of hydrochloric acid to pH = 5. The precipitated product was filtered and washed with H
2O. Yield 71% (1.51 g). White solid, mp 216–217 °C (dec.) (mp 220–221 °C (dec.) [
16]);
1H NMR (DMSO-
d6, 400 MHz)
δ 1.49 (s, 6H, C(CH
3)
2), 2.35 (s, 3H, CH
3), 5.09 (s, 2H, CH
2O), 8.51 (s, 1H, CH
pyr), 13.34 (br.s, 1H, C(O)OH);
13C NMR (DMSO-
d6, 100 MHz)
δ 18.9 (CH
3pyr), 24.4 (C(
CH
3)
2), 59.8 (CH
2O), 99.5 (
C(CH
3)
2), 120.7, 128.4, 141.9, 145.6, 151.1, (5 C
pyr), and 166.5 (C(O)OH).
2.1.2. Ethyl (E)-3-(2,2,8-trimethyl-4H-[1,3]dioxino[4,5-c]pyridin-5-yl)acrylate (6)
(2-Ethoxy-2-oxoethyl)triphenylphosphonium chloride (3.72 g, 9.66 mmol) and Et
3N (4.04 mL, 29.0 mmol) were added sequentially to a solution of compound
5 (2.00 g, 9.66 mmol) in 40 mL of CH
2Cl
2. The reaction mixture was stirred for 24 h at 75 °C in an autoclave under pressure. Then, the solvents were evaporated under reduced pressure, and the product was purified by column chromatography (eluent AcOEt). Yield 87% (2.33 g). The
1H NMR spectrum is fully described in the literature [
17].
2.1.3. (E)-3-(2,2,8-trimethyl-4H-[1,3]dioxino[4,5-c]pyridin-5-yl)acrylic acid (7)
A solution of K
2CO
3 (0.30 g, 2.17 mmol) in 3 mL of H
2O was added to a solution of compound
6 (0.60 g, 2.17 mmol) in 30 mL of MeOH. The reaction mixture was stirred at room temperature for 48 h. Then, the solvents were evaporated under reduced pressure at 45 °C. The dry residue was dissolved in H
2O, and the solution was acidified with an aqueous solution of hydrochloric acid to pH = 5. The precipitated product was filtered and washed with H
2O. Yield 88% (0.47 g). The
1H NMR spectrum is fully described in the literature [
17].
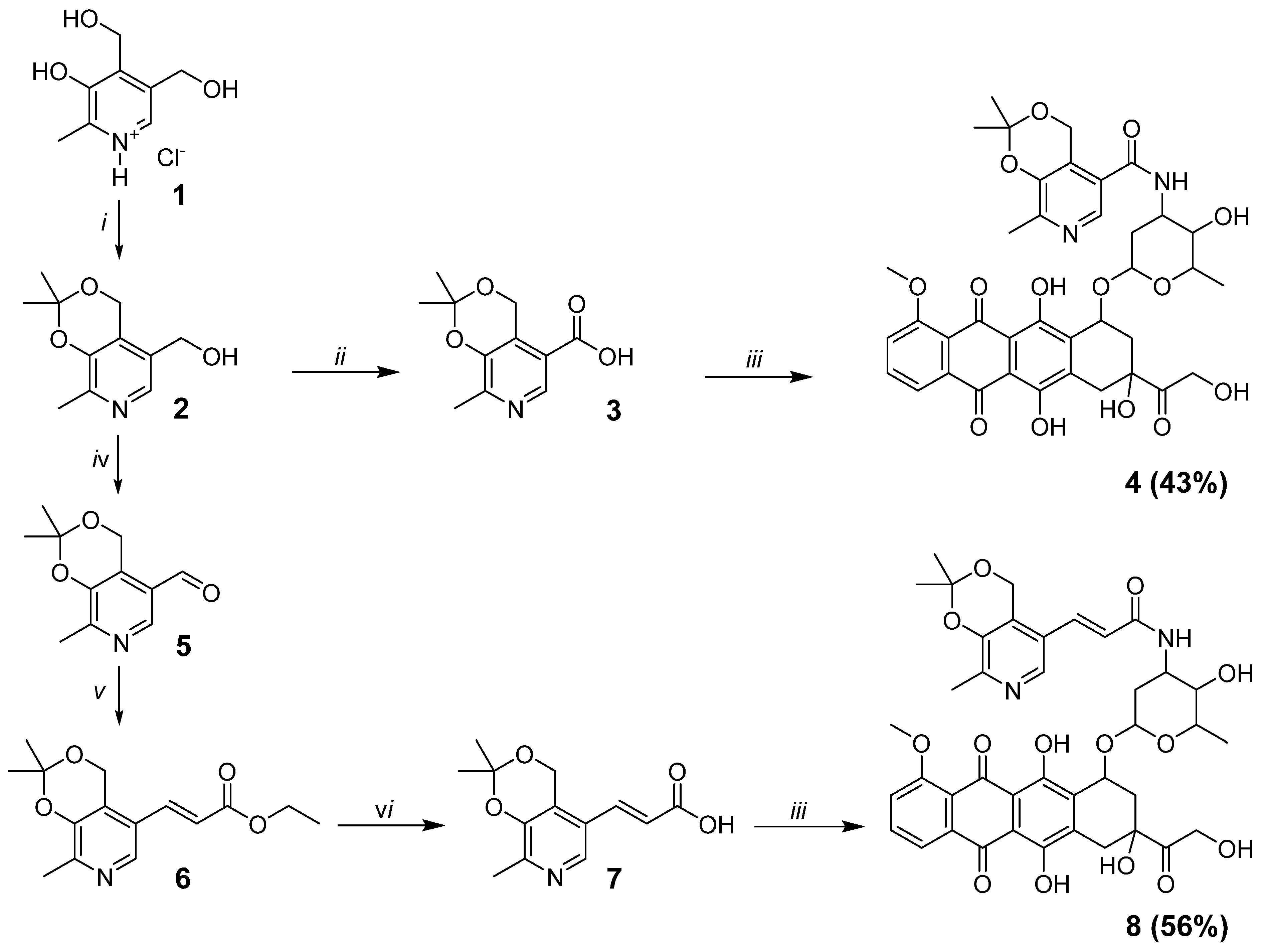
2.1.4. N-(3-hydroxy-2-methyl-6-((3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl)oxy)tetrahydro-2H-pyran-4-yl)-2,2,8-trimethyl-4H-[1,3]dioxino[4,5-c]pyridine-5-carboxamide (4)
HATU (1.70 g, 4.48 mmol) and DIPEA (0.31 mL, 1.79 mmol) were added sequentially to a solution of compound 3 (0.20 g, 0.90 mmol) and doxorubicin (0.52 g, 0.90 mmol) in 30 mL of CH2Cl2 and DMF (1:1, v/v). The reaction mixture was stirred for 12 h at room temperature. Then, the solvents were evaporated under reduced pressure at room temperature and the product was purified by column chromatography (eluent CHCl3—EtOH, 8:1, v/v); Yield 43% (0.29 g); dark red solid; mp 165–167 °C (dec.); 1H NMR (DMSO-d6, 400 MHz) δ 1.16 (d, 3H, J = 6.4, CH3), 1.44 (s, 3H, C(CH3)2), 1.45 (s, 3H, C(CH3)2), 1.51 (dd, 1H, J1 = 12.9, J2 = 4.0, CH2), 2.03 (td, 1H, J1 = 12.9, J2 = 12.9, J3 = 3.2, CH2), 2.12 (dd, 1H, J1 = 14.3, J2 = 5.5, CH2), 2.24 (dd, 1H, J1 = 14.3, J2 = 1.9, CH2), 2.30 (s, 3H, CH3), 2.88 (d, 1H, J = 18.2, CH2), 2.99 (d, 1H, J = 18.2, CH2), 3.56–3.60 (br.m, 1H, CH), 3.95 (s, 3H, OCH3), 4.08–4.20 (br.m, 1H, CH), 4.24 (q, 1H, J = 6.4, CH), 4.60 (br.s, 2H, CH2OH), 4.82–4.95 (m, 5H, CH2OH + CH2O + OH + CH), 5.25 (d, 1H, J = 3.2, OCHO), 5.44 (s, 1H, OH), 7.53–7.59 (m, 1H, CHar), 7.75–7.87 (m, 2H, 2CHar), 8.17 (d, 1H, J = 7.7, NH), 8.20 (s, 1H, CHpyr), 13.21 (s, 1H, OHar), 13.98 (s, 1H, OHar); 13C NMR (DMSO-d6, 100 MHz) δ 17.1 (CH3), 18.6 (CH3pyr), 24.4 (C(CH3)2), 24.5 (C(CH3)2), 29.3 (CH2), 32.0 (CH2), 36.4 (CH2), 46.0 (CHNH), 56.5 (CH3O), 58.9 (CH2O), 63.8 (CH2OH), 66.6 (CHO), 67.7 (CHO), 69.9 (CHO), 74.9 (C), 99.6 (C(CH3)2), 100.5 (OCHO), 110.5, 110.6, 118.9, 119.6, 119.8, 125.6, 125.9, 134.0, 134.5, 135.4, 136.1, 138.6, 145.3, 148.5, 154.5, 156.1, 160.7 (12 Car+ 5 Cpyr), 164.7 (C(O)NH), 186.2 (C(O)), 186.4 (C(O)), and 213.9 (C(O)). HRMS-ESI [M + H]+ 749.2551 (calculated for C38H41N2O14+, 749.2552).
2.1.5. (E)-N-(3-hydroxy-2-methyl-6-((3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl)oxy)tetrahydro-2H-pyran-4-yl)-3-(2,2,8-trimethyl-4H-[1,3]dioxino[4,5-c]pyridin-5-yl)acrylamide (8)
HATU (0.99 g, 2.61 mmol) and DIPEA (0.18 mL, 1.04 mmol) were added sequentially to a solution of compound 7 (0.13 g, 0.52 mmol) and doxorubicin (0.30 g, 0.52 mmol) in 15 mL of CH2Cl2 and DMF (2:1, v/v). The reaction mixture was stirred for 12 h at room temperature. Then, the solvents were evaporated under reduced pressure at room temperature, and the product was purified by column chromatography (eluent CHCl3—EtOH, 7:1, v/v); Yield 56% (0.23 g); dark red solid; mp 175–176 °C (dec.); 1H NMR (DMSO-d6, 400 MHz) δ 1.15 (d, 3H, J = 6.4, CH3), 1.46 (s, 6H, C(CH3)2), 1.51 (dd, 1H, J1 = 12.5, J2 = 4.1, CH2), 1.89 (td, 1H, J1 = 12.5, J2 = 12.5, J3 = 2.9, CH2), 2.08 (dd, 1H, J1 = 14.1, J2 = 5.6, CH2), 2.24 (dd, 1H, J1 = 14.1, J2 = 2.8, CH2), 2.27 (s, 3H, CH3), 2.82 (d, 1H, J = 18.1, CH2), 2.96 (d, 1H, J = 18.1, CH2), 3.43–3.47 (br.m, 1H, CH), 3.92 (s, 3H, OCH3), 4.06–4.16 (br.m, 1H, CH), 4.23 (q, 1H, J = 6.4, CH), 4.58 (br.s, 2H, CH2OH), 4.80–5.00 (br.m, 5H, CH2O + OH + CH2OH + CH), 5.24 (d, 1H, J = 2.9, OCHO), 5.44 (s, 1H, OH), 6.67 (d, 1H, J = 15.9, CH=CH), 7.20 (d, 1H, J = 15.9, CH=CH), 7.52–7.54 (m, 1H, CHar), 7.76–7.83 (m, 2H, 2CHar), 7.95 (d, 1H, J = 8.3, NH), 8.13 (s, 1H, CHpyr), 13.17 (s, 1H, OHar), 13.94 (s, 1H, OHar); 13C NMR (DMSO-d6, 100 MHz) δ 17.1 (CH3), 18.4 (CH3pyr), 24.40 (C(CH3)2), 24.44 (C(CH3)2), 29.8 (CH2), 32.0 (CH2), 36.5 (CH2), 45.2 (CHNH), 56.5 (CH3O), 58.6 (CH2O), 63.8 (CH2OH), 66.7 (CHO), 68.1 (CHO), 70.0 (CHO), 74.9 (C), 99.5 (C(CH3)2), 100.5 (OCHO), 110.5, 110.6, 118.9, 119.6, 119.7, 125.1, 125.4, 125.8, 131.7, 134.0, 134.4, 135.3, 136.1, 138.4, 145.2, 146.8, 154.5, 156.1, 160.7 (CH=CH + 12 Car + 5 Cpyr), 163.7 (C(O)NH), 186.1 (C(O)), 186.2 (C(O)), 213.9 (C(O)); HRMS-ESI [M + H]+ 775.2702 (calculated for C40H43N2O14+, 775.2709).
2.2. Cell Culture
The study utilized human cell lines including PC-3 (human prostate adenocarcinoma), HSF (primary human skin fibroblasts), MDA-MB-231 (estrogen-negative breast adenocarcinoma), MCF-7 (estrogen-positive breast adenocarcinoma), MSC (multipotent stem cells derived from adipose tissue), SF-539 (brain gliosarcoma), SNB-19 (glioblastoma), A-498 (kidney carcinoma), M-14 (human melanoma), NCI-H322-M (primary bronchioalveolar carcinoma), HCT-15 (colon adenocarcinoma), and HCT-116 (colorectal intestinal carcinoma). We also used immortalized C2C12 mouse myoblasts (ATCC—CRL-1772). Cancer cell lines were obtained from the ATCC collection and generously provided by the Fox Chase Cancer Center (Philadelphia, USA). Conditionally normal cells, including skin fibroblasts and multipotent stem cells, were isolated by our group from postoperative materials (skin and subcutaneous fat) obtained from a conditionally healthy donor. Reagents and consumables for cell culture work were purchased from PanEco (Russia).
Tumor cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 μg/mL penicillin, and 100 μg/mL streptomycin. The HSF, MSC, and C2C12 cells were grown in α-MEM with 10% fetal bovine serum, 2 mM L-glutamine, 100 μg/mL penicillin, and 100 μg/mL streptomycin. All cell cultures were maintained under aseptic conditions at 37 °C in a 5% CO2 atmosphere. Cells were grown in polystyrene flasks and upon reaching a monolayer, they were detached using a trypsin-EDTA solution (2.5% trypsin, 0.53 mM EDTA) in Dulbecco’s phosphate-buffered saline (DPBS).
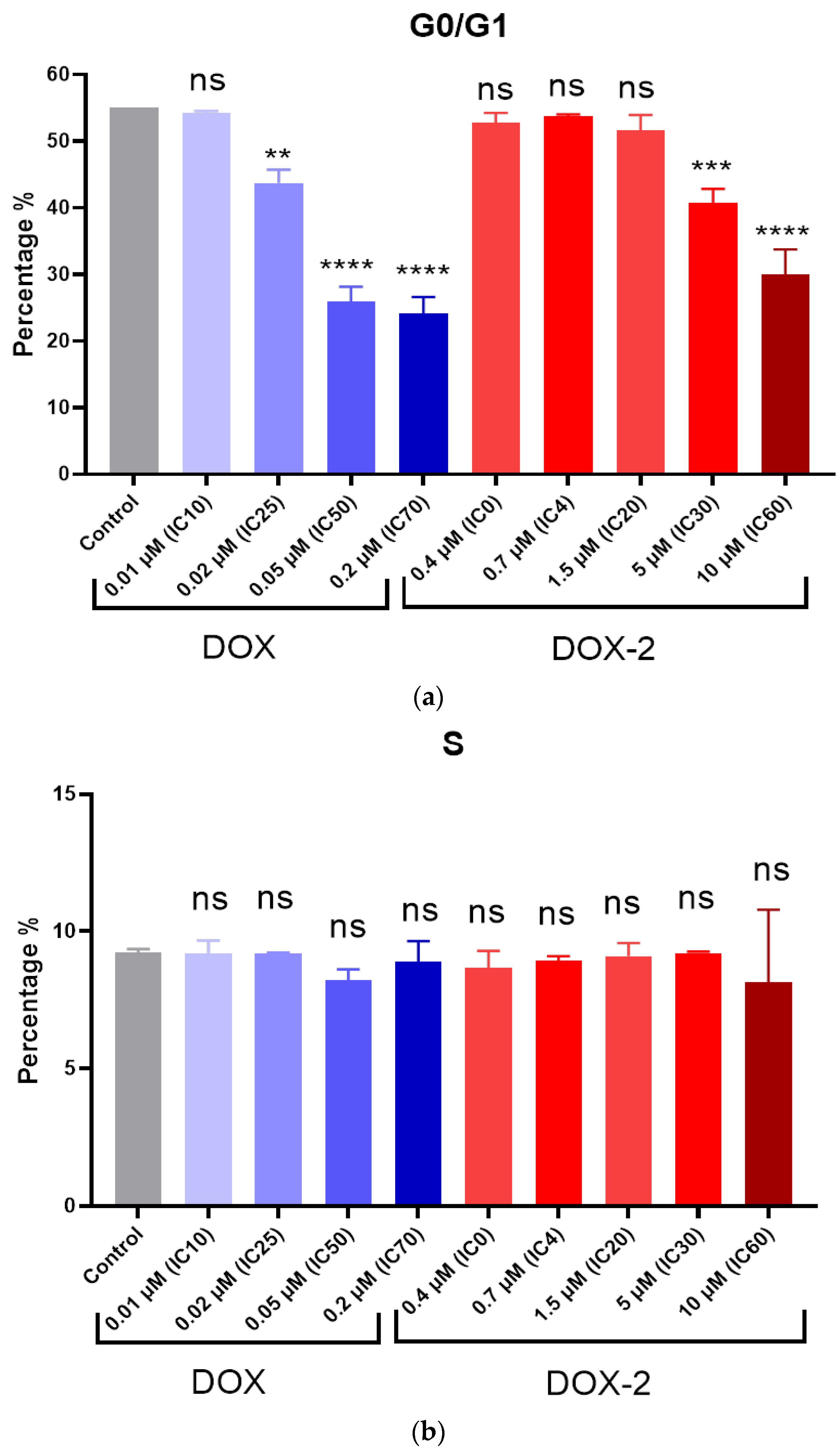
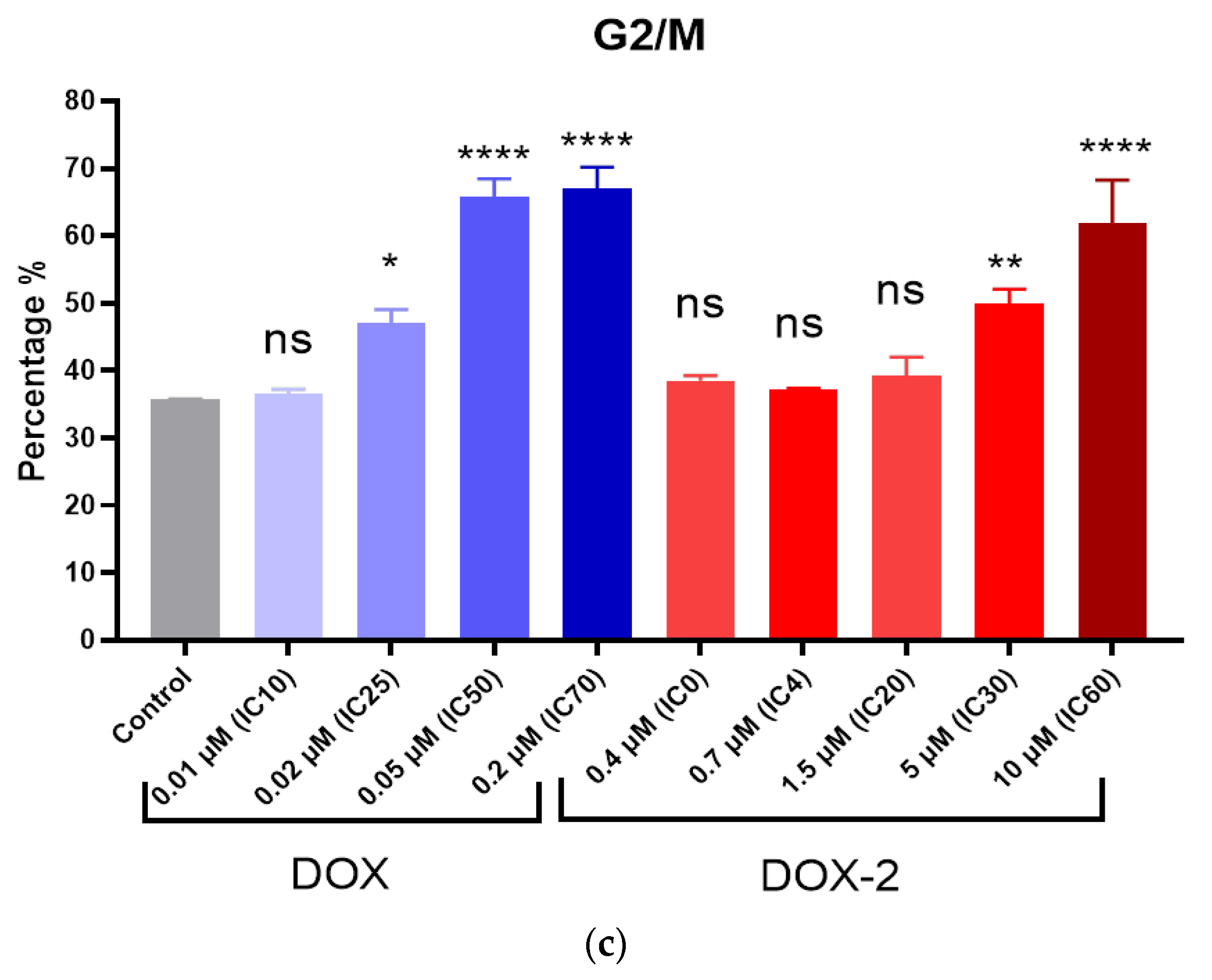
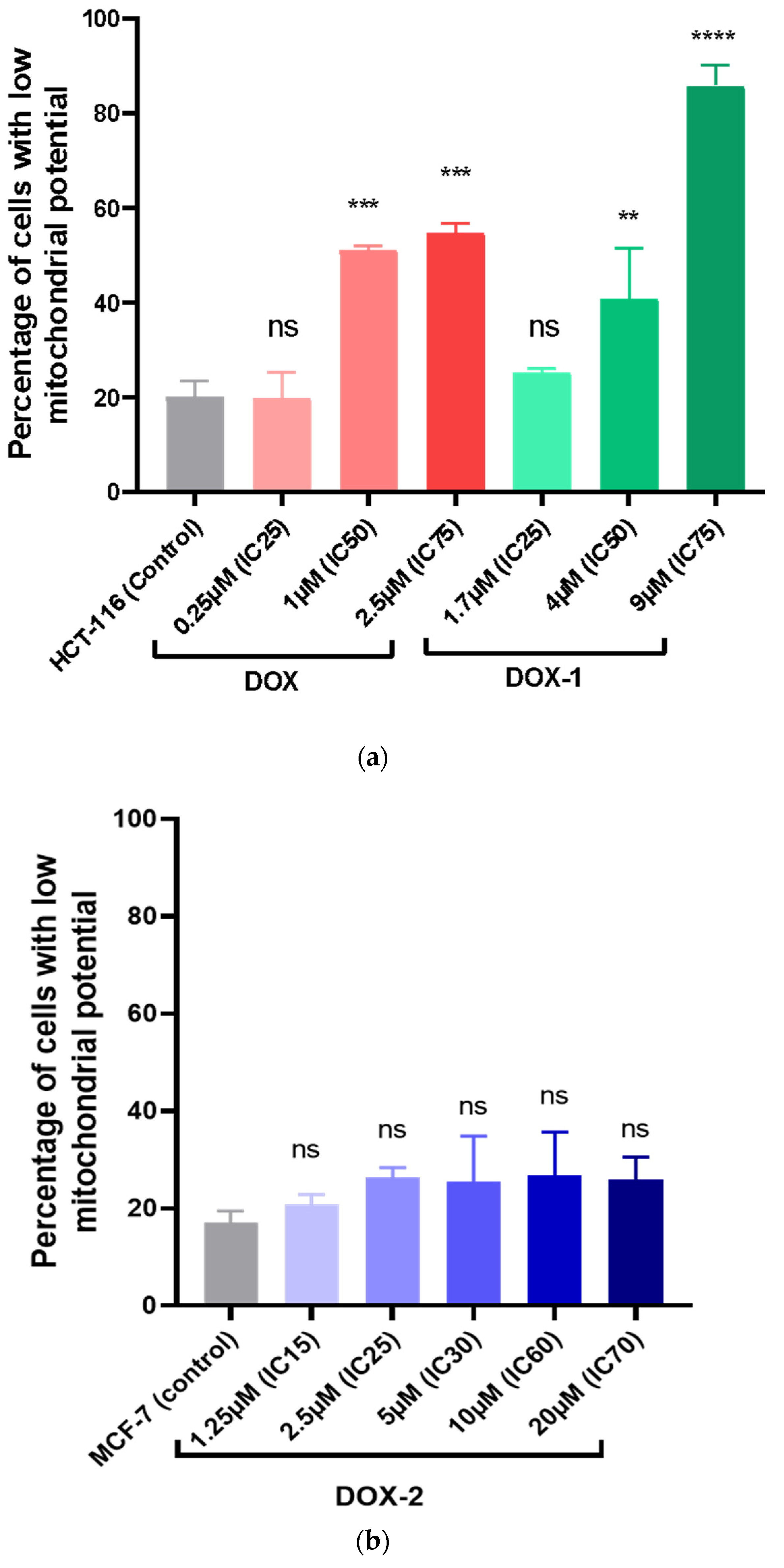
2.14. Comet Assay
PC-3 and MCF-7 cells were seeded into 6-well plates at a concentration of 15,000 cells/mL. After 24 h, DOX, DOX-1, and DOX-2 were added to the medium at concentrations corresponding to their IC25, IC50, and IC70 and incubated for 72 h. Cells were trypsinized and washed, then placed in 0.7% low melting point agarose and mounted on glass slides (200 000 cells/sample). Slides were treated with lysis solution (2.5 M NaCl, 0.1 M disodium EDTA, 1% Triton X-100, 10 mM Tris base, 10% DMSO, pH 10) for 1 h at 4 °C. Next, the samples were placed in an alkaline solution (12 g of NaOH brought to 1 L with water, disodium EDTA 0.2 M, pH >13) and incubated for 30 min at room temperature. Afterward, the slides were washed with electrophoresis 1X TBE buffer (10X TBE: 9.3 g disodium salt EDTA, 108 g Tris base, and 55 g boric acid per 1 L of water) twice and placed in the electrophoresis chamber. Electrophoresis was carried out under neutral conditions in 1X TBE buffer for 30 min at 31 V (at the rate of 1 V/cm). Afterward, the slides were washed in PBS (Ca free, Mg free) 2 times for 5 min, fixed in 96% ethanol for 5 min, and dried. Before analysis, 50 μL of SYBR Green solution (1μL stock + 10 mL TE buffer) was applied to the slides. Using an Observer Z1 fluorescent optical microscope (Carl Zeiss, Jena, Germany), photographs of cells were taken and analyzed in the ImageJ program (Plugin OpenComet).
“Tail moment” (TM) and “Olive moment” (OTM) were calculated using the following formula:


 ,
, 






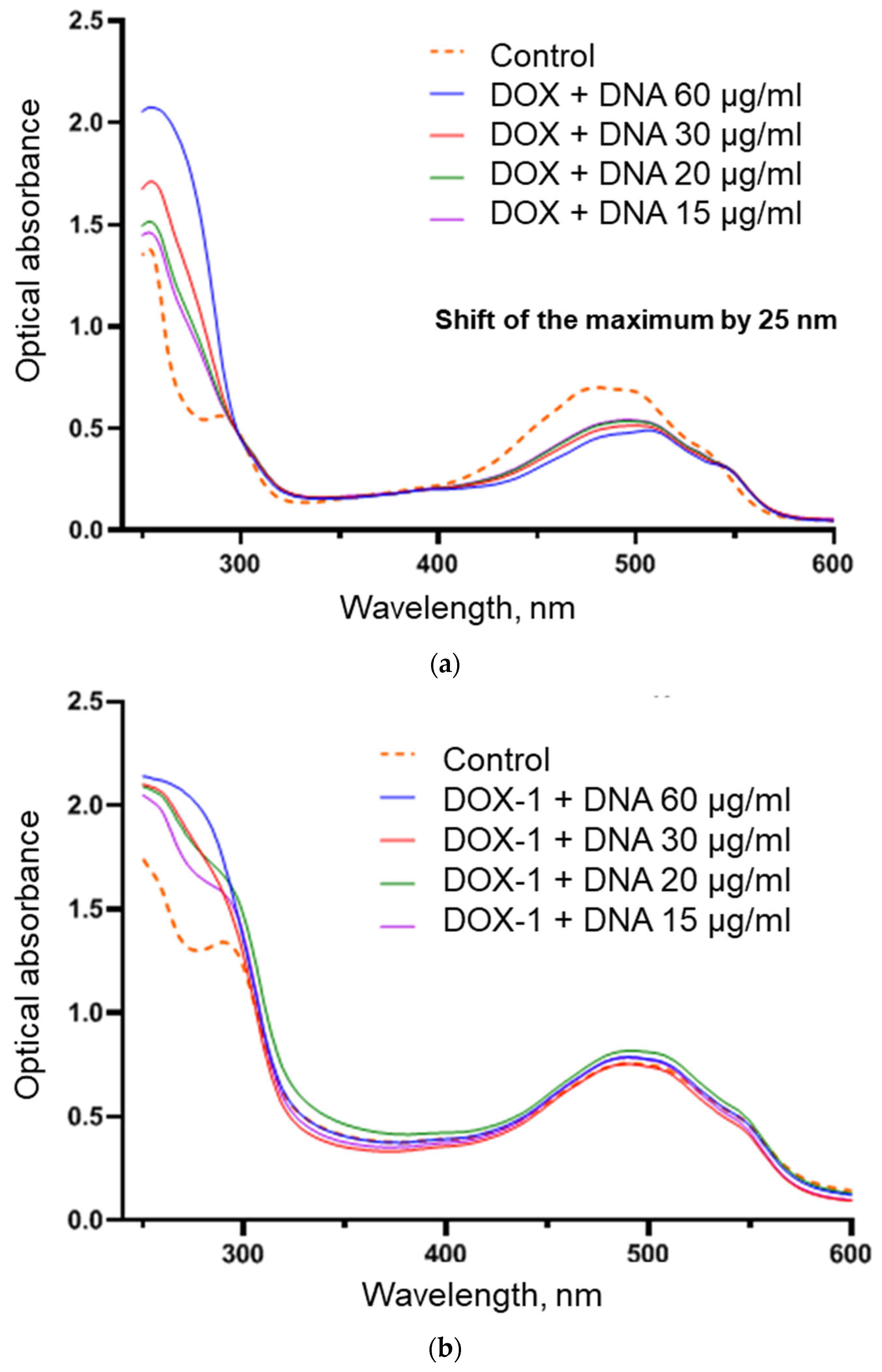
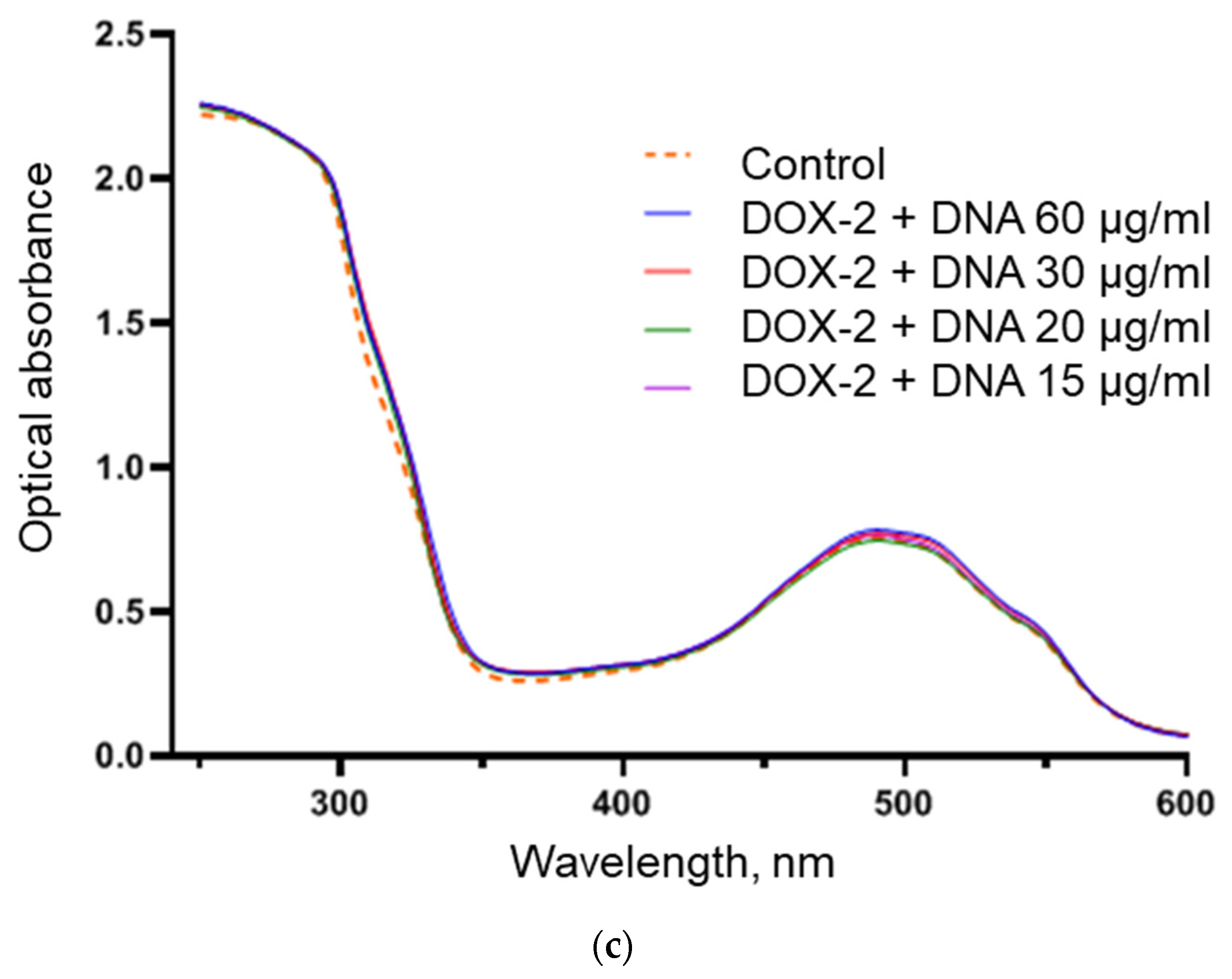


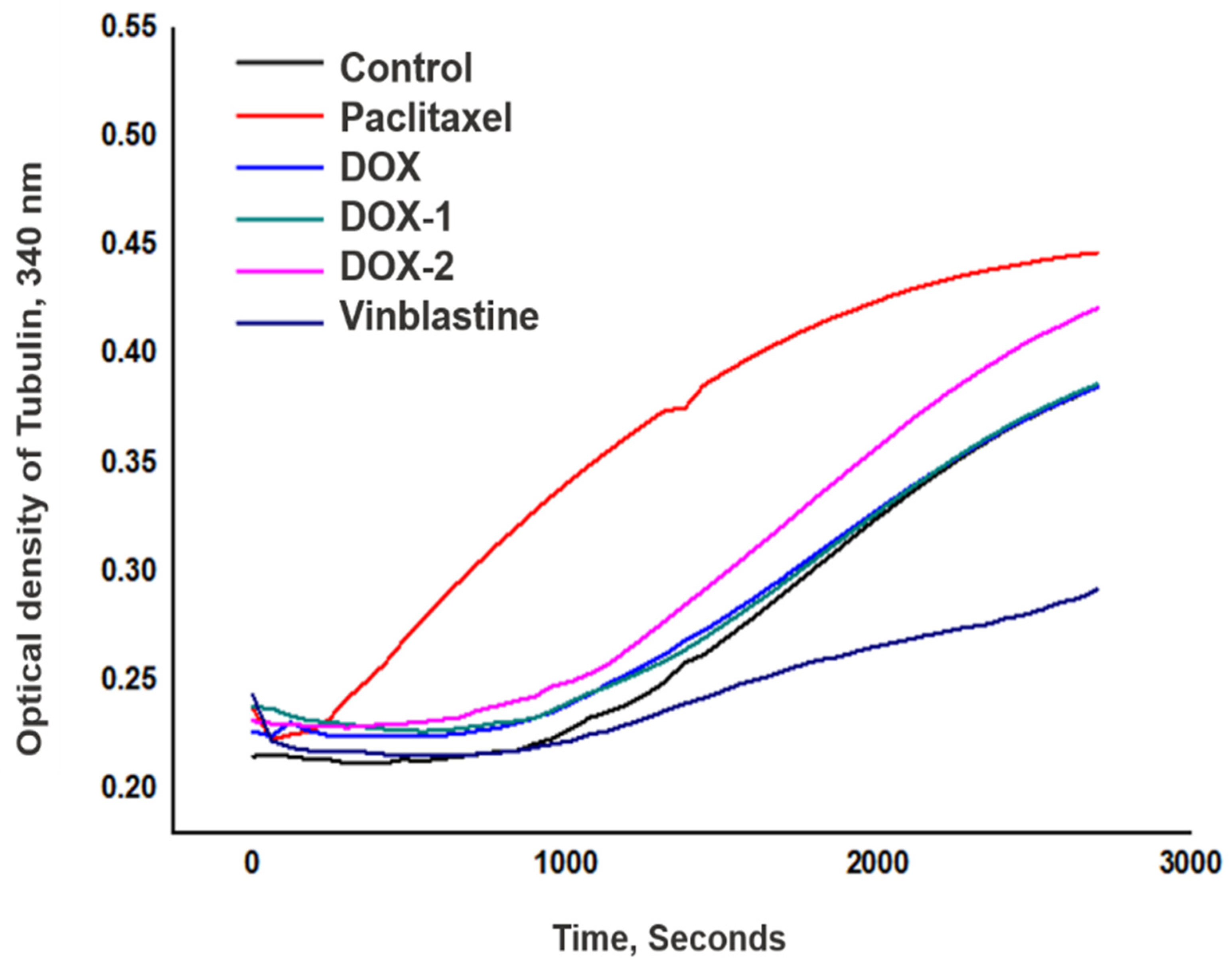

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}