

Novel Pharmacological Targets of Post-Traumatic Stress Disorders

 ,
,  ,
, 
 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
3. Pharmacological Treatment of PTSD
4. Novel Pharmacological Targets in PTSD
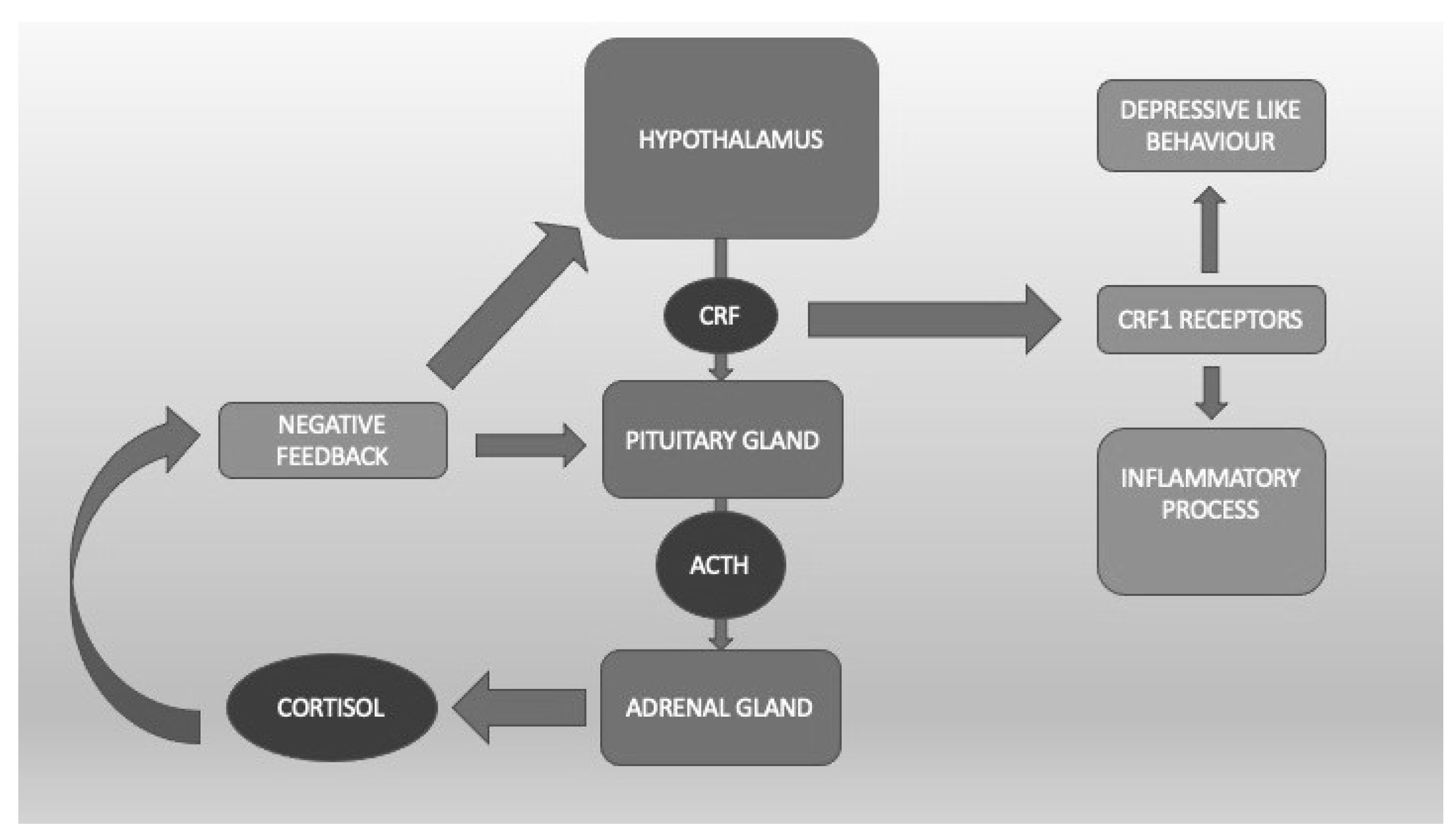
4.1. The Hypothalamic-Pituitary-Adrenal Axis
4.2. Opioid Peptides
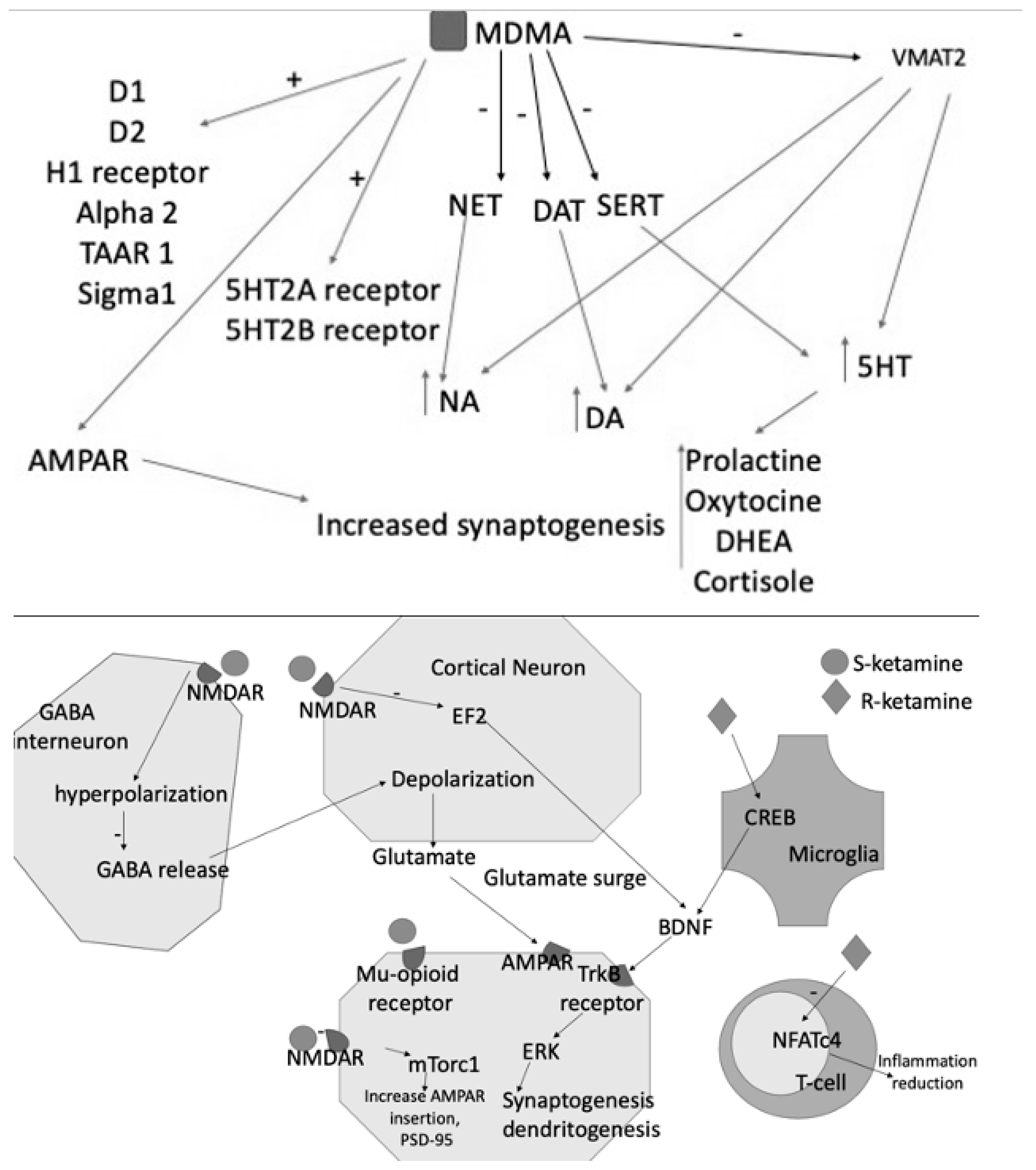
4.3. Glutamate
4.4. Cannabinoids
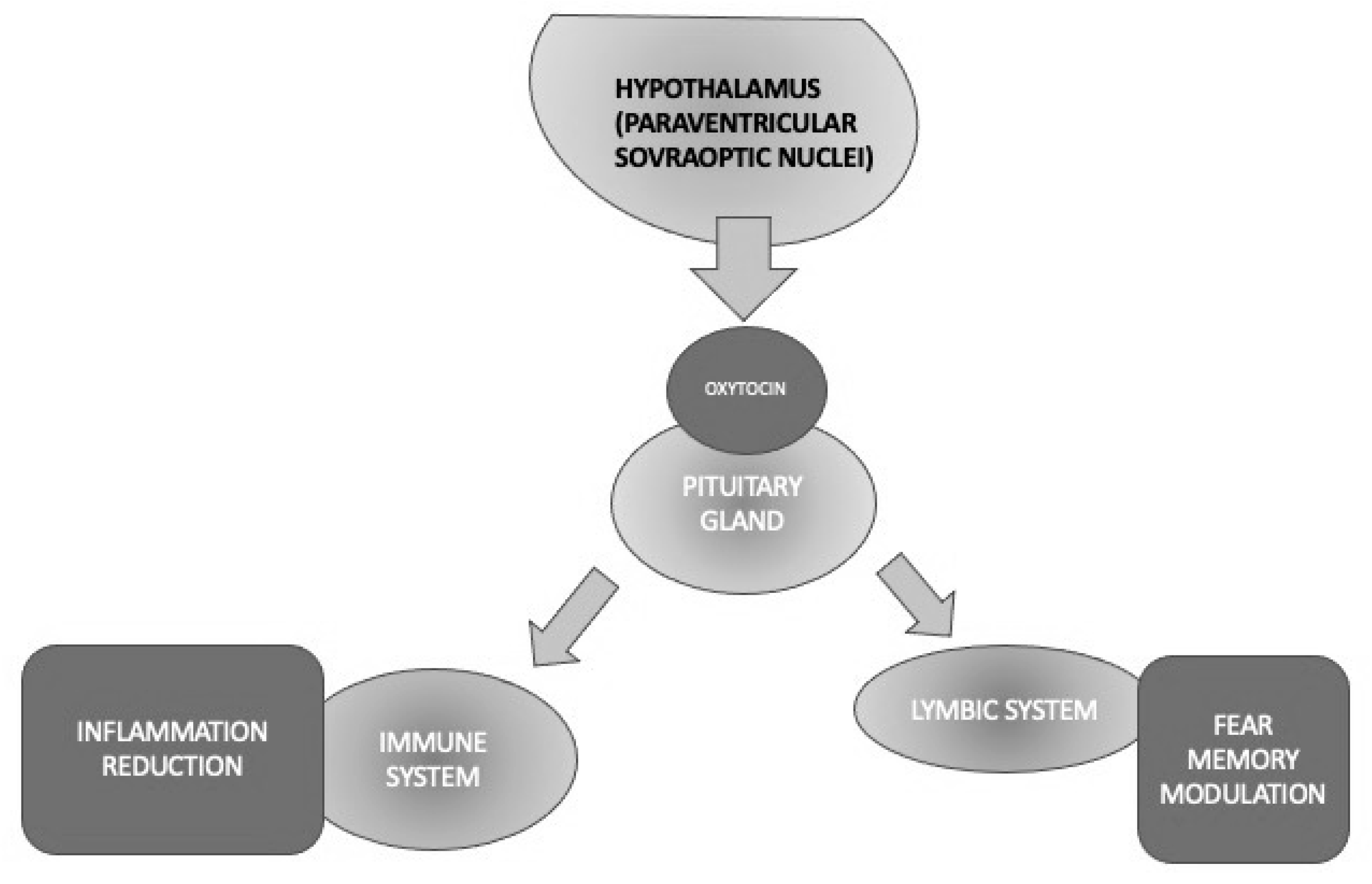
4.5. Oxytocin
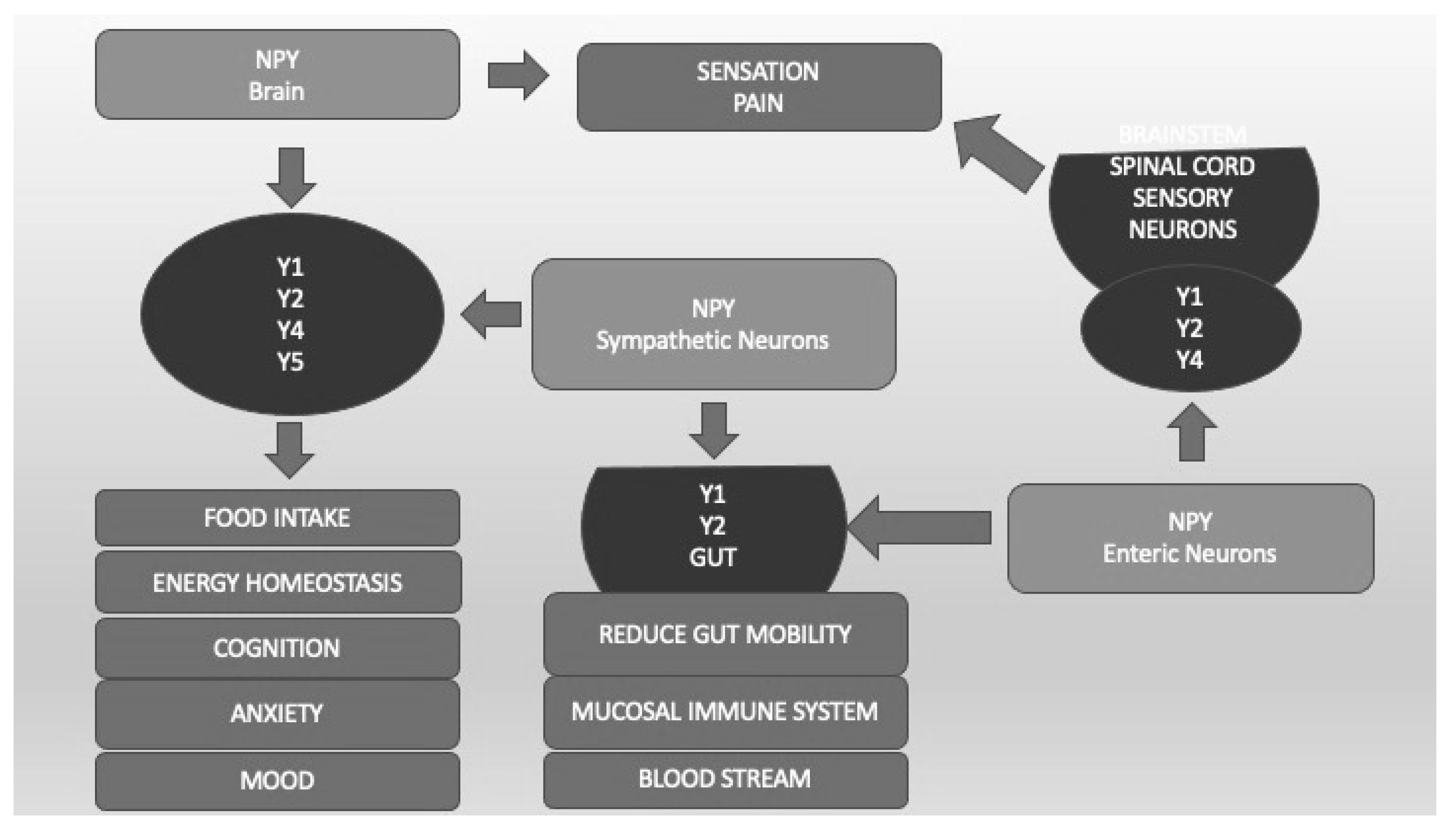
4.6. Neuropeptide Y
4.7. MicroRNA
4.8. Pipeline of Currently Studied Drugs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Text Revision DSM-5-TR, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2022. [Google Scholar]
- Barbara Young Welke. Recasting American Liberty: Gender, Race, Law, and the Railroad Revolution, 1865–1920; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2001. [Google Scholar]
- Birmes, P.; Hatton, L.; Brunet, A.; Schmitt, L. Early historical literature for post-traumatic symptomatology. Stress Health 2003, 19, 17–26. [Google Scholar] [CrossRef]
- Jones, E. Historical approaches to post-combat disorders. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Hoge, C.W.; McFarlane, A.C.; Vermetten, E.; Lanius, R.A.; Nievergelt, C.M.; Hobfoll, S.E.; Koenen, K.C.; Neylan, T.C.; Hyman, S.E. Post-traumatic stress disorder. Nat. Rev. Dis. Primers 2015, 1, 15057. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C. Posttraumatic stress disorder in the National Comorbidity Survey. Arch. Gen. Psychiatry 1995, 52, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Brewin, C.R.; Andrews, B.; Valentine, J.D. Meta-analysis of risk factors for posttraumatic stress disorder in trauma-exposed adults. J. Consult. Clin. Psychol. 2000, 68, 748–766. [Google Scholar] [CrossRef] [PubMed]
- Tull, M.T.; Forbes, C.N.; Weiss, N.H.; Gratz, K.L. An investigation of the effect of trauma script exposure on risk-taking among patients with substance use disorders and posttraumatic stress disorder. J. Anxiety Disord. 2019, 62, 77–85. [Google Scholar] [CrossRef]
- Sundin, J.; Herrell, R.K.; Hoge, C.W.; Fear, N.T.; Adler, A.B.; Greenberg, N.; Riviere, L.A.; Thomas, J.L.; Wessely, S.; Bliese, P.D. Mental health outcomes in US and UK military personnel returning from Iraq. Br. J. Psychiatry 2014, 204, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.; Frijling, J.L.; van Zuiden, M. Posttraumatische stressstoornis [Posttraumatic stress disorder: Current insights in diagnostics, treatment and prevention]. Ned Tijdschr Geneeskd. 2018, 161, D1905. (In Dutch) [Google Scholar]
- Fox, V.; Dalman, C.; Dal, H.; Hollander, A.-C.; Kirkbride, J.B.; Pitman, A. Suicide risk in people with post-traumatic stress disorder: A cohort study of 3.1 million people in Sweden. J. Affect. Disord. 2020, 279, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.S.; Gradus, J.L.; Keyes, K.M.; Calabrese, J.R.; Liberzon, I.; Tamburrino, M.B.; Cohen, G.H.; Sampson, L.; Galea, S. Subthreshold PTSD and PTSD in a prospective-longitudinal cohort of military personnel: Potential targets for preventive interventions. Depress. Anxiety 2018, 35, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Ressler, K.J.; Berretta, S.; Bolshakov, V.Y.; Rosso, I.M.; Meloni, E.G.; Rauch, S.L.; Carlezon, W.A. Post-traumatic stress disorder: Clinical and translational neuroscience from cells to circuits. Nat. Rev. Neurol. 2022, 18, 273–288. [Google Scholar] [CrossRef]
- de Silva, V.A.; Jayasekera, N.; Hanwella, R. Cannabis Use among Navy personnel in Sri Lanka: A cross sectional study. BMC Res. Notes 2016, 9, 174. [Google Scholar] [CrossRef]
- Kelmendi, B.; Adams, T.G.; Yarnell, S.; Southwick, S.; Abdallah, C.G.; Krystal, J.H. PTSD: From neurobiology to pharmacological treatments. Eur. J. Psychotraumatol. 2016, 7, 31858. [Google Scholar] [CrossRef] [PubMed]
- Snijders, C.; de Nijs, L.; Baker, D.G.; Hauger, R.L.; Hove, D.v.D.; Kenis, G.; Nievergelt, C.M.; Boks, M.P.; Vermetten, E.; Gage, F.H.; et al. MicroRNAs in post-traumatic stress disorder. Behav. Neurobiol. PTSD 2017, 38, 23–46. [Google Scholar] [CrossRef]
- Dunlop, B.W.; Wong, A. The hypothalamic-pituitary-adrenal axis in PTSD: Pathophysiology and treatment interventions. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 89, 361–379. [Google Scholar] [CrossRef]
- Nikbakhtzadeh, M.; Borzadaran, F.M.; Zamani, E.; Shabani, M. Protagonist role of opioidergic system on post-traumatic stress disorder and associated pain. Psychiatry Investig. 2020, 17, 506–516. [Google Scholar] [CrossRef]
- NICE. Overview|Post-Traumatic Stress Disorder|Guidance|NICE. Nice.org.uk. Available online: https://www.nice.org.uk/guidance/ng116 (accessed on 4 August 2023).
- Marazziti, D. Clinical Psychopharmacotherapy, 6th ed.; Giovanni Fioriti Editore: Roma, Italy, 2020. [Google Scholar]
- Harpaz-Rotem, I.; Rosenheck, R.A.; Mohamed, S.; Desai, R.A. Pharmacologic treatment of posttraumatic stress disorder among privately insured Americans. Psychiatr. Serv. 2008, 59, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, M.; Pearce, J.; Bethell, A.; Dankova, L.; Barbui, C.; Tol, W.A.; Van Ommeren, M.; De Jong, J.; Seedat, S.; Chen, H.; et al. Pharmacotherapy for post-traumatic stress disorder: Systematic review and meta-analysis. Br. J. Psychiatry 2015, 206, 93–100. [Google Scholar] [CrossRef] [PubMed]
- McRae, A.L.; Brady, K.T.; Mellman, T.A.; Sonne, S.C.; Killeen, T.K.; Timmerman, M.A.; Bayles-Dazet, W. Comparison of nefazodone and sertraline for the treatment of posttraumatic stress disorder. Depress. Anxiety 2004, 19, 190–196. [Google Scholar] [CrossRef]
- Jonas, D.E.; Cusack, K.; Forneris, C.A.; Wilkins, T.M.; Sonis, J.; Middleton, J.C.; Feltner, C.; Meredith, D.; Cavanaugh, J.; Brownley, K.A.; et al. Psychological and Pharmacological Treatments for Adults with Posttraumatic Stress Disorder (PTSD); Report No.: 13-EHC011-EF; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2013. [Google Scholar]
- Ehret, M. Treatment of posttraumatic stress disorder: Focus on pharmacotherapy. Ment. Health Clin. 2019, 9, 373–382. [Google Scholar] [CrossRef]
- Stein, D.; Zungu-Dirwayi, N.; van der Linden, G.; Seedat, S. Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database Syst. Rev. 2000, 4, CD002795. [Google Scholar] [CrossRef]
- Sripada, R.K.; Rauch, S.A.M.; Liberzon, I. Psychological mechanisms of PTSD and its treatment. Curr. Psychiatry Rep. 2016, 18, 99. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Abdallah, C.G.; Averill, L.A.; Kelmendi, B.; Harpaz-Rotem, I.; Sanacora, G.; Southwick, S.M.; Duman, R.S. Synaptic loss and the pathophysiology of PTSD: Implications for ketamine as a prototype novel therapeutic. Curr. Psychiatry Rep. 2017, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Asnis, G.M.; Kohn, S.R.; Henderson, M.; Brown, N.L. SSRIs versus non-SSRIs in post-traumatic stress disorder: An update with recommendations. Drugs 2004, 64, 383–404. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.E.; Hertzberg, M.A.; Moore, S.D.; Dennis, M.F.; Bukenya, D.S.; Beckham, J.C. A Placebo-controlled trial of bupropion sr in the treatment of chronic posttraumatic stress disorder. J. Clin. Psychopharmacol. 2007, 27, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Raskind, M.A.; Thompson, C.; Petrie, E.C.; Dobie, D.J.; Rein, R.J.; Hoff, D.J.; McFall, M.E.; Peskind, E.R. Prazosin reduces nightmares in combat veterans with posttraumatic stress disorder. J. Clin. Psychiatry 2002, 63, 565–568. [Google Scholar] [CrossRef]
- Raskind, M.A.; Peskind, E.R.; Kanter, E.D.; Petrie, E.C.; Radant, A.; Thompson, C.E.; Dobie, D.J.; Hoff, D.; Rein, R.J.; Straits-Tröster, K.; et al. Reduction of nightmares and other ptsd symptoms in combat veterans by prazosin: A placebo-controlled study. Am. J. Psychiatry 2003, 160, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.; Hamner, M.B.; Ulmer, H.G.; Lorberbaum, J.P.; Durkalsk, V.L. Open-label trial of escitalopram in the treatment of posttraumatic stress disorder. J. Clin. Psychiatry 2006, 67, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Pietrzak, R.H.; Rosenheck, R.A.; Cramer, J.A.; Vessicchio, J.; Jones, K.M.; Huang, G.D.; Vertrees, J.E.; Collins, J.; Krystal, A.D.; et al. Sleep disturbance in chronic military-related PTSD. J. Clin. Psychiatry 2016, 77, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Breen, A.; Blankley, K.; Fine, J. The efficacy of prazosin for the treatment of posttraumatic stress disorder nightmares in U.S. military veterans. J. Am. Assoc. Nurse Pract. 2017, 29, 65–69. [Google Scholar] [CrossRef]
- Gamache, K.; Pitman, R.K.; Nader, K. Preclinical evaluation of reconsolidation blockade by clonidine as a potential novel treatment for posttraumatic stress disorder. Neuropsychopharmacology 2012, 37, 2789–2796. [Google Scholar] [CrossRef]
- Burek, G.A.; Waite, M.R.; Heslin, K.; Liewen, A.K.; Yaqub, T.M.; Larsen, S.E. Low-dose clonidine in veterans with posttraumatic stress disorder. J. Psychiatr. Res. 2021, 137, 480–485. [Google Scholar] [CrossRef]
- Davis, L.L.; Davidson, J.R.T.; Ward, L.C.; Bartolucci, A.; Bowden, C.L.; Petty, F. Divalproex in the treatment of posttraumatic stress disorder. J. Clin. Psychopharmacol. 2008, 28, 84–88. [Google Scholar] [CrossRef]
- Wang, H.R.; Woo, Y.S.; Bahk, W.-M. anticonvulsants to treat post-traumatic stress disorder. Hum. Psychopharmacol. Clin. Exp. 2014, 29, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Guina, J.; Rossetter, S.R.; Derhodes, B.J.; Nahhas, R.W.; Welton, R.S. Benzodiazepines for PTSD. J. Psychiatr. Pract. 2015, 21, 281–303. [Google Scholar] [CrossRef]
- Charney, M.E.; Hellberg, S.N.; Bui, E.; Simon, N.M. Evidenced-based treatment of post-traumatic stress disorder: An updated review of validated psychotherapeutic and pharmacological approaches. Harv. Rev. Psychiatry 2018, 26, 99–115. [Google Scholar] [CrossRef]
- Mahan, A.L.; Ressler, K.J. Fear Conditioning, synaptic plasticity and the amygdala: Implications for posttraumatic stress disorder. Trends Neurosci. 2012, 35, 24–35. [Google Scholar] [CrossRef]
- Careaga, M.B.L.; Girardi, C.E.N.; Suchecki, D. Understanding posttraumatic stress disorder through fear conditioning, extinction and reconsolidation. Neurosci. Biobehav. Rev. 2016, 71, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kim, J.; Tonegawa, S. Amygdala reward neurons form and store fear extinction memory. Neuron 2020, 105, 1077–1093.e7. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.G.; Marazziti, D.; Diep, P.-T.; Carter, S. Oxytocin: An old hormone, a novel psychotropic drug and its possible use in treating psychiatric disorders. Curr. Med. Chem. 2022, 29, 5615–5687. [Google Scholar] [CrossRef]
- Heinrichs, S.C.; Lapsansky, J.; Lovenberg, T.W.; De Souza, E.B.; Chalmers, D.T. Corticotropin-releasing factor CRF1, but not CRF2, receptors mediate anxiogenic-like behavior. Regul. Pept. 1997, 71, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Cain, C.K.; Maynard, G.D.; Kehne, J.H. Targeting memory processes with drugs to prevent or cure PTSD. Expert Opin. Investig. Drugs 2012, 21, 1323–1350. [Google Scholar] [CrossRef]
- Juruena, M.F. Early-Life Stress and HPA axis trigger recurrent adulthood depression. Epilepsy Behav. 2014, 38, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Juruena, M.F.; Eror, F.; Cleare, A.J.; Young, A.H. The role of early life stress in HPA axis and anxiety. Adv. Exp. Med. Biol. 2020, 1191, 141–153. [Google Scholar] [CrossRef]
- Murphy, F.; Nasa, A.; Cullinane, D.; Raajakesary, K.; Gazzaz, A.; Sooknarine, V.; Haines, M.; Roman, E.; Kelly, L.; O’Neill, A.; et al. Childhood trauma, the HPA axis and psychiatric illnesses: A targeted literature synthesis. Front. Psychiatry 2022, 13, 748372. [Google Scholar] [CrossRef]
- Hauger, R.L.; Olivares-Reyes, J.A.; Dautzenberg, F.M.; Lohr, J.B.; Braun, S.; Oakley, R.H. Molecular and cell signaling targets for ptsd pathophysiology and pharmacotherapy. Neuropharmacology 2012, 62, 705–714. [Google Scholar] [CrossRef]
- Kehne, J. The CRF1 receptor, a novel target for the treatment of depression, anxiety, and stress-related disorders. CNS Neurol. Disord. Drug Targets 2007, 6, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Gutman, D.A.; Owens, M.J.; Thrivikraman, K.V.; Nemeroff, C.B. Persistent anxiolytic affects after chronic administration of the CRF1 receptor antagonist R121919 in rats. Neuropharmacology 2011, 60, 1135–1141. [Google Scholar] [CrossRef]
- Ising, M.; Zimmermann, U.S.; Künzel, H.E.; Uhr, M.; Foster, A.C.; Learned-Coughlin, S.M.; Holsboer, F.; Grigoriadis, D.E. High-Affinity CRF1 receptor antagonist NBI-34041: Preclinical and clinical data suggest safety and efficacy in attenuating elevated stress response. Neuropsychopharmacology 2007, 32, 1941–1949. [Google Scholar] [CrossRef]
- Almeida, F.B.; Pinna, G.; Barros, H.M.T. The Role of HPA Axis and Allopregnanolone on the Neurobiology of Major Depressive Disorders and PTSD. Int. J. Mol. Sci. 2021, 22, 5495. [Google Scholar] [CrossRef]
- Shang, Y.; Filizola, M. Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur. J. Pharmacol. 2015, 763, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Emery, M.A.; Eitan, S. Members of the same pharmacological family are not alike: Different opioids, different consequences, hope for the opioid crisis? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 428–449. [Google Scholar] [CrossRef]
- Moore, K.A.; Werner, C.; Zannelli, R.M.; Levine, B.; Smith, M.L. Screening postmortem blood and tissues for nine cases of drugs of abuse using automated microplate immunoassay. Forensic Sci. Int. 1999, 106, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Adem, A.; Madjid, N.; Kahl, U.; Holst, S.; Sadek, B.; Sandin, J.; Terenius, L.; Ögren, S.O. Nociceptin and the NOP receptor in aversive learning in mice. Eur. Neuropsychopharmacol. 2017, 27, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Saxe, G.; Stoddard, F.; Courtney, D.; Cunningham, K.; Chawla, N.; Sheridan, R.; King, D.; King, L. Relationship between acute morphine and the course of PTSD in children with burns. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, R.A.; Creamer, M.; O’Donnell, M.; Silove, D.; McFarlane, A.C. A study of the protective function of acute morphine administration on subsequent posttraumatic stress disorder. Biol. Psychiatry 2009, 65, 438–440. [Google Scholar] [CrossRef]
- Likhtik, E.; Popa, D.; Apergis-Schoute, J.; Fidacaro, G.A.; Paré, D. Amygdala intercalated neurons are required for expression of fear extinction. Nature 2008, 454, 642–645. [Google Scholar] [CrossRef]
- Seal, K.H.; Shi, Y.; Cohen, G.; Cohen, B.E.; Maguen, S.; Krebs, E.E.; Neylan, T.C. Association of mental health disorders with prescription opioids and high-risk opioid use in US veterans of Iraq and Afghanistan. JAMA 2012, 307, 940–947. [Google Scholar] [CrossRef]
- Shiner, B.; Leonard Westgate, C.; Bernardy, N.C.; Schnurr, P.P.; Watts, B.V. Trends in opioid use disorder diagnoses and medication treatment among veterans with posttraumatic stress disorder. J. Dual Diagn. 2017, 13, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Kelley, A.T.; Greenstone, C.L.; Kirsh, S.R. Defining access and the role of community care in the veterans health administration. J. Gen. Intern. Med. 2019, 35, 1584–1585. [Google Scholar] [CrossRef]
- Stoddard, F.J.; Sorrentino, E.A.; Ceranoglu, T.A.; Saxe, G.; Murphy, J.M.; Drake, J.E.; Ronfeldt, H.; White, G.W.; Kagan, J.; Snidman, N.; et al. Preliminary evidence for the effects of morphine on posttraumatic stress disorder symptoms in one- to four-year-olds with burns. J. Burn Care Res. 2009, 30, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Norman, S.B.; Inaba, R.K.; Smith, T.L.; Brown, S.A. Development of the PTSD-Alcohol Expectancy Questionnaire. Addict. Behav. 2008, 33, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Porto, G.P.; Milanesi, L.H.; Rubin, M.A.; Mello, C.F. Effect of morphine on the persistence of long-term memory in rats. Psychopharmacology 2014, 232, 1747–1753. [Google Scholar] [CrossRef]
- Madison, C.A.; Eitan, S. Buprenorphine: Prospective novel therapy for depression and PTSD. Psychol. Med. 2020, 50, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Takano, T.; Hansen, A.J. Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 2002, 3, 748–755. [Google Scholar] [CrossRef]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef] [PubMed]
- Riedel, G. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zhang, Y.; Zhang, J.; Wang, H.; Ren, B. Glutamate receptors and signal transduction in learning and memory. Mol. Biol. Rep. 2010, 38, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.B.; Singewald, N. Novel pharmacological targets in drug development for the treatment of anxiety and anxiety-related disorders. Pharmacol. Ther. 2019, 204, 107402. [Google Scholar] [CrossRef] [PubMed]
- Rasmusson, A.M.; Marx, C.E.; Pineles, S.L.; Locci, A.; Scioli-Salter, E.R.; Nillni, Y.I.; Liang, J.J.; Pinna, G. Neuroactive steroids and PTSD treatment. Neurosci. Lett. 2017, 649, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Canuso, C.M.; Singh, J.B.; Fedgchin, M.; Alphs, L.; Lane, R.; Lim, P.; Pinter, C.; Hough, D.; Sanacora, G.; Manji, H.; et al. Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: Results of a double-blind, randomized, placebo-controlled study. Am. J. Psychiatry 2018, 175, 620–630. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Roache, J.D.; Gueorguieva, R.; Averill, L.A.; Young-McCaughan, S.; Shiroma, P.R.; Purohit, P.; Brundige, A.; Murff, W.; Ahn, K.-H.; et al. Dose-related effects of ketamine for antidepressant-resistant symptoms of posttraumatic stress disorder in veterans and active duty military: A double-blind, randomized, placebo-controlled multi-center clinical trial. Neuropsychopharmacology 2022, 47, 1574–1581. [Google Scholar] [CrossRef]
- Asim, M.; Wang, B.; Hao, B.; Wang, X. Ketamine for post-traumatic stress disorders and it’s possible therapeutic mechanism. Neurochem. Int. 2021, 146, 105044. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Han, R.; Niu, K.; Xu, J.; Zhao, Z.; Lu, G.; Shang, Y. The multivariate effect of ketamine on PTSD: Systematic review and meta-analysis. Front. Psychiatry 2022, 13, 813803. [Google Scholar] [CrossRef]
- Bentley, S.; Artin, H.; Mehaffey, E.; Liu, F.; Sojourner, K.; Bismark, A.; Printz, D.; Lee, E.E.; Martis, B.; De Peralta, S.; et al. Response to intravenous racemic ketamine after switch from intranasal (S)-ketamine on symptoms of treatment-resistant depression and post-traumatic stress disorder in Veterans: A retrospective case series. Pharmacotherapy 2022, 42, 272–279. [Google Scholar]
- Garcia, M.A.; Junglen, A.; Ceroni, T.; Johnson, D.; Ciesla, J.; Delahanty, D.L. The mediating impact of PTSD symptoms on cortisol awakening response in the context of intimate partner violence. Biol. Psychol. 2020, 152, 107873. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Bogenschutz, M.; Lilienstein, A.; Harrison, C.; Kleiman, S.; Parker-Guilbert, K.; Ot’alora, M.; Garas, W.; Paleos, C.; Gorman, I.; et al. MDMA-assisted therapy for severe PTSD: A randomized, double-blind, placebo-controlled phase 3 study. Nat. Med. 2021, 27, 1025–1033. [Google Scholar] [CrossRef]
- Anneken, J.H.; Cunningham, J.I.; Collins, S.A.; Yamamoto, B.K.; Gudelsky, G.A. MDMA increases glutamate release and reduces parvalbumin-positive gabaergic cells in the dorsal hippocampus of the rat: Role of cyclooxygenase. J. Neuroimmune Pharmacol. 2012, 8, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Reiff, C.M.; McDonald, W.M. MDMA-assisted psychotherapy for the treatment of PTSD. Braz. J. Psychiatry 2021, 43, 123–124. [Google Scholar] [CrossRef]
- Vargas, A.S.; Luís, Â.; Barroso, M.; Gallardo, E.; Pereira, L. Psilocybin as a New Approach to Treat Depression and Anxiety in the Context of Life-Threatening Diseases-A Systematic Review and Meta-Analysis of Clinical Trials. Biomedicines 2020, 8, 331. [Google Scholar] [PubMed]
- Zanikov, T.; Gerasymchuk, M.; Ghasemi Gojani, E.; Robinson, G.I.; Asghari, S.; Groves, A.; Haselhorst, L.; Nandakumar, S.; Stahl, C.; Cameron, M.; et al. The Effect of Combined Treatment of Psilocybin and Eugenol on Lipopolysaccharide-Induced Brain Inflammation in Mice. Molecules 2023, 28, 2624. [Google Scholar] [PubMed]
- Davis, A.K.; Levin, A.W.; Nagib, P.B.; Armstrong, S.B.; Lancelotta, R.L. Study Protocol of an Open-Label Proof-of-Concept Trial Examining the Safety and Clinical Efficacy of Psilocybin-Assisted Therapy for Veterans with PTSD. BMJ Open 2023, 13, e068884. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.; Mackie, K. An Introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Hauer, D.; Schelling, G.; Gola, H.; Campolongo, P.; Morath, J.; Roozendaal, B.; Hamuni, G.; Karabatsiakis, A.; Atsak, P.; Vogeser, M.; et al. Plasma concentrations of endocannabinoids and related primary fatty acid amides in patients with post-traumatic stress disorder. PLoS ONE 2013, 8, e62741. [Google Scholar] [CrossRef]
- Morena, M.; Patel, S.; Bains, J.S.; Hill, M.N. Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 2016, 41, 80–102. [Google Scholar] [CrossRef] [PubMed]
- Leen, N.A.; de Weijer, A.D.; van Rooij, S.J.H.; Kennis, M.; Baas, J.M.P.; Geuze, E. The role of the endocannabinoids 2-AG and anandamide in clinical symptoms and treatment outcome in veterans with PTSD. Chronic Stress 2022, 6, 24705470221107290. [Google Scholar] [CrossRef]
- Bitencourt, R.M.; Takahashi, R.N. cannabidiol as a therapeutic alternative for post-traumatic stress disorder: From bench research to confirmation in human trials. Front. Neurosci. 2018, 12, 502. [Google Scholar] [CrossRef]
- Jurkus, R.; Day, H.L.L.; Guimarães, F.S.; Lee, J.L.C.; Bertoglio, L.J.; Stevenson, C.W. Cannabidiol regulation of learned fear: Implications for treating anxiety-related disorders. Front. Pharmacol. 2016, 7, 454. [Google Scholar] [CrossRef]
- Korem, N.; Zer-Aviv, T.M.; Ganon-Elazar, E.; Abush, H.; Akirav, I. Targeting the endocannabinoid system to treat anxiety-related disorders. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 193–202. [Google Scholar] [CrossRef]
- El-Solh, A.A. Management of nightmares in patients with posttraumatic stress disorder: Current perspectives. Nat. Sci. Sleep 2018, 10, 409–420. [Google Scholar] [CrossRef]
- Cowling, T.; MacDougall, D. Nabilone for the Treatment of Post-Traumatic Stress Disorder: A Review of Clinical Effectiveness and Guidelines [Internet]; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2019. [Google Scholar]
- Ney, L.J.; Matthews, A.; Bruno, R.; Felmingham, K.L. Cannabinoid interventions for PTSD: Where to next? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 93, 124–140. [Google Scholar] [CrossRef]
- Orsolini, L.; Chiappini, S.; Volpe, U.; De Berardis, D.; Latini, R.; Papanti, G.D.; Corkery, J.M. Use of medicinal cannabis and synthetic cannabinoids in post-traumatic stress disorder (PTSD): A systematic review. Medicina 2019, 55, 525. [Google Scholar] [CrossRef] [PubMed]
- Bolsoni, L.M.; Crippa, J.A.S.; Hallak, J.E.C.; Guimarães, F.S.; Zuardi, A.W. Effects of cannabidiol on symptoms induced by the recall of traumatic events in patients with posttraumatic stress disorder. Psychopharmacology 2022, 239, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Froemke, R.C.; Young, L.J. Oxytocin, neural plasticity, and social behavior. Annu. Rev. Neurosci. 2021, 44, 359–381. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S. Oxytocin pathways and the evolution of human behavior. Annu. Rev. Psychol. 2014, 65, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Marsh, N.; Marsh, A.A.; Lee, M.R.; Hurlemann, R. Oxytocin and the neurobiology of prosocial behavior. Neuroscientist 2020, 27, 604–619. [Google Scholar] [CrossRef]
- Petersson, M.; Hulting, A.-L.; Uvnäs-Moberg, K. oxytocin causes a sustained decrease in plasma levels of corticosterone in rats. Neurosci. Lett. 1999, 264, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Petersson, M.; Uvnäs-Moberg, K. Systemic oxytocin treatment modulates glucocorticoid and mineralocorticoid receptor mRNA in the rat hippocampus. Neurosci. Lett. 2003, 343, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Young, L.J.; Newport, D.J.; Mletzko, T.; Miller, A.H.; Nemeroff, C.B. Lower CSF oxytocin concentrations in women with a history of childhood abuse. Mol. Psychiatry 2008, 14, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Chatzittofis, A.; Nordström, P.; Uvnäs-Moberg, K.; Asberg, M.; Jokinen, J. CSF and plasma oxytocin levels in suicide attempters, the role of childhood trauma and revictimization. Neuro Endocrinol. Lett. 2014, 35, 213–217. [Google Scholar] [PubMed]
- Mohiyeddini, C.; Opacka-Juffry, J.; Gross, J.J. Emotional suppression explains the link between early life stress and plasma oxytocin. Anxiety Stress Coping 2014, 27, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, R.; Fujiwara, T. Association of oxytocin level and less severe forms of childhood maltreatment history among healthy Japanese adults involved with child care. Front. Behav. Neurosci. 2015, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Carmassi, C.; Marazziti, D.; Mucci, F.; Della Vecchia, A.; Barberi, F.M.; Baroni, S.; Giannaccini, G.; Palego, L.; Massimetti, G.; Dell’osso, L. Decreased plasma oxytocin levels in patients with PTSD. Front. Psychol. 2021, 12, 612338. [Google Scholar] [CrossRef] [PubMed]
- Marazziti, D.; Baroni, S.; Mucci, F.; Piccinni, A.; Moroni, I.; Giannaccini, G.; Carmassi, C.; Massimetti, E.; Dell’osso, L. Sex-related differences in plasma oxytocin levels in humans. Clin. Pract. Epidemiol. Ment. Health 2019, 15, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Frijling, J.L.; van Zuiden, M.; Nawijn, L.; Koch, S.B.J.; Neumann, I.D.; Veltman, D.J.; Olff, M. Salivary oxytocin and vasopressin levels in police officers with and without post-traumatic stress disorder. J. Neuroendocrinol. 2015, 27, 743–751. [Google Scholar] [CrossRef]
- Preti, A.; Melis, M.; Siddi, S.; Vellante, M.; Doneddu, G.; Fadda, R. Oxytocin and autism: A systematic review of randomized controlled trials. J. Child Adolesc. Psychopharmacol. 2014, 24, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, K.; Herpertz, S.C. Oxytocin and borderline personality disorder. Curr. Top. Behav. Neurosci. 2017, 35, 499–514. [Google Scholar] [CrossRef]
- Yamasue, H.; Domes, G. Oxytocin and autism spectrum disorders. Behav. Pharmacol. Neuropept. Oxytocin 2017, 35, 449–465. [Google Scholar] [CrossRef]
- King, C.E.; Gano, A.; Becker, H.C. The role of oxytocin in alcohol and drug abuse. Brain Res. 2020, 1736, 146761. [Google Scholar] [CrossRef]
- Goh, K.K.; Chen, C.-H.; Lane, H.-Y. Oxytocin in schizophrenia: Pathophysiology and implications for future treatment. Int. J. Mol. Sci. 2021, 22, 2146. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.B.; Marathe, P.A.; van Eijk, L.; Eri, R.; Ravindran, M.; Benedek, D.M.; Ursano, R.J.; Canales, J.J.; Johnson, L.R. Diverse therapeutic developments for post-traumatic stress disorder (PTSD) indicate common mechanisms of memory modulation. Pharmacol. Ther. 2022, 239, 108195. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.B.J.; van Zuiden, M.; Nawijn, L.; Frijling, J.L.; Veltman, D.J.; Olff, M. Intranasal oxytocin as strategy for medication-enhanced psychotherapy of PTSD: Salience processing and fear inhibition processes. Psychoneuroendocrinology 2014, 40, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.C.; Hand, A.; Jarnecke, A.M.; Moran-Santa Maria, M.M.; Brady, K.T.; Joseph, J.E. Effects of oxytocin on working memory and executive control system connectivity in posttraumatic stress disorder. Exp. Clin. Psychopharmacol. 2018, 26, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Sippel, L.M.; Flanagan, J.C.; Holtzheimer, P.E.; Moran-Santa-Maria, M.M.; Brady, K.T.; Joseph, J.E. Effects of intranasal oxytocin on threat- and reward-related functional connectivity in men and women with and without childhood abuse-related PTSD. Psychiatry Res. Neuroimaging 2021, 317, 111368. [Google Scholar] [CrossRef] [PubMed]
- Schultebraucks, K.; Maslahati, T.; Wingenfeld, K.; Hellmann-Regen, J.; Kraft, J.; Kownatzki, M.; Behnia, B.; Ripke, S.; Otte, C.; Roepke, S. Intranasal oxytocin administration impacts the acquisition and consolidation of trauma-associated memories: A double-blind randomized placebo-controlled experimental study in healthy women. Neuropsychopharmacology 2022, 47, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Olff, M.; Langeland, W.; Witteveen, A.; Denys, D. A psychobiological rationale for oxytocin in the treatment of posttraumatic stress disorder. CNS Spectr. 2010, 15, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Kautz, M.; Charney, D.S.; Murrough, J.W. Neuropeptide Y, resilience, and PTSD therapeutics. Neurosci. Lett. 2017, 649, 164–169. [Google Scholar] [CrossRef]
- Tasan, R.; Verma, D.; Wood, J.; Lach, G.; Hörmer, B.; de Lima, T.; Herzog, H.; Sperk, G. The role of neuropeptide y in fear conditioning and extinction. Neuropeptides 2016, 55, 111–126. [Google Scholar] [CrossRef]
- Roth, T.L.; Zoladz, P.R.; Sweatt, J.D.; Diamond, D.M. Epigenetic modification of hippocampal BDNF DNA in adult rats in an animal model of post-traumatic stress disorder. J. Psychiatr. Res. 2011, 45, 919–926. [Google Scholar] [CrossRef]
- Mehta, D.; Bruenig, D.; Carrillo-Roa, T.; Lawford, B.; Harvey, W.; Morris, C.P.; Smith, A.K.; Binder, E.B.; Young, R.M.; Voisey, J. Genomewide DNA methylation analysis in combat veterans reveals a novel locus for PTSD. Acta Psychiatr. Scand. 2017, 136, 493–505. [Google Scholar] [CrossRef]
- Rutten, B.P.F.; Vermetten, E.; Vinkers, C.H.; Ursini, G.; Daskalakis, N.P.; Pishva, E.; De Nijs, L.; Houtepen, L.C.; Eijssen, L.; Jaffe, A.E.; et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol. Psychiatry 2017, 23, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Al Jowf, G.I.; Snijders, C.; Rutten, B.P.F.; de Nijs, L.; Eijssen, L.M.T. The molecular biology of susceptibility to post-traumatic stress disorder: Highlights of epigenetics and epigenomics. Int. J. Mol. Sci. 2021, 22, 10743. [Google Scholar] [CrossRef] [PubMed]
- Maurel, O.M.; Torrisi, S.A.; Barbagallo, C.; Purrello, M.; Salomone, S.; Drago, F.; Ragusa, M.; Leggio, G.M. Dysregulation of MiR-15a-5p, MiR-497a-5p and MiR-511-5p is associated with modulation of BDNF and FKBP5 in brain areas of PTSD-related susceptible and resilient mice. Int. J. Mol. Sci. 2021, 22, 5157. [Google Scholar] [CrossRef]
- Klengel, T.; Binder, E.B. Allele-specific epigenetic modification: A molecular mechanism for gene–environment interactions in stress-related psychiatric disorders? Epigenomics 2013, 5, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Li, L.P.; Dustrude, E.T.; Haulcomb, M.M.; Abreu, A.R.; Fitz, S.D.; Johnson, P.L.; Thakur, G.A.; Molosh, A.I.; Lai, Y.; Shekhar, A. PSD95 and NNOS interaction as a novel molecular target to modulate conditioned fear: Relevance to PTSD. Transl. Psychiatry 2018, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.P.; Li, X.; Maurer, V.; Oberhauser, M.; Gstir, R.; Wearick-Silva, L.E.; Viola, T.W.; Schafferer, S.; Grassi-Oliveira, R.; Whittle, N.; et al. MicroRNA-mediated rescue of fear extinction memory by miR-144-3p in extinction-impaired mice. BiolPsychiatry 2017, 81, 979–989. [Google Scholar]
- Griggs, E.M.; Young, E.J.; Rumbaugh, G.; Miller, C.A. MicroRNA-182 regu-lates amygdala-dependent memory formation. J. Neurosci. 2013, 33, 1734–1740. [Google Scholar] [PubMed]
- Chen, Y.-L.; Tong, L.; Chen, Y.; Fu, C.-H.; Peng, J.-B.; Ji, L.-L. MiR-153 downregulation alleviates PTSD-like behaviors and reduces cell apoptosis by upregulating the Sigma-1 receptor in the hippocampus of rats exposed to single-prolonged stress. Exp. Neurol. 2022, 352, 114034. [Google Scholar]
- Chen, Y.; An, Q.; Yang, S.-T.; Chen, Y.-L.; Tong, L.; Ji, L.-L. MicroRNA-124 attenuates PTSD like behaviors and reduces the level of inflammatory cytokines by downregulating the expression of TRAF6 in the hippocampus of rats following single prolonged stress. Exp. Neurol. 2022, 356, 114154. [Google Scholar]
- Zhou, J.; Nagarkatti, P.; Zhong, Y.; Ginsberg, J.P.; Singh, N.P.; Zhang, J.; Nagarkatti, M. Dysregulation in MicroRNA expression is associated with alterations in immune functions in combat veterans with post-traumatic stress disorder. PLoS ONE 2014, 9, e94075. [Google Scholar] [CrossRef]
- Gupta, S.; Guleria, R.S.; Szabo, Y.Z. MicroRNAs as biomarker and novel therapeutic target for posttraumatic stress disorder in veterans. Psychiatry Res. 2021, 305, 114252. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.P.; Singewald, N. Potential of microRNAs as novel targets in the alleviation of pathological fear. Genes Brain Behav. 2018, 17, e12427. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Start Date | NCT Number | Status | Study Title | Interventions | Sponsor |
|---|---|---|---|---|---|
| November 2017 | NCT03339258 | Recruiting | A Randomized Controlled Trial of Doxazosin for Nightmares, Sleep Disturbance, and Non-Nightmare Clinical Symptoms in PTSD | Drug: Doxazosin Mesylate, Extended Release, Drug: Placebo | San Francisco Veterans Affairs Medical Center |
| June 2020 | NCT04448808 | Recruiting | Treating Nightmares in Posttraumatic Stress Disorder with Dronabinol | Drug: BX-1, Drug: Placebo | Charite University, Berlin, Germany |
| May 2021 | NCT04877093 | Recruiting | Repurposing Low-Dose Clonidine for PTSD in Veterans | Drug: Clonidine, Drug: Placebo | Aurora Health Care |
| July 2021 | NCT04951076 | Active, not recruiting | A Phase 2b Study of BNC210 Tablet Formulation in Adults with Post-Traumatic Stress Disorder (PTSD) | Drug: BNC210, Drug: Placebo | Bionomics Limited |
| November 2021 | NCT05103657 | Recruiting | A Study to Test Whether Taking BI 1358894 for 8 Weeks Helps Adults with Post-traumatic Stress Disorder | Drug: BI 1358894, Drug: Placebo | Boehringer Ingelheim |
| January 2022 | NCT05178316 | Recruiting | A Study of JZP150 in Adults with Posttraumatic Stress Disorder | Drug: JZP150, Drug: Placebo | Jazz Pharmaceutical |
| February 2022 | NCT05254405 | Recruiting | An Open Label Pilot Study of IV Brexanolone for the Treatment of Post-Traumatic Stress Disorder | Drug: Brexanolone Injection [Zulresso] | Donald Jeffrey Newport |
| May 2022 | NCT05360953 | Recruiting | Treating Nightmares in Posttraumatic Stress Disorder with Clonidine and Doxazosin | Drug: Clonidine, Drug: Doxazosin, Drug: Placebo | Charite University, Berlin, Germany |
| May 2022 | NCT05391971 | Recruiting | Effects of Stellate Ganglion Block in Post-traumatic Stress Disorder | Drug: Bupivacaine, Drug: Saline | NYU Langone Health |
| June 2022 | NCT05401565 | Recruiting | Study To Evaluate The Efficacy And Safety Of Balovaptan In Adults with Post-Traumatic Stress Disorder (PTSD) | Drug: Balovaptan, Drug: Placebo | Hoffmann-La Roche |
| July 2022 | NCT05446857 | Recruiting | Glecaprevir/Pibrentasvir for the Treatment of PTSD | Drug: Glecaprevir/Pibrentasvir Pill | White River Junction Veterans Affairs Medical Center |
| February 2023 | NCT05741710 | Recruiting | A Study to Assess the Use of Methylone in the Treatment of PTSD | Drug: Methylone, Drug: Placebo | Transcend Therapeutics |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marazziti, D.; Carmassi, C.; Cappellato, G.; Chiarantini, I.; Massoni, L.; Mucci, F.; Arone, A.; Violi, M.; Palermo, S.; De Iorio, G.; et al. Novel Pharmacological Targets of Post-Traumatic Stress Disorders. Life 2023, 13, 1731. https://doi.org/10.3390/life13081731
Marazziti D, Carmassi C, Cappellato G, Chiarantini I, Massoni L, Mucci F, Arone A, Violi M, Palermo S, De Iorio G, et al. Novel Pharmacological Targets of Post-Traumatic Stress Disorders. Life. 2023; 13(8):1731. https://doi.org/10.3390/life13081731
Chicago/Turabian StyleMarazziti, Donatella, Claudia Carmassi, Gabriele Cappellato, Ilaria Chiarantini, Leonardo Massoni, Federico Mucci, Alessandro Arone, Miriam Violi, Stefania Palermo, Giovanni De Iorio, and et al. 2023. "Novel Pharmacological Targets of Post-Traumatic Stress Disorders" Life 13, no. 8: 1731. https://doi.org/10.3390/life13081731
APA StyleMarazziti, D., Carmassi, C., Cappellato, G., Chiarantini, I., Massoni, L., Mucci, F., Arone, A., Violi, M., Palermo, S., De Iorio, G., & Dell’Osso, L. (2023). Novel Pharmacological Targets of Post-Traumatic Stress Disorders. Life, 13(8), 1731. https://doi.org/10.3390/life13081731