


Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease


Abstract
1. Introduction
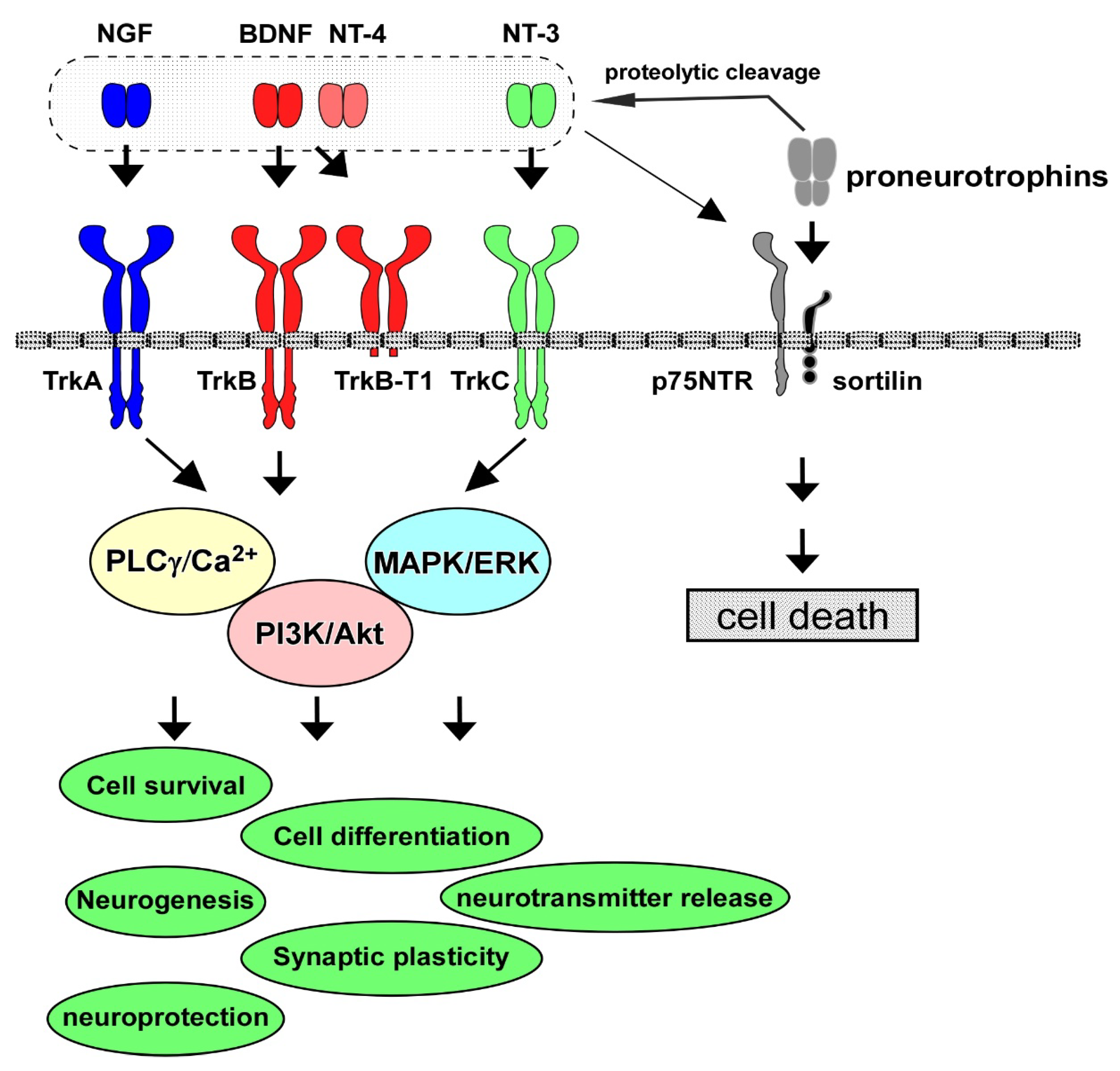
2. Neurotrophins, Receptors, and Intracellular Signaling
3. Roles of BDNF/TrkB System in CNS Neurons
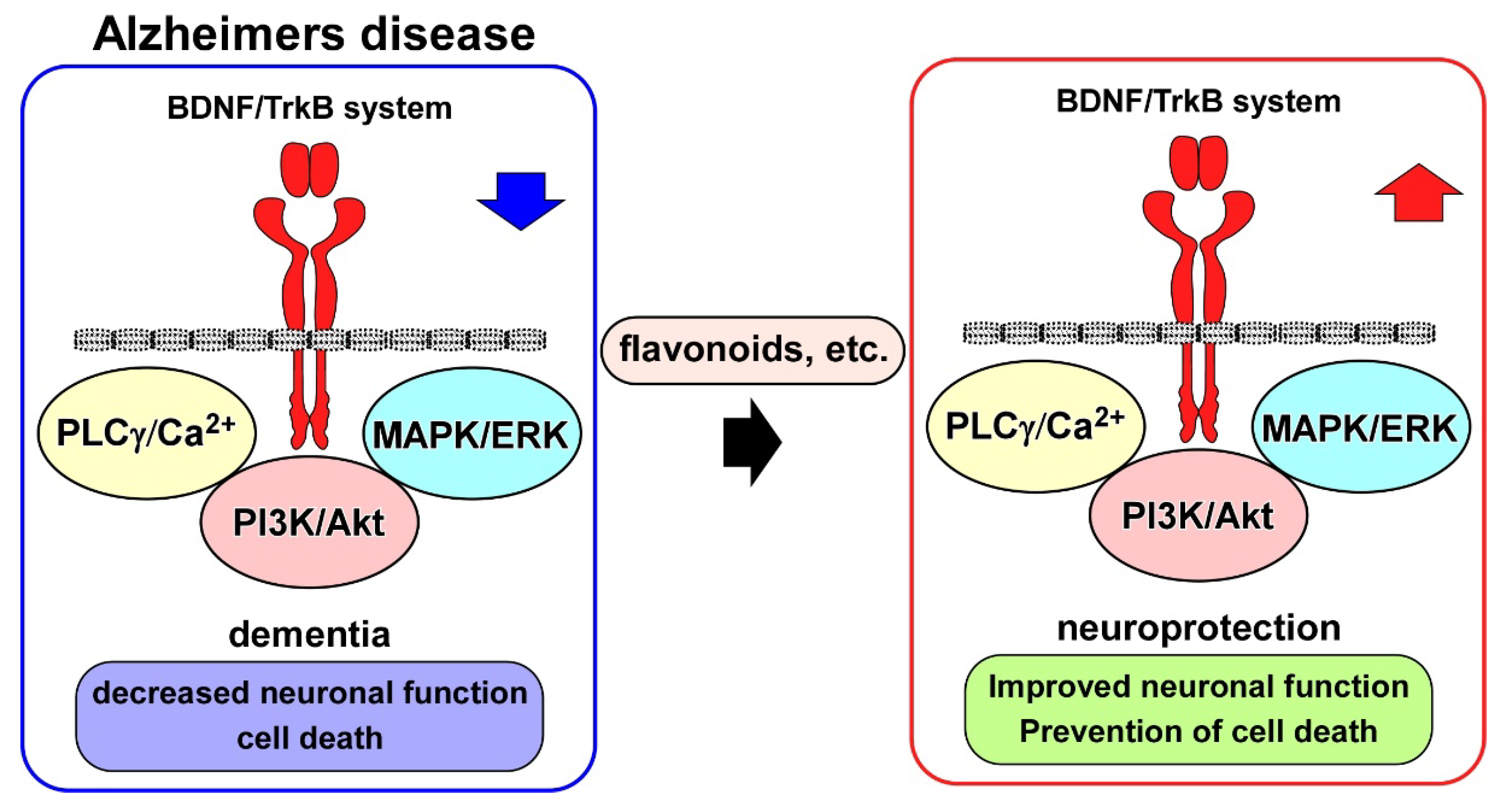
4. BDNF/TrkB System and AD Models
5. BDNF/TrkB System and Neuroprotection Drug Candidates for AD
6. Other Neurotrophic Factors in AD
6.1. Glial Cell Line-Derived Neurotrophic Factor (GDNF)
6.2. Basic Fibroblast Growth Factor (bFGF, or FGF-2)
6.3. Insulin-like Growth Factor 1 (IGF-1)

{kind=link}
{kind=link}
| Target Factors | Experimental AD Models | Treatments | Neurotrophic Effects | References |
|---|---|---|---|---|
| GDNF | Intracisternal injection of aluminum complexes into rabbit brain | Intracisternal injection of 100 μL of 500 ng/mL GDNF | Inhibition of neuronal death by upregulating bcl-XL and abolishing caspase-3 activity | [89] |
| 3xTgAD mouse | 6 months of GDNF overexpression using recombinant lentiviral vectors in hippocampal astrocytes | Preservation of learning and memory Upregulation of BDNF | [100] | |
| bFGF | Rat primary cortical neurons treated with glutamate (10 μM, 30 μM) and Aβ25–35 (1.0 μM) | Application of bFGF (0.3, 1, and 3 ng/mL) to the culture at 24 h before Aβ25–35 and glutamate stimulation | Decreased neuronal damage elicited by Aβ25–35 and glutamate | [111] |
| Rat hippocampal neurons treated with Aβ25–35 (20 μM) | Pre-treatment for 16 h with bFGF (10 ng/mL) | Suppression of reactive oxygen species (ROS) accumulation | [112] | |
| Rat primary hippocampal neurons treated with 20 μM Aβ1–42 | Treatment with LMW and HMW bFGF (10 μg/mL, 50 μg/mL, and 200 μg/mL) for 24 h | Protection against Aβ1–42-induced neurotoxicity Activation of the ERK and Akt signaling pathways | [120] | |
| APP23 mouse | Subcutaneous injection of recombinant bFGF (20 μg/kg per day) for 21 days | Attenuation of spatial memory deficits and Aβ and tau pathologies by downregulating BACE1 | [121] | |
| APP+PS1 mouse | Hippocampal AAV2/1(AAV serotype 2/1 hybrid virus)-bFGF injection | Reduced Aβ synthesis, and restored spatial learning in the radial arm water maze test | [122] | |
| APP+PS1 mouse | Intranasal administration of bFGF (0.5 μg/g) | Improvement of spatial learning ability, hippocampal LTP, and Aβ species accumulation in hippocampus | [126] | |
| IGF-1 | SH-SY5Y cells treated with Aβ25–35 (25 μM) or Aβ1–42 (1.5 μM) for 24 h | Application of IGF-1 (100 ng/mL) to the culture | Prevention of Aβ-induced cell death through activation of the PI3K/Akt pathway | [145] |
| Aging rats (>18-mo-old) | Chronic administration of IGF-1 using minipumps (50 μg/kg) | Downregulation of brain Aβ levels by enhancing transport of Aβ carrier proteins | [146] |
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nordvall, G.; Forsell, P.; Sandin, J. Neurotrophin-targeted therapeutics: A gateway to cognition and more? Drug Discov. Today 2022, 27, 103318. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, C.; Lang, U.E. Pathways connecting late-life depression and dementia. Front. Pharmacol. 2020, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Salton, S.R. Neurotrophin crosstalk in the etiology and treatment of neuropsychiatric and neurodegenerative disease. Front. Mol. Neurosci. 2022, 15, 932497. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Cheng, Y.Z.; Wang, Y.T.; Wei, R.M.; Ge, Y.J.; Kong, X.Y.; Li, X.Y. Environmental Enrichment Reverses Maternal Sleep Deprivation-Induced Anxiety-Like Behavior and Cognitive Impairment in CD-1 Mice. Front. Behav. Neurosci. 2022, 16, 943900. [Google Scholar] [CrossRef]
- Abdolahi, S.; Zare-Chahoki, A.; Noorbakhsh, F.; Gorji, A. A Review of Molecular Interplay between Neurotrophins and miRNAs in Neuropsychological Disorders. Mol. Neurobiol. 2022, 59, 6260–6280. [Google Scholar] [CrossRef]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Kunugi, H. Glucocorticoid suppresses BDNF-stimulated MAPK/ERK pathway via inhibiting interaction of Shp2 with TrkB. FEBS Lett. 2011, 585, 3224–3228. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; You, Y.; Gupta, V.B.; Klistorner, A.; Graham, S.L. TrkB receptor signalling: Implications in neurodegenerative, psychiatric and proliferative disorders. Int. J. Mol. Sci. 2013, 14, 10122–10142. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, K.; Zagrebelsky, M.; Berndt-Huch, J.; Polack, M.; Buschler, A.; Sendtner, M.; Korte, M. Neurotrophin receptors TrkB. T1 and p75NTR cooperate in modulating both functional and structural plasticity in mature hippocampal neurons. Eur. J. Neurosci. 2010, 32, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, S.G.; Lovering, R.M.; Renn, C.L.; Leitch, C.C.; Liu, X.; Tallon, L.J.; Sadzewicz, L.D.; Pratap, A.; Ott, S.; Sengamalay, N. Genetic deletion of trkB. T1 increases neuromuscular function. Am. J. Physiol.-Cell Physiol. 2012, 302, C141–C153. [Google Scholar] [CrossRef] [PubMed]
- Teng, K.K.; Felice, S.; Kim, T.; Hempstead, B.L. Understanding proneurotrophin actions: Recent advances and challenges. Dev. Neurobiol. 2010, 70, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Volosin, M.; Song, W.; Almeida, R.D.; Kaplan, D.R.; Hempstead, B.L.; Friedman, W.J. Interaction of survival and death signaling in basal forebrain neurons: Roles of neurotrophins and proneurotrophins. J. Neurosci. 2006, 26, 7756–7766. [Google Scholar] [CrossRef]
- Liepinsh, E.; Ilag, L.L.; Otting, G.; Ibáñez, C.F. NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J. 1997, 16, 4999–5005. [Google Scholar] [CrossRef]
- Becker, K.; Cana, A.; Baumgärtner, W.; Spitzbarth, I. p75 neurotrophin receptor: A double-edged sword in pathology and regeneration of the central nervous system. Vet. Pathol. 2018, 55, 786–801. [Google Scholar] [CrossRef]
- Hempstead, B.L. The many faces of p75NTR. Curr. Opin. Neurobiol. 2002, 12, 260–267. [Google Scholar] [CrossRef]
- Mukai, J.; Hachiya, T.; Shoji-Hoshino, S.; Kimura, M.T.; Nadano, D.; Suvanto, P.; Hanaoka, T.; Li, Y.; Irie, S.; Greene, L.A. NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J. Biol. Chem. 2000, 275, 17566–17570. [Google Scholar] [CrossRef]
- Ye, X.; Mehlen, P.; Rabizadeh, S.; VanArsdale, T.; Zhang, H.; Shin, H.; Wang, J.J.; Leo, E.; Zapata, J.; Hauser, C.A. TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J. Biol. Chem. 1999, 274, 30202–30208. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Tucker, K.L.; Barde, Y.-A. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 1999, 24, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef] [PubMed]
- He, X.-L.; Garcia, K.C. Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 2004, 304, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Willnow, T.E.; Petersen, C.M. p75NTR–live or let die. Curr. Opin. Neurobiol. 2005, 15, 49–57. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Luikart, B.W.; Nef, S.; Virmani, T.; Lush, M.E.; Liu, Y.; Kavalali, E.T.; Parada, L.F. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J. Neurosci. 2005, 25, 3774–3786. [Google Scholar] [CrossRef]
- Ji, Y.; Lu, Y.; Yang, F.; Shen, W.; Tang, T.T.-T.; Feng, L.; Duan, S.; Lu, B. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat. Neurosci. 2010, 13, 302–309. [Google Scholar] [CrossRef]
- Alonso, M.; Medina, J.H.; Pozzo-Miller, L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn. Mem. 2004, 11, 172–178. [Google Scholar] [CrossRef]
- Südhof, T.C. The synaptic vesicle cycle: A cascade of protein–protein interactions. Nature 1995, 375, 645–653. [Google Scholar] [CrossRef]
- Matsumoto, T.; Numakawa, T.; Yokomaku, D.; Adachi, N.; Yamagishi, S.; Numakawa, Y.; Kunugi, H.; Taguchi, T. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: Different dependency on signaling pathways and neuronal activity. Mol. Cell. Neurosci. 2006, 31, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, M.V.; Melo, C.V.; Pereira, D.B.; Carvalho, R.; Correia, S.S.; Backos, D.S.; Carvalho, A.L.; Esteban, J.A.; Duarte, C.B. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J. Biol. Chem. 2007, 282, 12619–12628. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, M.V.; Melo, C.V.; Pereira, D.B.; Carvalho, R.F.; Carvalho, A.L.; Duarte, C.B. BDNF regulates the expression and traffic of NMDA receptors in cultured hippocampal neurons. Mol. Cell. Neurosci. 2007, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Niyaz, M.; Kudo, M.; Kunugi, H. Glucocorticoid prevents brain-derived neurotrophic factor-mediated maturation of synaptic function in developing hippocampal neurons through reduction in the activity of mitogen-activated protein kinase. Mol. Endocrinol. 2008, 22, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Schuman, E.M. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 1995, 267, 1658–1662. [Google Scholar] [CrossRef] [PubMed]
- Levine, E.S.; Dreyfus, C.F.; Black, I.B.; Plummer, M.R. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc. Natl. Acad. Sci. USA 1995, 92, 8074–8077. [Google Scholar] [CrossRef]
- Numakawa, T.; Kumamaru, E.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Kunugi, H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc. Natl. Acad. Sci. USA 2009, 106, 647–652. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Kavalali, E.T.; Monteggia, L.M. Genetic dissection of presynaptic and postsynaptic BDNF-TrkB signaling in synaptic efficacy of CA3-CA1 synapses. Cell Rep. 2018, 24, 1550–1561. [Google Scholar] [CrossRef]
- Scharfman, H.; Goodman, J.; Macleod, A.; Phani, S.; Antonelli, C.; Croll, S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 2005, 192, 348–356. [Google Scholar] [CrossRef]
- Taliaz, D.; Stall, N.; Dar, D.; Zangen, A. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol. Psychiatry 2010, 15, 80–92. [Google Scholar] [CrossRef]
- Ben-Zeev, T.; Shoenfeld, Y.; Hoffman, J.R. The Effect of Exercise on Neurogenesis in the Brain. Isr. Med. Assoc. J. 2022, 24, 533–538. [Google Scholar] [PubMed]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.N.; Kramár, E.A.; Beardwood, J.H.; Al-Shammari, A.; Wood, M.A.; Keiser, A.A. Temporal endurance of exercise-induced benefits on hippocampus-dependent memory and synaptic plasticity in female mice. Neurobiol. Learn. Mem. 2022, 194, 107658. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Gao, J.Y.; Lin, S.Y.; Pan, S.T.; Xiao, B.; Ma, Y.T.; Xie, K.; Shen, W.; Liu, Z.T.; Li, G.Y.; et al. Effects of Involuntary and Voluntary Exercise in Combination with Acousto-Optic Stimulation on Adult Neurogenesis in an Alzheimer’s Mouse Model. Mol. Neurobiol. 2022, 59, 3254–3279. [Google Scholar] [CrossRef]
- Rahmati, M.; Keshvari, M.; Xie, W.; Yang, G.; Jin, H.; Li, H.; Chehelcheraghi, F.; Li, Y. Resistance training and Urtica dioica increase neurotrophin levels and improve cognitive function by increasing age in the hippocampus of rats. Biomed. Pharmacother. 2022, 153, 113306. [Google Scholar] [CrossRef]
- Zadeh, H.J.; Roholamini, Z.; Aminizadeh, S.; Deh-Ahmadi, M.A. Endurance training and MitoQ supplementation improve spatial memory, VEGF expression, and neurogenic factors in hippocampal tissue of rats. J. Clin. Transl. Res. 2023, 9, 1. [Google Scholar]
- Kim, T.W.; Park, S.S.; Park, H.S. Physical exercise ameliorates memory impairment in offspring of old mice. J. Exerc. Rehabil. 2022, 18, 155–161. [Google Scholar] [CrossRef]
- Yabuno, S.; Yasuhara, T.; Nagase, T.; Kawauchi, S.; Sugahara, C.; Okazaki, Y.; Hosomoto, K.; Sasada, S.; Sasaki, T.; Tajiri, N.; et al. Synergistic therapeutic effects of intracerebral transplantation of human modified bone marrow-derived stromal cells (SB623) and voluntary exercise with running wheel in a rat model of ischemic stroke. Stem Cell Res. Ther. 2023, 14, 10. [Google Scholar] [CrossRef]
- Leem, Y.H.; Park, J.S.; Park, J.E.; Kim, D.Y.; Kim, H.S. Neurogenic effects of rotarod walking exercise in subventricular zone, subgranular zone, and substantia nigra in MPTP-induced Parkinson’s disease mice. Sci. Rep. 2022, 12, 10544. [Google Scholar] [CrossRef] [PubMed]
- Reddy, I.; Yadav, Y.; Dey, C.S. Cellular and Molecular Regulation of Exercise-A Neuronal Perspective. Cell. Mol. Neurobiol. 2022, in press. [CrossRef] [PubMed]
- Deshpande, S.S.; Malik, S.C.; Conforti, P.; Lin, J.-D.; Chu, Y.-H.; Nath, S.; Greulich, F.; Dumbach, M.-A.; Uhlenhaut, N.H.; Schachtrup, C. P75 neurotrophin receptor controls subventricular zone neural stem cell migration after stroke. Cell Tissue Res. 2022, 387, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef] [PubMed]
- Rosa, E.; Fahnestock, M. CREB expression mediates amyloid β-induced basal BDNF downregulation. Neurobiol. Aging 2015, 36, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.K. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef]
- Xia, D.Y.; Huang, X.; Bi, C.F.; Mao, L.L.; Peng, L.J.; Qian, H.R. PGC-1α or FNDC5 Is Involved in Modulating the Effects of Aβ(1-42) Oligomers on Suppressing the Expression of BDNF, a Beneficial Factor for Inhibiting Neuronal Apoptosis, Aβ Deposition and Cognitive Decline of APP/PS1 Tg Mice. Front. Aging Neurosci. 2017, 9, 65. [Google Scholar] [CrossRef]
- Do Carmo, S.; Cuello, A.C. Modeling Alzheimer’s disease in transgenic rats. Mol. Neurodegener. 2013, 8, 37. [Google Scholar] [CrossRef]
- Leon, W.C.; Canneva, F.; Partridge, V.; Allard, S.; Ferretti, M.T.; DeWilde, A.; Vercauteren, F.; Atifeh, R.; Ducatenzeiler, A.; Klein, W.; et al. A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J. Alzheimers Dis. 2010, 20, 113–126. [Google Scholar] [CrossRef]
- Iulita, M.F.; Bistué Millón, M.B.; Pentz, R.; Aguilar, L.F.; Do Carmo, S.; Allard, S.; Michalski, B.; Wilson, E.N.; Ducatenzeiler, A.; Bruno, M.A.; et al. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol. Dis. 2017, 108, 307–323. [Google Scholar] [CrossRef]
- Bharani, K.L.; Ledreux, A.; Gilmore, A.; Carroll, S.L.; Granholm, A.C. Serum pro-BDNF levels correlate with phospho-tau staining in Alzheimer’s disease. Neurobiol. Aging 2020, 87, 49–59. [Google Scholar] [CrossRef]
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e655. [Google Scholar] [CrossRef]
- Pak, M.E.; Yang, H.J.; Li, W.; Kim, J.K.; Go, Y. Yuk-Gunja-Tang attenuates neuronal death and memory impairment via ERK/CREB/BDNF signaling in the hippocampi of experimental Alzheimer’s disease model. Front. Pharmacol. 2022, 13, 1014840. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Chang, K.H.; Chiu, Y.J.; Weng, Z.K.; Sun, Y.C.; Lin, W.; Lee-Chen, G.J.; Chen, C.M. Neuroprotective Action of Coumarin Derivatives through Activation of TRKB-CREB-BDNF Pathway and Reduction of Caspase Activity in Neuronal Cells Expressing Pro-Aggregated Tau Protein. Int. J. Mol. Sci. 2022, 23, 12734. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, I.; Poorkaj, P.; Hong, M.; Nochlin, D.; Lee, V.M.; Bird, T.D.; Schellenberg, G.D. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. USA 1999, 96, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Van Swieten, J.C.; Joosse, M.; Hasegawa, M.; Stevens, M.; Tibben, A.; Niermeijer, M.F.; Hillebrand, M.; Ravid, R.; Oostra, B.A.; et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Hum. Genet. 1999, 64, 414–421. [Google Scholar] [CrossRef]
- Chiu, Y.J.; Lin, T.H.; Chang, K.H.; Lin, W.; Hsieh-Li, H.M.; Su, M.T.; Chen, C.M.; Sun, Y.C.; Lee-Chen, G.J. Novel TRKB agonists activate TRKB and downstream ERK and AKT signaling to protect Aβ-GFP SH-SY5Y cells against Aβ toxicity. Aging 2022, 14, 7568–7586. [Google Scholar] [CrossRef]
- Rosa, E.; Mahendram, S.; Ke, Y.D.; Ittner, L.M.; Ginsberg, S.D.; Fahnestock, M. Tau downregulates BDNF expression in animal and cellular models of Alzheimer’s disease. Neurobiol. Aging 2016, 48, 135–142. [Google Scholar] [CrossRef]
- Liu, S.; Fan, M.; Xu, J.X.; Yang, L.J.; Qi, C.C.; Xia, Q.R.; Ge, J.F. Exosomes derived from bone-marrow mesenchymal stem cells alleviate cognitive decline in AD-like mice by improving BDNF-related neuropathology. J. Neuroinflammation 2022, 19, 35. [Google Scholar] [CrossRef]
- Salem, M.A.; Budzyńska, B.; Kowalczyk, J.; El Sayed, N.S.; Mansour, S.M. Tadalafil and bergapten mitigate streptozotocin-induced sporadic Alzheimer’s disease in mice via modulating neuroinflammation, PI3K/Akt, Wnt/β-catenin, AMPK/mTOR signaling pathways. Toxicol. Appl. Pharmacol. 2021, 429, 115697. [Google Scholar] [CrossRef]
- Bawari, S.; Tewari, D.; Argüelles, S.; Sah, A.N.; Nabavi, S.F.; Xu, S.; Vacca, R.A.; Nabavi, S.M.; Shirooie, S. Targeting BDNF signaling by natural products: Novel synaptic repair therapeutics for neurodegeneration and behavior disorders. Pharmacol. Res. 2019, 148, 104458. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H. Brain-Derived Neurotrophic Factor Signaling in the Pathophysiology of Alzheimer’s Disease: Beneficial Effects of Flavonoids for Neuroprotection. Int. J. Mol. Sci. 2021, 22, 5719. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, X.; Huang, X.; Yu, H.; Li, S.; Zhang, Z.; Xie, Y.; Song, X.; Liu, J.; Yang, X.; et al. Mitochondriomics reveals the underlying neuroprotective mechanism of TrkB receptor agonist R13 in the 5×FAD mice. Neuropharmacology 2022, 204, 108899. [Google Scholar] [CrossRef]
- Zhu, X.; Perry, G.; Moreira, P.I.; Aliev, G.; Cash, A.D.; Hirai, K.; Smith, M.A. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J. Alzheimers Dis. 2006, 9, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, V.C.; Araujo, S.M.; Pinheiro, F.C.; Poetini, M.R.; Meichtry, L.B.; Fronza, M.G.; Boeira, S.P.; Savegnago, L.; Prigol, M. Chrysin restores memory deficit in hypothyroidism mice: Behavioral, neurochemical and computational approaches involving the neurotrophinergic system. J. Psychiatr. Res. 2021, 144, 225–233. [Google Scholar] [CrossRef]
- Yan, T.; He, B.; Xu, M.; Wu, B.; Xiao, F.; Bi, K.; Jia, Y. Kaempferide prevents cognitive decline via attenuation of oxidative stress and enhancement of brain-derived neurotrophic factor/tropomyosin receptor kinase B/cAMP response element-binding signaling pathway. Phytother. Res. 2019, 33, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Deng, Y.; Liu, Y.; Gao, J.; Yan, L.; Gong, Q. Icariside II Ameliorates Cognitive Impairments Induced by Chronic Cerebral Hypoperfusion by Inhibiting the Amyloidogenic Pathway: Involvement of BDNF/TrkB/CREB Signaling and Up-Regulation of PPARα and PPARγ in Rats. Front. Pharmacol. 2018, 9, 1211. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Duarte Azevedo, M.; Sander, S.; Tenenbaum, L. GDNF, A Neuron-Derived Factor Upregulated in Glial Cells during Disease. J. Clin. Med. 2020, 9, 456. [Google Scholar] [CrossRef]
- Lübke, J.H.; Idoon, F.; Mohasel-Roodi, M.; Alipour, F.; Hami, J.; Ehteshampour, A.; Mostafaee, H.; Sadeghi, A. Neurotrophic factors in Alzheimer’s disease: Pathogenesis and therapy. Acta Neurobiol. Exp. 2021, 81, 314–327. [Google Scholar] [CrossRef]
- Pickens, C.L.; Calu, D.J. Alcohol reward, dopamine depletion, and GDNF. J. Neurosci. 2011, 31, 14833–14834. [Google Scholar] [CrossRef] [PubMed]
- Barak, S.; Ahmadiantehrani, S.; Logrip, M.L.; Ron, D. GDNF and alcohol use disorder. Addict. Biol. 2019, 24, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Bonafina, A.; Trinchero, M.F.; Ríos, A.S.; Bekinschtein, P.; Schinder, A.F.; Paratcha, G.; Ledda, F. GDNF and GFRα1 Are Required for Proper Integration of Adult-Born Hippocampal Neurons. Cell Rep. 2019, 29, 4308–4319.e4304. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, D.J. GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance. Int. J. Mol. Sci. 2018, 19, 1078. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Takahashi, M. Intracellular RET signaling pathways activated by GDNF. Cell Tissue Res. 2020, 382, 113–123. [Google Scholar] [CrossRef]
- Garcia-Lavandeira, M.; Diaz-Rodriguez, E.; Garcia-Rendueles, M.E.R.; Rodrigues, J.S.; Perez-Romero, S.; Bravo, S.B.; Alvarez, C.V. Functional role of the RET dependence receptor, GFRa co-receptors and ligands in the pituitary. Front. Horm. Res. 2010, 38, 127–138. [Google Scholar]
- Huang, H.; Liu, H.; Yan, R.; Hu, M. PI3K/Akt and ERK/MAPK Signaling Promote Different Aspects of Neuron Survival and Axonal Regrowth Following Rat Facial Nerve Axotomy. Neurochem. Res. 2017, 42, 3515–3524. [Google Scholar] [CrossRef]
- Ibáñez, C.F.; Paratcha, G.; Ledda, F. RET-independent signaling by GDNF ligands and GFRα receptors. Cell Tissue Res. 2020, 382, 71–82. [Google Scholar] [CrossRef]
- Ghribi, O.; Herman, M.M.; Forbes, M.S.; DeWitt, D.A.; Savory, J. GDNF protects against aluminum-induced apoptosis in rabbits by upregulating Bcl-2 and Bcl-XL and inhibiting mitochondrial Bax translocation. Neurobiol. Dis. 2001, 8, 764–773. [Google Scholar] [CrossRef]
- Cheng, H.; Fu, Y.S.; Guo, J.W. Ability of GDNF to diminish free radical production leads to protection against kainate-induced excitotoxicity in hippocampus. Hippocampus 2004, 14, 77–86. [Google Scholar] [CrossRef]
- Marksteiner, J.; Kemmler, G.; Weiss, E.M.; Knaus, G.; Ullrich, C.; Mechtcheriakov, S.; Oberbauer, H.; Auffinger, S.; Hinterhölzl, J.; Hinterhuber, H.; et al. Five out of 16 plasma signaling proteins are enhanced in plasma of patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2011, 32, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Straten, G.; Saur, R.; Laske, C.; Gasser, T.; Annas, P.; Basun, H.; Leyhe, T. Influence of lithium treatment on GDNF serum and CSF concentrations in patients with early Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Miranda, A.S.; Guimar, I.; Talib, L.L.; Diniz, B.S.; Gattaz, W.F.; Teixeira, A.L. Decreased Neurotrophic Support is Associated with Cognitive Decline in Non-Demented Subjects. J. Alzheimers Dis. 2015, 46, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.Q.; Gregory, R.A.; Scofield, M.D.; Helke, K.L.; Boger, H.A. Effects of aging on glutamate neurotransmission in the substantia nigra of Gdnf heterozygous mice. Neurobiol. Aging 2015, 36, 1569–1576. [Google Scholar] [CrossRef]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.L.; Zugno, A.I. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341. [Google Scholar]
- Krashia, P.; Nobili, A.; D’Amelio, M. Unifying Hypothesis of Dopamine Neuron Loss in Neurodegenerative Diseases: Focusing on Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 12, 123. [Google Scholar] [CrossRef]
- Mitra, S.; Turconi, G.; Darreh-Shori, T.; Mätlik, K.; Aquilino, M.; Eriksdotter, M.; Andressoo, J.O. Increased Endogenous GDNF in Mice Protects Against Age-Related Decline in Neuronal Cholinergic Markers. Front. Aging Neurosci. 2021, 13, 714186. [Google Scholar] [CrossRef]
- Airavaara, M.; Pletnikova, O.; Doyle, M.E.; Zhang, Y.E.; Troncoso, J.C.; Liu, Q.R. Identification of novel GDNF isoforms and cis-antisense GDNFOS gene and their regulation in human middle temporal gyrus of Alzheimer disease. J. Biol. Chem. 2011, 286, 45093–45102. [Google Scholar] [CrossRef]
- Revilla, S.; Suñol, C.; García-Mesa, Y.; Giménez-Llort, L.; Sanfeliu, C.; Cristòfol, R. Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 2014, 81, 55–63. [Google Scholar] [CrossRef]
- Revilla, S.; Ursulet, S.; Álvarez-López, M.J.; Castro-Freire, M.; Perpiñá, U.; García-Mesa, Y.; Bortolozzi, A.; Giménez-Llort, L.; Kaliman, P.; Cristòfol, R.; et al. Lenti-GDNF gene therapy protects against Alzheimer’s disease-like neuropathology in 3xTg-AD mice and MC65 cells. CNS Neurosci. Ther. 2014, 20, 961–972. [Google Scholar] [CrossRef]
- Gospodarowicz, D. Localisation of a fibroblast growth factor and its effect alone and with hydrocortisone on 3T3 cell growth. Nature 1974, 249, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014, 1, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, X.M.; van Kuppevelt, T.H. Heparan sulfate in angiogenesis: A target for therapy. Angiogenesis 2014, 17, 443–462. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.X.; Chen, X.; Qin, X.Y.; Kardami, E.; Cheng, Y. Antidepressant-Like Effects of Low- and High-Molecular Weight FGF-2 on Chronic Unpredictable Mild Stress Mice. Front. Mol. Neurosci. 2018, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Z.; Cheng, Y.; Kardami, E.; Loh, Y.P. Low and High Molecular Weight FGF-2 Have Differential Effects on Astrocyte Proliferation, but Are Both Protective Against Aβ-Induced Cytotoxicity. Front. Mol. Neurosci. 2019, 12, 328. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.J.; Bruce, C.; Chioni, A.M.; Kocher, H.M.; Grose, R.P. The ins and outs of fibroblast growth factor receptor signalling. Clin. Sci. 2014, 127, 217–231. [Google Scholar] [CrossRef]
- Xie, M.; Li, J.P. Heparan sulfate proteoglycan—A common receptor for diverse cytokines. Cell. Signal. 2019, 54, 115–121. [Google Scholar] [CrossRef]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Silva, A.; Montague, J.R.; Lopez, T.F.; Mudd, L.M. Growth factor effects on survival and development of calbindin immunopositive cultured septal neurons. Brain Res. Bull. 2000, 51, 35–42. [Google Scholar] [CrossRef]
- Alam, R.; Mrad, Y.; Hammoud, H.; Saker, Z.; Fares, Y.; Estephan, E.; Bahmad, H.F.; Harati, H.; Nabha, S. New insights into the role of fibroblast growth factors in Alzheimer’s disease. Mol. Biol. Rep. 2022, 49, 1413–1427. [Google Scholar] [CrossRef]
- Noshita, T.; Murayama, N.; Oka, T.; Ogino, R.; Nakamura, S.; Inoue, T. Effect of bFGF on neuronal damage induced by sequential treatment of amyloid β and excitatory amino acid in vitro and in vivo. Eur. J. Pharmacol. 2012, 695, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Keller, J.N.; Kruman, I.; Mattson, M.P. Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 1997, 756, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.H.; Mattson, M.P. Neurotrophic factors protect cortical synaptic terminals against amyloid and oxidative stress-induced impairment of glucose transport, glutamate transport and mitochondrial function. Cereb. Cortex 2000, 10, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Sebastian, L.; Sopher, B.L.; Miller, M.W.; Glazner, G.W.; Ware, C.B.; Martin, G.M.; Mattson, M.P. Neurotrophic factors [activity-dependent neurotrophic factor (ADNF) and basic fibroblast growth factor (bFGF)] interrupt excitotoxic neurodegenerative cascades promoted by a PS1 mutation. Proc. Natl. Acad. Sci. USA 1999, 96, 4125–4130. [Google Scholar] [CrossRef]
- Gray, C.W.; Patel, A.J. Neurodegeneration mediated by glutamate and beta-amyloid peptide: A comparison and possible interaction. Brain Res. 1995, 691, 169–179. [Google Scholar] [CrossRef]
- Mattson, M.P.; Tomaselli, K.J.; Rydel, R.E. Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res. 1993, 621, 35–49. [Google Scholar] [CrossRef]
- Assis-Nascimento, P.; Jarvis, K.M.; Montague, J.R.; Mudd, L.M. Beta-amyloid toxicity in embryonic rat astrocytes. Neurochem. Res. 2007, 32, 1476–1482. [Google Scholar] [CrossRef]
- Emmett, C.J.; Aswani, S.P.; Stewart, G.R.; Fairchild, D.; Johnson, R.M. Dose-response comparison of recombinant human nerve growth factor and recombinant human basic fibroblast growth factor in the fimbria fornix model of acute cholinergic degeneration. Brain Res. 1995, 673, 199–207. [Google Scholar] [CrossRef]
- Pankratova, S.; Bjornsdottir, H.; Christensen, C.; Zhang, L.; Li, S.; Dmytriyeva, O.; Bock, E.; Berezin, V. Immunomodulator CD200 Promotes Neurotrophic Activity by Interacting with and Activating the Fibroblast Growth Factor Receptor. Mol. Neurobiol. 2016, 53, 584–594. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, Z.; Kardami, E.; Loh, Y.P. Neuroprotective effects of LMW and HMW FGF2 against amyloid beta toxicity in primary cultured hippocampal neurons. Neurosci. Lett. 2016, 632, 109–113. [Google Scholar] [CrossRef]
- Katsouri, L.; Ashraf, A.; Birch, A.M.; Lee, K.K.; Mirzaei, N.; Sastre, M. Systemic administration of fibroblast growth factor-2 (FGF2) reduces BACE1 expression and amyloid pathology in APP23 mice. Neurobiol. Aging 2015, 36, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Ingraham, K.L.; Jacobsen, M.T.; Xiong, H.; Ikezu, T. FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc. Natl. Acad. Sci. USA 2011, 108, E1339–E1348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, J.; Feng, C.; Shao, X.; Liu, Q.; Zhang, Q.; Pang, Z.; Jiang, X. Intranasal nanoparticles of basic fibroblast growth factor for brain delivery to treat Alzheimer’s disease. Int. J. Pharm. 2014, 461, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Frey, W.H., 2nd. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9 (Suppl. 3), S5. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Zhang, C.; Shao, X.; Liu, Q.; Qian, Y.; Feng, L.; Chen, J.; Zha, Y.; Zhang, Q.; Jiang, X. Enhancement of nose-to-brain delivery of basic fibroblast growth factor for improving rat memory impairments induced by co-injection of β-amyloid and ibotenic acid into the bilateral hippocampus. Int. J. Pharm. 2012, 423, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zheng, Y.; Xiao, J.; Lin, S.; Chen, D.; Li, S. FGF2 shows therapeutic effects in Alzheimer’s disease animal model via suppressing PI3K/Akt mediated ER stress. Int. J. Clin. Exp. Med. 2016, 9, 2130–2138. [Google Scholar]
- Russo, V.C.; Gluckman, P.D.; Feldman, E.L.; Werther, G.A. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 2005, 26, 916–943. [Google Scholar] [CrossRef]
- Bhalla, S.; Mehan, S.; Khan, A.; Rehman, M.U. Protective role of IGF-1 and GLP-1 signaling activation in neurological dysfunctions. Neurosci. Biobehav. Rev. 2022, 142, 104896. [Google Scholar] [CrossRef]
- Taguchi, A.; White, M.F. Insulin-like signaling, nutrient homeostasis, and life span. Annu. Rev. Physiol. 2008, 70, 191–212. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Sadagurski, M.; Dong, X.C.; Myers, M.G., Jr.; White, M.F. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol. Metab. 2014, 3, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 3306. [Google Scholar] [CrossRef] [PubMed]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Cohen, E.; Paulsson, J.F.; Blinder, P.; Burstyn-Cohen, T.; Du, D.; Estepa, G.; Adame, A.; Pham, H.M.; Holzenberger, M.; Kelly, J.W.; et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 2009, 139, 1157–1169. [Google Scholar] [CrossRef]
- Gontier, G.; George, C.; Chaker, Z.; Holzenberger, M.; Aïd, S. Blocking IGF Signaling in Adult Neurons Alleviates Alzheimer’s Disease Pathology through Amyloid-β Clearance. J. Neurosci. 2015, 35, 11500–11513. [Google Scholar] [CrossRef]
- Stöhr, O.; Schilbach, K.; Moll, L.; Hettich, M.M.; Freude, S.; Wunderlich, F.T.; Ernst, M.; Zemva, J.; Brüning, J.C.; Krone, W.; et al. Insulin receptor signaling mediates APP processing and β-amyloid accumulation without altering survival in a transgenic mouse model of Alzheimer’s disease. Age 2013, 35, 83–101. [Google Scholar] [CrossRef]
- Sohrabi, M.; Floden, A.M.; Manocha, G.D.; Klug, M.G.; Combs, C.K. IGF-1R Inhibitor Ameliorates Neuroinflammation in an Alzheimer’s Disease Transgenic Mouse Model. Front. Cell Neurosci. 2020, 14, 200. [Google Scholar] [CrossRef]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.D.; Baker, L.D.; Borson, S.; Jensen, J.E.; Barsness, S.M.; Craft, S.; Merriam, G.R.; Otto, R.K.; Novotny, E.J.; Vitiello, M.V. Growth hormone-releasing hormone effects on brain γ-aminobutyric acid levels in mild cognitive impairment and healthy aging. JAMA Neurol. 2013, 70, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.D.; Barsness, S.M.; Borson, S.; Merriam, G.R.; Friedman, S.D.; Craft, S.; Vitiello, M.V. Effects of growth hormone–releasing hormone on cognitive function in adults with mild cognitive impairment and healthy older adults: Results of a controlled trial. Arch. Neurol. 2012, 69, 1420–1429. [Google Scholar] [CrossRef]
- Hou, X.; Jin, Y.; Chen, J.; Hong, Y.; Luo, D.; Yin, Q.; Liu, X. IGF-1 protects against Aβ(25-35)-induced neuronal cell death via inhibition of PUMA expression and Bax activation. Neurosci. Lett. 2017, 637, 188–194. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Gomez-Isla, T.; LeRoith, D.; Torres-Aleman, I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat. Med. 2002, 8, 1390–1397. [Google Scholar] [CrossRef]
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhang-Fallah, J.; Dikkes, P.; Warot, X.M.; Rio, C.; et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 2003, 23, 7084–7092. [Google Scholar] [CrossRef]
- Åberg, D. Role of the growth hormone/insulin-like growth factor 1 axis in neurogenesis. Endocr. Dev. 2010, 17, 63–76. [Google Scholar]
- Morel, G.R.; León, M.L.; Uriarte, M.; Reggiani, P.C.; Goya, R.G. Therapeutic potential of IGF-I on hippocampal neurogenesis and function during aging. Neurogenesis 2017, 4, e1259709. [Google Scholar] [CrossRef]
- Madathil, S.K.; Saatman, K.E. IGF-1/IGF-R signaling in traumatic brain injury. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Abingdon, UK, 2015. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numakawa, T.; Kajihara, R. Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease. Life 2023, 13, 647. https://doi.org/10.3390/life13030647
Numakawa T, Kajihara R. Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease. Life. 2023; 13(3):647. https://doi.org/10.3390/life13030647
Chicago/Turabian StyleNumakawa, Tadahiro, and Ryutaro Kajihara. 2023. "Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease" Life 13, no. 3: 647. https://doi.org/10.3390/life13030647
APA StyleNumakawa, T., & Kajihara, R. (2023). Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease. Life, 13(3), 647. https://doi.org/10.3390/life13030647