
Synthesis and Characterization of Gd-Functionalized B4C Nanoparticles for BNCT Applications

 ,
,  ,
,  , ,
, ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. FGdBNP Syntesis
2.2. FGdBNP Characterization
2.3. Interaction of GdBNP with Cells
3. Results and Discussion
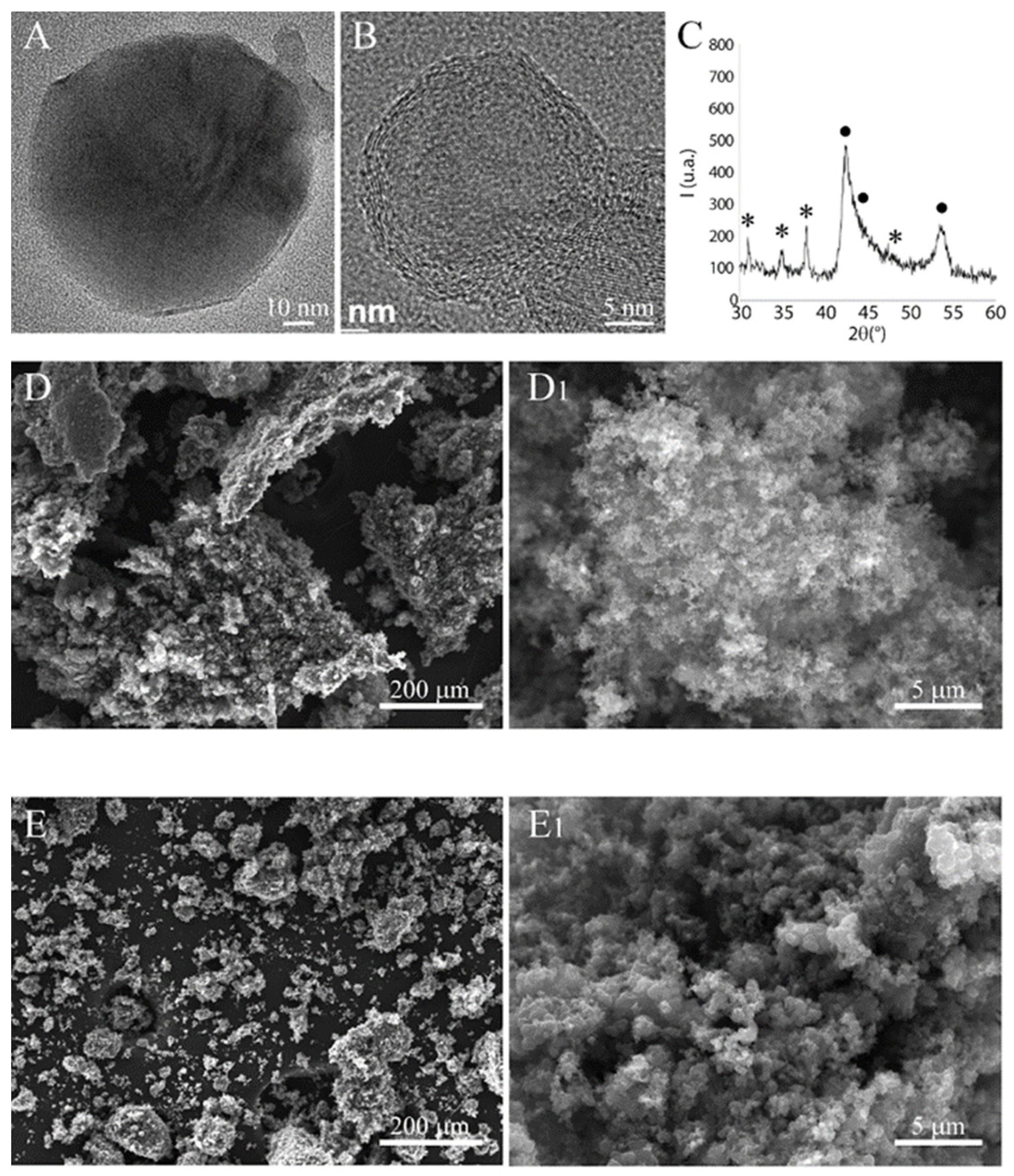
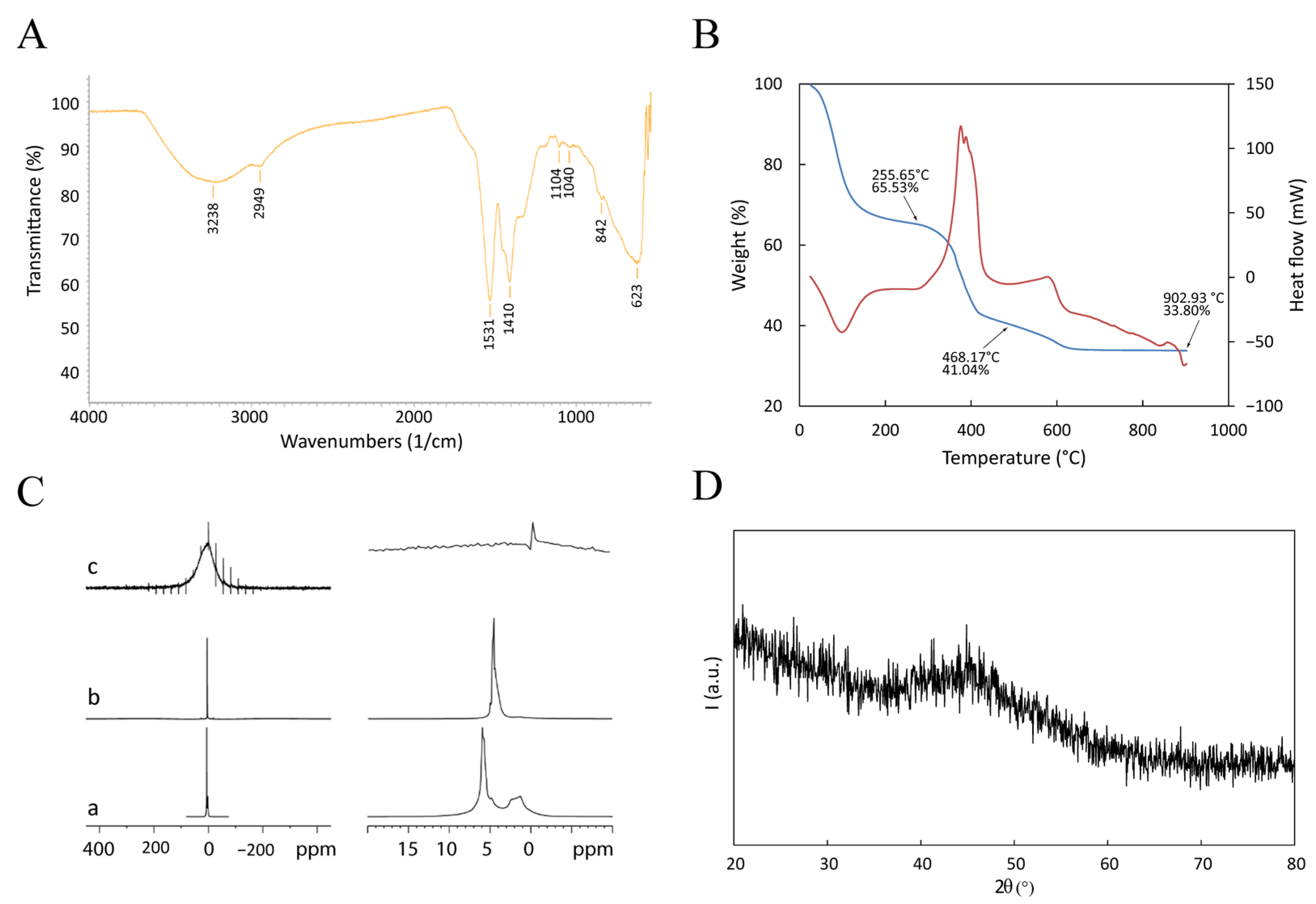
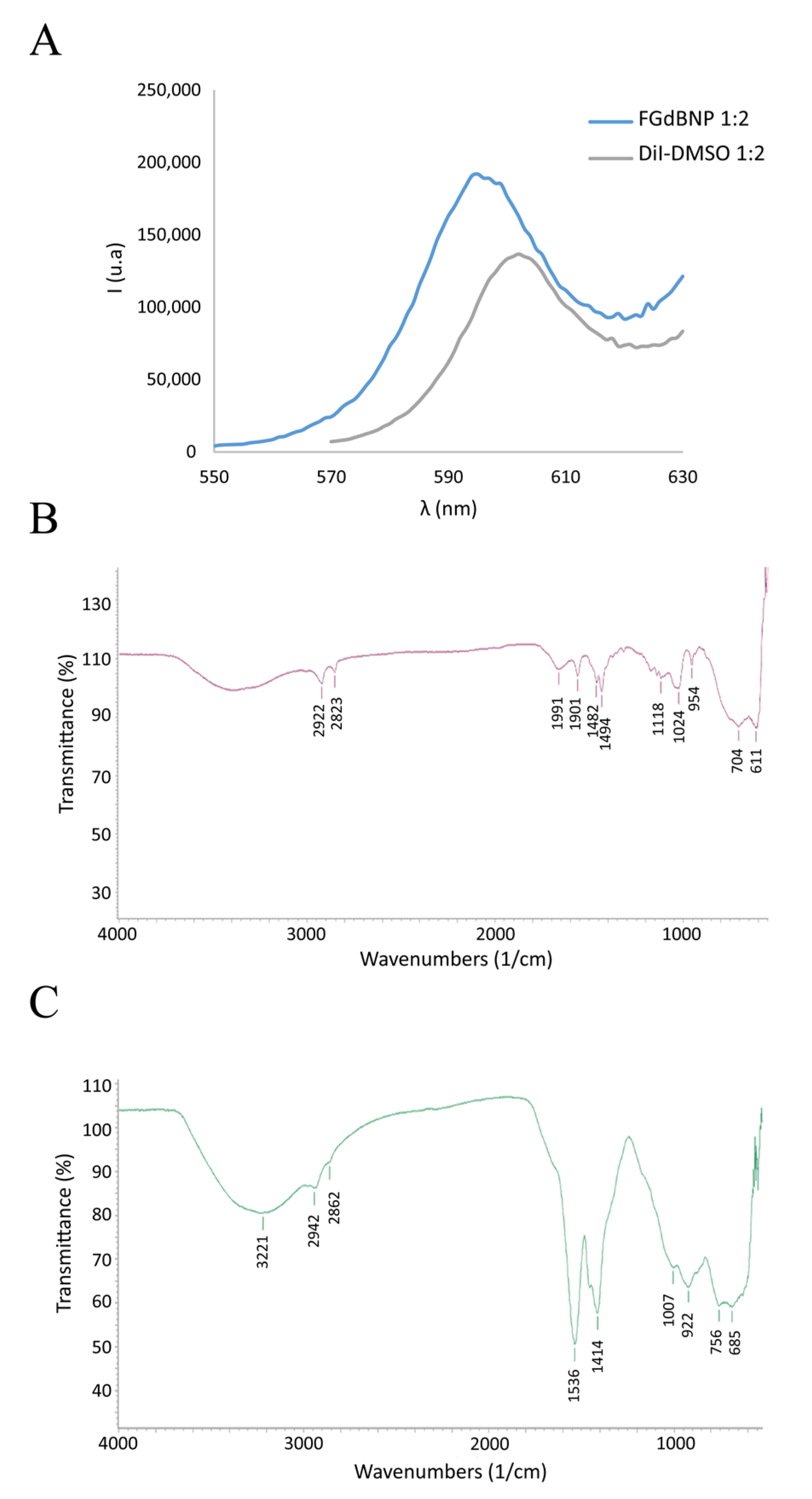
3.1. Nanoparticles Characterization
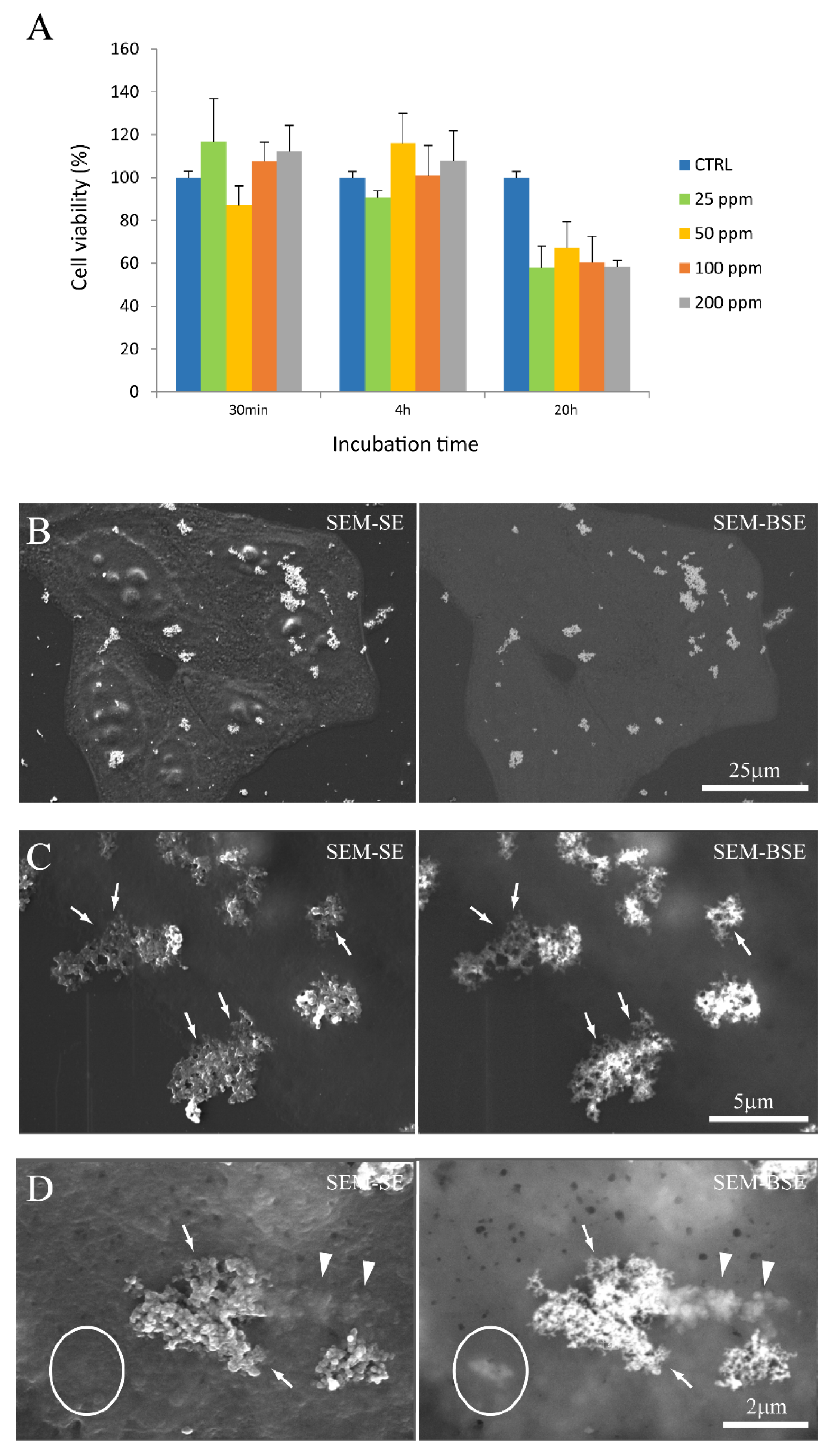
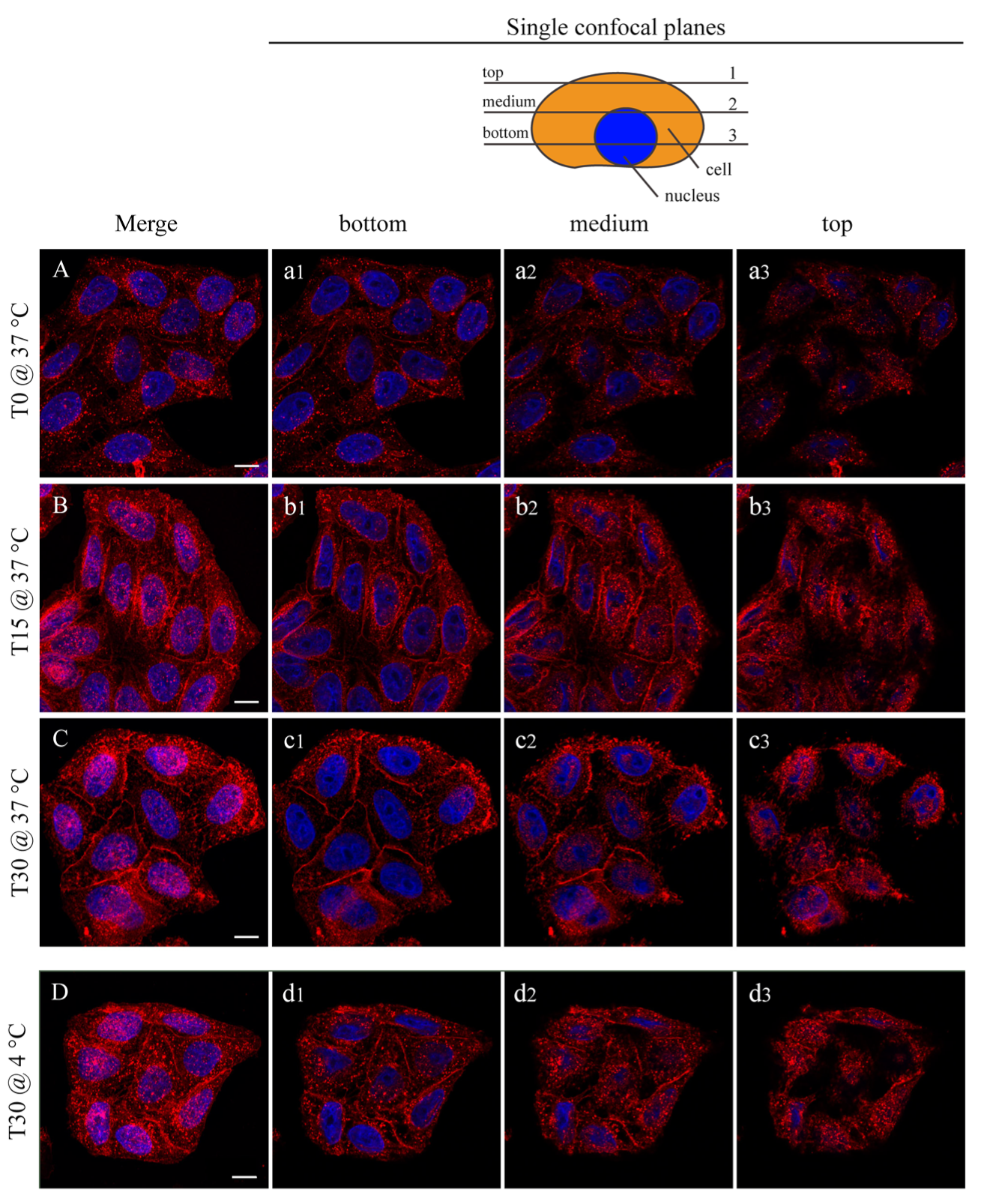
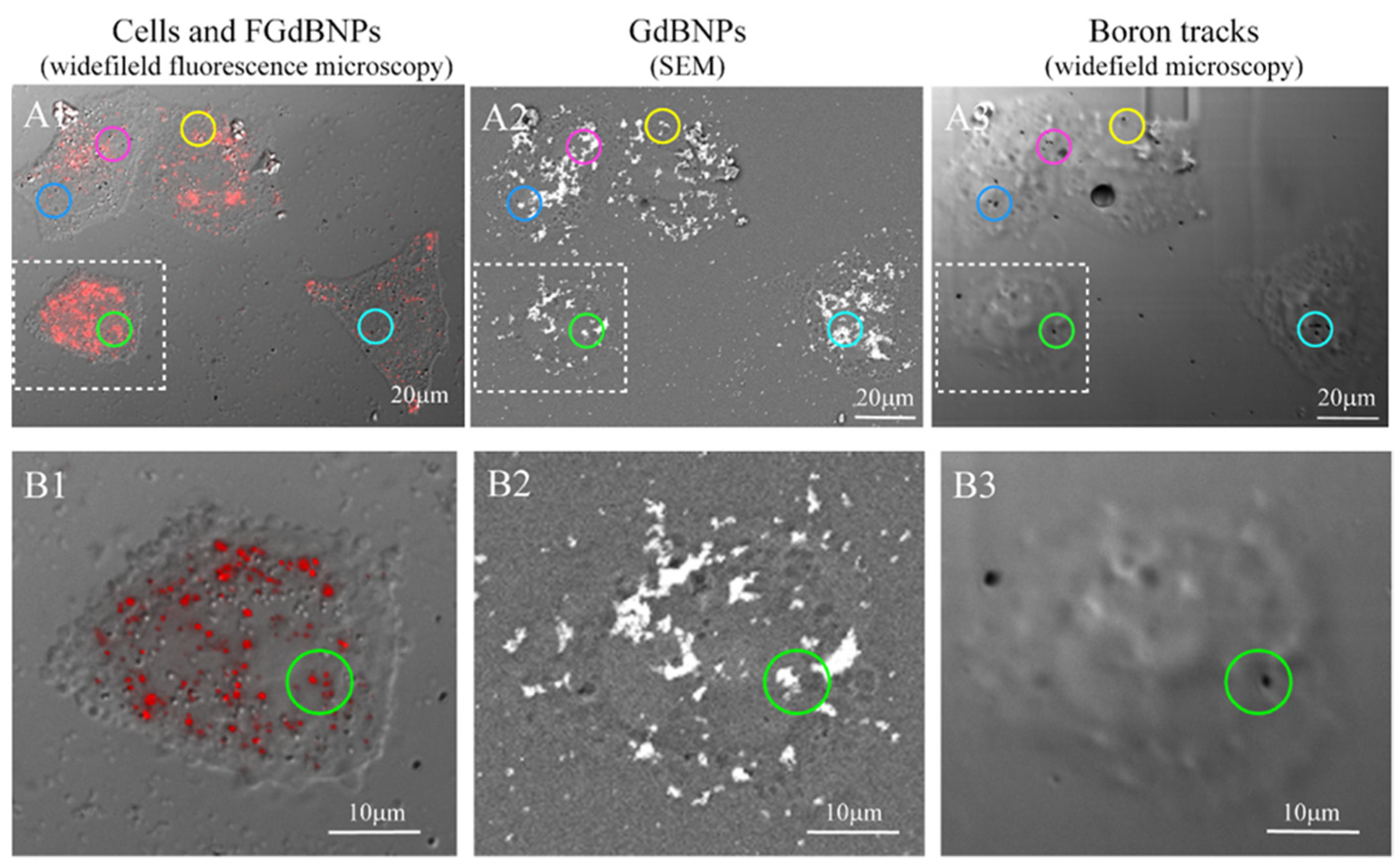
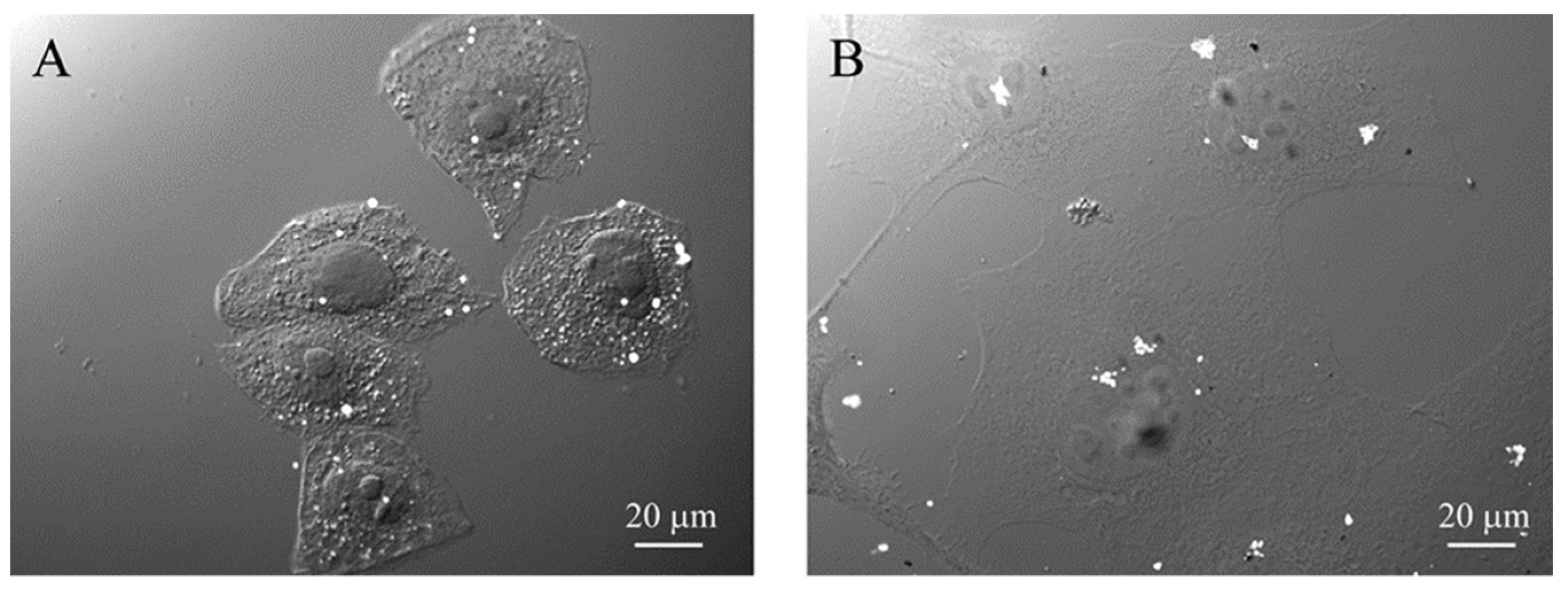
3.2. Nanoparticle Interactions with Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.G.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The Chemistry of Neutron Capture Therapy. Chem. Rev. 1998, 98, 1515–1562. [Google Scholar] [CrossRef]
- Coderre, J.A.; Turcotte, J.C.; Riley, K.J.; Binns, P.J.; Harling, O.K.; Kiger, W.S. Boron Neutron Capture Therapy: Cellular Targeting of High Linear Energy Transfer Radiation. Technol. Cancer Res. Treat. 2003, 2, 355–375. [Google Scholar] [CrossRef]
- Takai, S.; Wanibuchi, M.; Kawabata, S.; Takeuchi, K.; Sakurai, Y.; Suzuki, M.; Ono, K.; Miyatake, S.I. Reactor-Based Boron Neutron Capture Therapy for 44 Cases of Recurrent and Refractory High-Grade Meningiomas with Long-Term Follow-Up. Neuro-Oncology 2022, 24, 90–98. [Google Scholar] [CrossRef]
- Barth, R.F.; Mi, P.; Yang, W. Boron Delivery Agents for Neutron Capture Therapy of Cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef]
- Suzuki, M.; Kato, I.; Aihara, T.; Hiratsuka, J.; Yoshimura, K.; Niimi, M.; Kimura, Y.; Ariyoshi, Y.; Haginomori, S.I.; Sakurai, Y.; et al. Boron Neutron Capture Therapy Outcomes for Advanced or Recurrent Head and Neck Cancer. J. Radiat. Res. 2014, 55, 146–153. [Google Scholar] [CrossRef]
- Malouff, T.D.; Seneviratne, D.S.; Ebner, D.K.; Stross, W.C.; Waddle, M.R.; Trifiletti, D.M.; Krishnan, S. Boron Neutron Capture Therapy: A Review of Clinical Applications. Front. Oncol. 2021, 11, 601820. [Google Scholar] [CrossRef]
- Yinghuai, Z.; Maguire, J.A.; Hosmane, N.S. Recent Developments in Boron Neutron Capture Therapy Driven by Nanotechnology. In Boron Science: New Technologies and Applications; CRC Press: Boca Raton, FL, USA, 2016; pp. 147–163. [Google Scholar] [CrossRef]
- Mortensen, M.W.; Sørensen, P.G.; Björkdahl, O.; Jensen, M.R.; Gundersen, H.J.G.; Bjørnholm, T. Preparation and Characterization of Boron Carbide Nanoparticles for Use as a Novel Agent in T Cell-Guided Boron Neutron Capture Therapy. Appl. Radiat. Isot. 2006, 64, 315–324. [Google Scholar] [CrossRef]
- Mortensen, M.W.; Björkdahl, O.; Sørensen, P.G.; Hansen, T.; Jensen, M.R.; Gundersen, H.J.G.; Bjørnholm, T. Functionalization and Cellular Uptake of Boron Carbide Naraoparticles. The First Step toward T Cell-Guided Boron Neutron Capture Therapy. Bioconjug. Chem. 2006, 17, 284–290. [Google Scholar] [CrossRef]
- Hu, K.; Yang, Z.; Zhang, L.; Xie, L.; Wang, L.; Xu, H.; Josephson, L.; Liang, S.H.; Zhang, M.R. Boron Agents for Neutron Capture Therapy. Coord. Chem. Rev. 2020, 405, 213139. [Google Scholar] [CrossRef]
- Ailuno, G.; Balboni, A.; Caviglioli, G.; Lai, F.; Barbieri, F.; Dellacasagrande, I.; Florio, T.; Baldassari, S. Boron Vehiculating Nanosystems for Neutron Capture Therapy in Cancer Treatment. Cells 2022, 11, 4029. [Google Scholar] [CrossRef]
- Iwagami, T.; Ishikawa, Y.; Koshizaki, N.; Yamamoto, N.; Tanaka, H.; Masunaga, S.I.; Sakurai, Y.; Kato, I.; Iwai, S.; Suzuki, M.; et al. Boron Carbide Particle as a Boron Compound for Boron Neutron Capture Therapy. J. Nucl. Med. Radiat. Ther. 2014, 5, 177. [Google Scholar] [CrossRef]
- Watts, J.L.; Spratt, H.J.; Talbot, P.C.; Alarco, J.A.; Raftery, N.A.; Mackinnon, I.D.R. Precision Structural and Phase Analysis of Boron Carbide. Ceram. Int. 2020, 46, 11033–11040. [Google Scholar] [CrossRef]
- Gosset, D. Basic Properties of Boron Carbide. In Comprehensive Nuclear Materials, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 539–553. [Google Scholar] [CrossRef]
- Thévenot, F. Boron Carbide-A Comprehensive Review. J. Eur. Ceram. Soc. 1990, 6, 205–225. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Balakrishnarajan, M.M.; Pancharatna, P.D.; Hoffmann, R. Structure and Bonding in Boron Carbide: The Invincibility of Imperfections. New J. Chem. 2007, 31, 473–485. [Google Scholar] [CrossRef]
- Flores-Rojas, G.G.; López-Saucedo, F.; Vera-Graziano, R.; Mendizabal, E.; Bucio, E. Magnetic Nanoparticles for Medical Applications: Updated Review. Macromol 2022, 2, 374–390. [Google Scholar] [CrossRef]
- Ho, S.L.; Yue, H.; Tegafaw, T.; Ahmad, M.Y.; Liu, S.; Nam, S.W.; Chang, Y.; Lee, G.H. Gadolinium Neutron Capture Therapy (GdNCT) Agents from Molecular to Nano: Current Status and Perspectives. ACS Omega 2022, 7, 2533–2553. [Google Scholar] [CrossRef]
- Li, H.; Meade, T.J. Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances. J. Am. Chem. Soc. 2019, 141, 17025–17041. [Google Scholar] [CrossRef]
- Alberti, D.; Protti, N.; Toppino, A.; Deagostino, A.; Lanzardo, S.; Bortolussi, S.; Altieri, S.; Voena, C.; Chiarle, R.; Geninatti Crich, S.; et al. A Theranostic Approach Based on the Use of a Dual Boron/Gd Agent to Improve the Efficacy of Boron Neutron Capture Therapy in the Lung Cancer Treatment. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 741–750. [Google Scholar] [CrossRef]
- Brugger, R.M. Gadolinium as Xda Neutron Capture Therapy Agent. Med. Phys. 1992, 19, 733–744. [Google Scholar] [CrossRef]
- Goorley, T.; Nikjoo, H. Electron and Photon Spectra for Three Gadolinium-Based Cancer Therapy Approaches. Radiat. Res. 2000, 154, 556–563. [Google Scholar] [CrossRef]
- Postuma, I.; Sommi, P.; Vitali, A.; Shu, D.; di Martino, G.; Cansolino, L.; Ferrari, C.; Ricci, V.; Magni, C.; Protti, N.; et al. Colocalization of Tracks from Boron Neutron Capture Reactions and Images of Isolated Cells. Appl. Radiat. Isot. 2021, 167, 109353. [Google Scholar] [CrossRef]
- Diaz-Cano, A.; Trice, R.W.; Youngblood, J.P. Stabilization of Highly-Loaded Boron Carbide Aqueous Suspensions. Ceram. Int. 2017, 43, 8572–8578. [Google Scholar] [CrossRef]
- Ferraro, D.; Tredici, I.G.; Ghigna, P.; Castillio-Michel, H.; Falqui, A.; Di Benedetto, C.; Alberti, G.; Ricci, V.; Anselmi-Tamburini, U.; Sommi, P. Dependence of the Ce(III)/Ce(IV) Ratio on Intracellular Localization in Ceria Nanoparticles Internalized by Human Cells. Nanoscale 2017, 9, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Swift, T.; Swanson, L.; Geoghegan, M.; Rimmer, S. The PH-Responsive Behaviour of Poly(Acrylic Acid) in Aqueous Solution Is Dependent on Molar Mass. Soft Matter 2016, 12, 2542–2549. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Kaittanis, C.; Grimm, J.; Perez, J.M. Drug/Dye-Loaded, Multifunctional Iron Oxide Nanoparticles for Combined Targeted Cancer Therapy and Dual Optical/Magnetic Resonance Imaging. Small 2009, 5, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Postuma, I.; Bortolussi, S.; Protti, N.; Ballarini, F.; Bruschi, P.; Ciani, L.; Ristori, S.; Panza, L.; Ferrari, C.; Cansolino, L.; et al. An Improved Neutron Autoradiography Set-up for 10B Concentration Measurements in Biological Samples. Rep. Pract. Oncol. Radiother. 2016, 21, 123–128. [Google Scholar] [CrossRef]
- Aghazadeh, M. Preparation of Gd2O3 Ultrafine Nanoparticles by Pulse Electrodeposition Followed by Heat-Treatment Method. J. Ultrafine Grained Nanostructured Mater. 2016, 49, 80–86. [Google Scholar] [CrossRef]
- Lee, W.; Kim, K.W.; Lim, J.E.; Sarkar, S.; Kim, J.Y.; Chang, Y.; Yoo, J. In vivo evaluation of the effects of combined boron and gadolinium neutron capture therapy in mouse models. Sci. Rep. 2022, 12, 13360. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitali, A.; Demichelis, M.P.; Di Martino, G.; Postuma, I.; Bortolussi, S.; Falqui, A.; Milanese, C.; Ferrara, C.; Sommi, P.; Anselmi-Tamburini, U. Synthesis and Characterization of Gd-Functionalized B4C Nanoparticles for BNCT Applications. Life 2023, 13, 429. https://doi.org/10.3390/life13020429
Vitali A, Demichelis MP, Di Martino G, Postuma I, Bortolussi S, Falqui A, Milanese C, Ferrara C, Sommi P, Anselmi-Tamburini U. Synthesis and Characterization of Gd-Functionalized B4C Nanoparticles for BNCT Applications. Life. 2023; 13(2):429. https://doi.org/10.3390/life13020429
Chicago/Turabian StyleVitali, Agostina, Maria Paola Demichelis, Greta Di Martino, Ian Postuma, Silva Bortolussi, Andrea Falqui, Chiara Milanese, Chiara Ferrara, Patrizia Sommi, and Umberto Anselmi-Tamburini. 2023. "Synthesis and Characterization of Gd-Functionalized B4C Nanoparticles for BNCT Applications" Life 13, no. 2: 429. https://doi.org/10.3390/life13020429
APA StyleVitali, A., Demichelis, M. P., Di Martino, G., Postuma, I., Bortolussi, S., Falqui, A., Milanese, C., Ferrara, C., Sommi, P., & Anselmi-Tamburini, U. (2023). Synthesis and Characterization of Gd-Functionalized B4C Nanoparticles for BNCT Applications. Life, 13(2), 429. https://doi.org/10.3390/life13020429