
Chronic Neuroinflammation and Cognitive Decline in Patients with Cardiac Disease: Evidence, Relevance, and Therapeutic Implications



Abstract
1. Introduction
2. Cognitive Decline and Dementia in Common Cardiac Disorders
2.1. Myocardial Infarction and Coronary Artery Disease
2.2. Heart Failure
2.3. Hypertension
2.4. Atrial Fibrillation
2.5. Aortic Valve Stenosis
2.6. Cardiac Arrest
3. Acute vs. Chronic Neuroinflammation
4. Chronic Neuroinflammation in Neurodegenerative Disorders
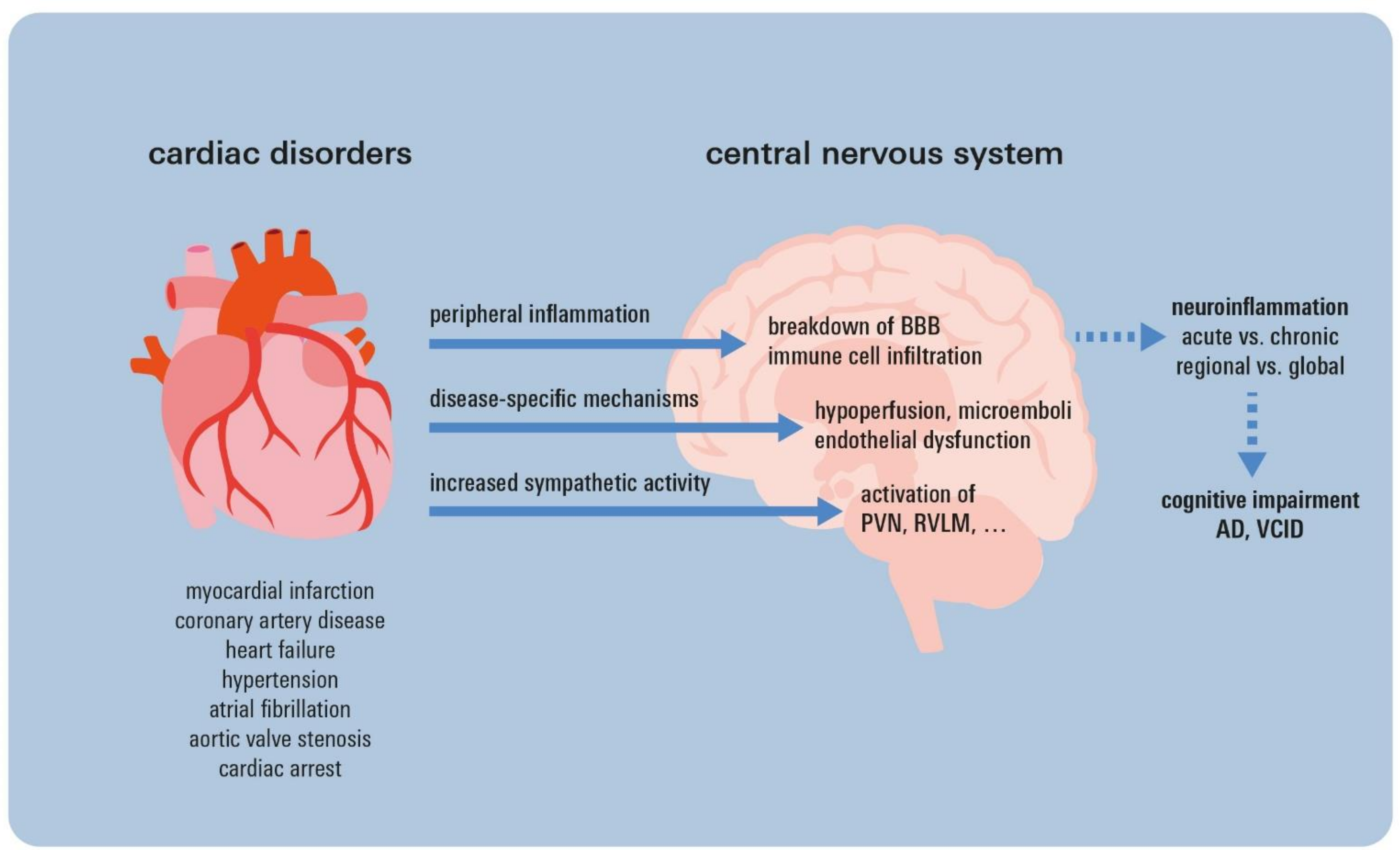
5. Neuroinflammatory Sequelae of Cardiac Disorders
5.1. Myocardial Infarction and Coronary Artery Disease
5.2. Heart Failure
5.3. Hypertension
5.4. Atrial Fibrillation
5.5. Aortic Valve Stenosis
5.6. Cardiac Arrest
6. Potential Biomarkers
7. Therapeutic Implications
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef]
- Kobayashi, M.; Voors, A.A.; Girerd, N.; Billotte, M.; Anker, S.D.; Cleland, J.G.; Lang, C.C.; Ng, L.L.; van Veldhuisen, D.J.; Dickstein, K.; et al. Heart failure etiologies and clinical factors precipitating for worsening heart failure: Findings from BIOSTAT-CHF. Eur. J. Intern. Med. 2020, 71, 62–69. [Google Scholar] [CrossRef]
- Zhang, W.; Luo, P. Myocardial Infarction Predisposes Neurodegenerative Diseases. J. Alzheimers Dis. 2020, 74, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Wolters, F.J.; Segufa, R.A.; Darweesh, S.K.L.; Bos, D.; Ikram, M.A.; Sabayan, B.; Hofman, A.; Sedaghat, S. Coronary heart disease, heart failure, and the risk of dementia: A systematic review and meta-analysis. Alzheimers Dement. 2018, 14, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Zuidersma, M.; Thombs, B.D.; de Jonge, P. Onset and recurrence of depression as predictors of cardiovascular prognosis in depressed acute coronary syndrome patients: A systematic review. Psychother. Psychosom. 2011, 80, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Sun, D.; Wang, Y.; Yan, M.; Zheng, J.; Ren, J. Cognitive Impairment in Heart Failure: Landscape, Challenges, and Future Directions. Front. Cardiovasc. Med. 2021, 8, 831734. [Google Scholar] [CrossRef]
- Frantz, S.; Hundertmark, M.J.; Schulz-Menger, J.; Bengel, F.M.; Bauersachs, J. Left ventricular remodelling post-myocardial infarction: Pathophysiology, imaging, and novel therapies. Eur. Heart J. 2022, 43, 2549–2561. [Google Scholar] [CrossRef]
- Thorp, E.B.; Flanagan, M.E.; Popko, B.; DeBerge, M. Resolving inflammatory links between myocardial infarction and vascular dementia. Semin. Immunol. 2022, 59, 101600. [Google Scholar] [CrossRef]
- Breteler, M.M.; Claus, J.J.; Grobbee, D.E.; Hofman, A. Cardiovascular disease and distribution of cognitive function in elderly people: The Rotterdam Study. BMJ 1994, 308, 1604–1608. [Google Scholar] [CrossRef]
- Ikram, M.A.; van Oijen, M.; de Jong, F.J.; Kors, J.A.; Koudstaal, P.J.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Unrecognized myocardial infarction in relation to risk of dementia and cerebral small vessel disease. Stroke 2008, 39, 1421–1426. [Google Scholar] [CrossRef]
- Aronson, M.K.; Ooi, W.L.; Morgenstern, H.; Hafner, A.; Masur, D.; Crystal, H.; Frishman, W.H.; Fisher, D.; Katzman, R. Women, myocardial infarction, and dementia in the very old. Neurology 1990, 40, 1102–1106. [Google Scholar] [CrossRef]
- Xie, W.; Zheng, F.; Yan, L.; Zhong, B. Cognitive Decline Before and After Incident Coronary Events. J. Am. Coll. Cardiol. 2019, 73, 3041–3050. [Google Scholar] [CrossRef]
- Gu, S.Z.; Beska, B.; Chan, D.; Neely, D.; Batty, J.A.; Adams-Hall, J.; Mossop, H.; Qiu, W.; Kunadian, V. Cognitive Decline in Older Patients With Non- ST Elevation Acute Coronary Syndrome. J. Am. Heart Assoc. 2019, 8, e011218. [Google Scholar] [CrossRef] [PubMed]
- Haring, B.; Leng, X.; Robinson, J.; Johnson, K.C.; Jackson, R.D.; Beyth, R.; Wactawski-Wende, J.; von Ballmoos, M.W.; Goveas, J.S.; Kuller, L.H.; et al. Cardiovascular disease and cognitive decline in postmenopausal women: Results from the Women’s Health Initiative Memory Study. J. Am. Heart Assoc. 2013, 2, e000369. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Panza, F.; Colacicco, A.M.; D’Introno, A.; Capurso, C.; Torres, F.; Grigoletto, F.; Maggi, S.; Del Parigi, A.; Reiman, E.M.; et al. Vascular risk factors, incidence of MCI, and rates of progression to dementia. Neurology 2004, 63, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Sundboll, J.; Horvath-Puho, E.; Adelborg, K.; Schmidt, M.; Pedersen, L.; Botker, H.E.; Henderson, V.W.; Sorensen, H.T. Higher Risk of Vascular Dementia in Myocardial Infarction Survivors. Circulation 2018, 137, 567–577. [Google Scholar] [CrossRef]
- Zheng, L.; Mack, W.J.; Chui, H.C.; Heflin, L.; Mungas, D.; Reed, B.; DeCarli, C.; Weiner, M.W.; Kramer, J.H. Coronary artery disease is associated with cognitive decline independent of changes on magnetic resonance imaging in cognitively normal elderly adults. J. Am. Geriatr. Soc. 2012, 60, 499–504. [Google Scholar] [CrossRef]
- Lima, L.M.; Carvalho, M.; Ferreira, C.N.; Fernandes, A.P.; Neto, C.P.; Garcia, J.C.; Reis, H.J.; Janka, Z.; Palotas, A.; Sousa, M. Atheromatosis extent in coronary artery disease is not correlated with apolipoprotein-E polymorphism and its plasma levels, but associated with cognitive decline. Curr. Alzheimer Res. 2010, 7, 556–563. [Google Scholar] [CrossRef]
- Slawson, D.C. No Difference in Cognitive Decline in Older Patients with Coronary Artery Disease Undergoing CABG or PCI. Am. Fam. Physician 2021, 104, 422. [Google Scholar]
- Toyama, K.; Sugiyama, S.; Oka, H.; Hamada, M.; Iwasaki, Y.; Horio, E.; Rokutanda, T.; Nakamura, S.; Spin, J.M.; Tsao, P.S.; et al. A Pilot Study: The Beneficial Effects of Combined Statin-exercise Therapy on Cognitive Function in Patients with Coronary Artery Disease and Mild Cognitive Decline. Intern. Med. 2017, 56, 641–649. [Google Scholar] [CrossRef]
- Lutski, M.; Weinstein, G.; Goldbourt, U.; Tanne, D. Cardiovascular Health and Cognitive Decline 2 Decades Later in Men with Preexisting Coronary Artery Disease. Am. J. Cardiol. 2018, 121, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Zuccala, G.; Marzetti, E.; Cesari, M.; Lo Monaco, M.R.; Antonica, L.; Cocchi, A.; Carbonin, P.; Bernabei, R. Correlates of cognitive impairment among patients with heart failure: Results of a multicenter survey. Am. J. Med. 2005, 118, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Teo, S.Y.; Kang, K.; Tan, M.; Ling, L.H.; Yeo, P.S.D.; Sim, D.; Jaufeerally, F.; Leong, K.T.G.; Ong, H.Y.; et al. Cognitive impairment in Asian patients with heart failure: Prevalence, biomarkers, clinical correlates, and outcomes. Eur. J. Heart Fail. 2019, 21, 688–690. [Google Scholar] [CrossRef]
- Frey, A.; Sell, R.; Homola, G.A.; Malsch, C.; Kraft, P.; Gunreben, I.; Morbach, C.; Alkonyi, B.; Schmid, E.; Colonna, I.; et al. Cognitive Deficits and Related Brain Lesions in Patients With Chronic Heart Failure. JACC Heart Fail. 2018, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Festa, J.R.; Jia, X.; Cheung, K.; Marchidann, A.; Schmidt, M.; Shapiro, P.A.; Mancini, D.M.; Naka, Y.; Deng, M.; Lantz, E.R.; et al. Association of low ejection fraction with impaired verbal memory in older patients with heart failure. Arch. Neurol. 2011, 68, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, A.M.; Kiefe, C.I.; Person, S.D.; Gore, J.G.; Saczynski, J.S. Cognitive change in heart failure: A systematic review. Circ. Cardiovasc. Qual. Outcomes 2013, 6, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Ampadu, J.; Morley, J.E. Heart failure and cognitive dysfunction. Int. J. Cardiol. 2015, 178, 12–23. [Google Scholar] [CrossRef]
- Qiu, C.; Winblad, B.; Marengoni, A.; Klarin, I.; Fastbom, J.; Fratiglioni, L. Heart failure and risk of dementia and Alzheimer disease: A population-based cohort study. Arch. Intern. Med. 2006, 166, 1003–1008. [Google Scholar] [CrossRef]
- Adelborg, K.; Horvath-Puho, E.; Ording, A.; Pedersen, L.; Sorensen, H.T.; Henderson, V.W. Heart failure and risk of dementia: A Danish nationwide population-based cohort study. Eur. J. Heart Fail. 2017, 19, 253–260. [Google Scholar] [CrossRef]
- Geer, J.H.; Jeon, S.; O’Connell, M.; Linsky, S.; Conley, S.; Hollenbeak, C.S.; Jacoby, D.; Yaggi, H.K.; Redeker, N.S. Correlates of cognition among people with chronic heart failure and insomnia. Sleep Breath, 2022; online ahead of print. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, R.; Dong, J. Effect of Chronic Heart Failure Complicated with Type 2 Diabetes Mellitus on Cognitive Function in the Elderly. Evid. Based Complement. Alternat. Med. 2022, 2022, 4841205. [Google Scholar] [CrossRef] [PubMed]
- Ventoulis, I.; Arfaras-Melainis, A.; Parissis, J.; Polyzogopoulou, E. Cognitive Impairment in Acute Heart Failure: Narrative Review. J. Cardiovasc. Dev. Dis. 2021, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Mansukhani, M.P.; Kolla, B.P.; Somers, V.K. Hypertension and Cognitive Decline: Implications of Obstructive Sleep Apnea. Front. Cardiovasc. Med. 2019, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Hanon, O.; Seux, M.L.; Lenoir, H.; Rigaud, A.S.; Forette, F. Hypertension and dementia. Curr. Cardiol. Rep. 2003, 5, 435–440. [Google Scholar] [CrossRef]
- Vicario, A.; Martinez, C.D.; Baretto, D.; Diaz Casale, A.; Nicolosi, L. Hypertension and cognitive decline: Impact on executive function. J. Clin. Hypertens. 2005, 7, 598–604. [Google Scholar] [CrossRef]
- Cheon, E.J. Hypertension and cognitive dysfunction: A narrative review. J. Yeungnam. Med. Sci. 2022; epub ahead of print. [Google Scholar] [CrossRef]
- Ding, L.; Zhu, X.; Xiong, Z.; Yang, F.; Zhang, X. The Association of Age at Diagnosis of Hypertension with Cognitive Decline: The China Health and Retirement Longitudinal Study (CHARLS). J. Gen. Intern. Med. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Turana, Y.; Tengkawan, J.; Chia, Y.C.; Hoshide, S.; Shin, J.; Chen, C.H.; Buranakitjaroen, P.; Nailes, J.; Park, S.; Siddique, S.; et al. Hypertension and Dementia: A comprehensive review from the HOPE Asia Network. J. Clin. Hypertens. 2019, 21, 1091–1098. [Google Scholar] [CrossRef]
- Islam, M.M.; Poly, T.N.; Walther, B.A.; Yang, H.C.; Wu, C.C.; Lin, M.C.; Chien, S.C.; Li, Y.C. Association Between Atrial Fibrillation and Dementia: A Meta-Analysis. Front. Aging Neurosci. 2019, 11, 305. [Google Scholar] [CrossRef]
- Morales-Bacas, E.; Duque-Holguera, M.; Portilla-Cuenca, J.C.; Casado-Naranjo, I. Atrial fibrillation and cognitive impairment: A narrative review. Rev. Neurol. 2022, 75, 311–318. [Google Scholar] [CrossRef]
- Kim, D.; Yang, P.S.; Joung, B. Prevention of Dementia in Patients with Atrial Fibrillation. Korean Circ J 2021, 51, 308–319. [Google Scholar] [CrossRef]
- Kim, D.; Yang, P.S.; Sung, J.H.; Jang, E.; Yu, H.T.; Kim, T.H.; Uhm, J.S.; Kim, J.Y.; Pak, H.N.; Lee, M.H.; et al. Less dementia after catheter ablation for atrial fibrillation: A nationwide cohort study. Eur. Heart J. 2020, 41, 4483–4493. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Jiang, C.; Lai, Y.; Wang, Y.; Li, S.; He, L.; Tang, R.; Sang, C.; Long, D.; Du, X.; et al. Association Between Atrial Fibrillation and Domain-Specific Cognitive Decline—Insights From the Systolic Blood Pressure Intervention Trial. Circ. J. 2022, 87, 20–26. [Google Scholar] [CrossRef]
- Myers, S.J.; Jimenez-Ruiz, A.; Sposato, L.A.; Whitehead, S.N. Atrial cardiopathy and cognitive impairment. Front. Aging Neurosci. 2022, 14, 914360. [Google Scholar] [CrossRef] [PubMed]
- Ndunda, P.M.; Vindhyal, M.R.; Muutu, T.M.; Fanari, Z. Clinical Outcomes of Sentinel Cerebral Protection System Use During Transcatheter Aortic Valve Replacement: A Systematic Review and Meta-Analysis. Cardiovasc. Revasc. Med. 2020, 21, 717–722. [Google Scholar] [CrossRef]
- Wolters, F.J.; Bos, D.; Vernooij, M.W.; Franco, O.H.; Heart-Brain Connection collaborative research group; Hofman, A.; Koudstaal, P.J.; van der Lugt, A.; Ikram, M.A. Aortic Valve Calcification and the Risk of dementia: A Population-Based Study. J. Alzheimers Dis. 2017, 55, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, S.; Matsumoto, Y.; Suzuki, H.; Takanami, K.; Kikuchi, Y.; Takahashi, J.; Miyata, S.; Tomita, N.; Kumagai, K.; Taki, Y.; et al. Transcatheter aortic valve implantation and cognitive function in elderly patients with severe aortic stenosis. EuroIntervention 2020, 15, e1580–e1587. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.S.; Herrmann, N.; Saleem, M.; Lanctot, K.L. Cognitive Outcomes following Transcatheter Aortic Valve Implantation: A Systematic Review. Cardiovasc. Psychiatry Neurol. 2015, 2015, 209569. [Google Scholar] [CrossRef]
- Volpp, K.G.; Abella, B.S. Improving Out-of-Hospital Cardiac Arrest Survival Rates-Optimization Given Constraints. JAMA Cardiol 2022, 8, 8–9. [Google Scholar] [CrossRef]
- Moulaert, V.R.; Verbunt, J.A.; van Heugten, C.M.; Wade, D.T. Cognitive impairments in survivors of out-of-hospital cardiac arrest: A systematic review. Resuscitation 2009, 80, 297–305. [Google Scholar] [CrossRef]
- Buanes, E.A.; Gramstad, A.; Sovig, K.K.; Hufthammer, K.O.; Flaatten, H.; Husby, T.; Langorgen, J.; Heltne, J.K. Cognitive function and health-related quality of life four years after cardiac arrest. Resuscitation 2015, 89, 13–18. [Google Scholar] [CrossRef]
- Caro-Codon, J.; Rey, J.R.; Lopez-de-Sa, E.; Gonzalez Fernandez, O.; Rosillo, S.O.; Armada, E.; Iniesta, A.M.; Fernandez de Bobadilla, J.; Ruiz Cantador, J.; Rodriguez Sotelo, L.; et al. Long-term neurological outcomes in out-of-hospital cardiac arrest patients treated with targeted-temperature management. Resuscitation 2018, 133, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Byron-Alhassan, A.; Collins, B.; Bedard, M.; Quinlan, B.; Le May, M.; Duchesne, L.; Osborne, C.; Wells, G.; Smith, A.M.; Tulloch, H.E. Cognitive dysfunction after out-of-hospital cardiac arrest: Rate of impairment and clinical predictors. Resuscitation 2021, 165, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Orbo, M.; Aslaksen, P.M.; Larsby, K.; Norli, L.; Schafer, C.; Tande, P.M.; Vangberg, T.R.; Anke, A. Determinants of cognitive outcome in survivors of out-of-hospital cardiac arrest. Resuscitation 2014, 85, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Mayer, C.L.; Huber, B.R.; Peskind, E. Traumatic brain injury, neuroinflammation, and post-traumatic headaches. Headache 2013, 53, 1523–1530. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Wang, M.; Pan, W.; Xu, Y.; Zhang, J.; Wan, J.; Jiang, H. Microglia-Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases. J. Inflamm. Res. 2022, 15, 3083–3094. [Google Scholar] [CrossRef]
- Cheng, K.; Wang, J.; Chen, Q.; Zhao, G.; Pang, Y.; Xu, Y.; Ge, J.; Zhu, W. Inflammasome-mediated neurodegeneration following heart disease. Ann. Transl. Med. 2021, 9, 1560. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S. Glia and their cytokines in progression of neurodegeneration. Neurobiol. Aging 2005, 26, 349–354. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef]
- Ayyubova, G. Dysfunctional microglia and tau pathology in Alzheimer’s disease. Rev. Neurosci. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Tian, Z.; Ji, X.; Liu, J. Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects. Int. J. Mol. Sci. 2022, 23, 6224. [Google Scholar] [CrossRef]
- Simpson, J.E.; Fernando, M.S.; Clark, L.; Ince, P.G.; Matthews, F.; Forster, G.; O’Brien, J.T.; Barber, R.; Kalaria, R.N.; Brayne, C.; et al. White matter lesions in an unselected cohort of the elderly: Astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol. Appl. Neurobiol. 2007, 33, 410–419. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Hupe, H.C.; Wang, Y.; Bankstahl, J.P.; Berding, G.; Ross, T.L.; Bauersachs, J.; Wollert, K.C.; Bengel, F.M. Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 263–275. [Google Scholar] [CrossRef]
- Bascunana, P.; Hess, A.; Borchert, T.; Wang, Y.; Wollert, K.C.; Bengel, F.M.; Thackeray, J.T. (11)C-Methionine PET Identifies Astroglia Involvement in Heart-Brain Inflammation Networking After Acute Myocardial Infarction. J. Nucl. Med. 2020, 61, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Dworak, M.; Stebbing, M.; Kompa, A.R.; Rana, I.; Krum, H.; Badoer, E. Attenuation of microglial and neuronal activation in the brain by ICV minocycline following myocardial infarction. Auton. Neurosci. 2014, 185, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Rana, I.; Stebbing, M.; Kompa, A.; Kelly, D.J.; Krum, H.; Badoer, E. Microglia activation in the hypothalamic PVN following myocardial infarction. Brain Res. 2010, 1326, 96–104. [Google Scholar] [CrossRef]
- Dworak, M.; Stebbing, M.; Kompa, A.R.; Rana, I.; Krum, H.; Badoer, E. Sustained activation of microglia in the hypothalamic PVN following myocardial infarction. Auton. Neurosci. 2012, 169, 70–76. [Google Scholar] [CrossRef]
- Rinaldi, B.; Guida, F.; Furiano, A.; Donniacuo, M.; Luongo, L.; Gritti, G.; Urbanek, K.; Messina, G.; Maione, S.; Rossi, F.; et al. Effect of Prolonged Moderate Exercise on the Changes of Nonneuronal Cells in Early Myocardial Infarction. Neural Plast. 2015, 2015, 265967. [Google Scholar] [CrossRef]
- Yuan, S.; Zhang, X.; Bo, Y.; Li, W.; Zhang, H.; Jiang, Q. The effects of electroacupuncture treatment on the postoperative cognitive function in aged rats with acute myocardial ischemia-reperfusion. Brain Res. 2014, 1593, 19–29. [Google Scholar] [CrossRef]
- Frick, T.; Springe, D.; Grandgirard, D.; Leib, S.L.; Haenggi, M. An improved simple rat model for global cerebral ischaemia by induced cardiac arrest. Neurol. Res. 2016, 38, 373–380. [Google Scholar] [CrossRef]
- Wang, H.W.; Ahmad, M.; Jadayel, R.; Najjar, F.; Lagace, D.; Leenen, F.H.H. Inhibition of inflammation by minocycline improves heart failure and depression-like behaviour in rats after myocardial infarction. PLoS ONE 2019, 14, e0217437. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yin, D.; He, X.; Gao, M.; Choi, Y.; Luo, G.; Wang, H.; Qu, X. Modulation of activated astrocytes in the hypothalamus paraventricular nucleus to prevent ventricular arrhythmia complicating acute myocardial infarction. Int. J. Cardiol. 2020, 308, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Mei, Y.; Hu, Z.; Xing, W.; Lv, K.; Hu, N.; Zhang, T.; Wang, D. Ghrelin attenuates depressive-like behavior, heart failure, and neuroinflammation in postmyocardial infarction rat model. Eur. J. Pharmacol. 2021, 901, 174096. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, F.F.; Naxerova, K.; Schloss, M.J.; Hulsmans, M.; Nair, A.V.; Dutta, P.; Calcagno, D.M.; Herisson, F.; Anzai, A.; Sun, Y.; et al. Tissue-Specific Macrophage Responses to Remote Injury Impact the Outcome of Subsequent Local Immune Challenge. Immunity 2019, 51, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Shemer, A.; Grozovski, J.; Tay, T.L.; Tao, J.; Volaski, A.; Suss, P.; Ardura-Fabregat, A.; Gross-Vered, M.; Kim, J.S.; David, E.; et al. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat. Commun. 2018, 9, 5206. [Google Scholar] [CrossRef] [PubMed]
- Cugurra, A.; Mamuladze, T.; Rustenhoven, J.; Dykstra, T.; Beroshvili, G.; Greenberg, Z.J.; Baker, W.; Papadopoulos, Z.; Drieu, A.; Blackburn, S.; et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 2021, 373, eabf7844. [Google Scholar] [CrossRef]
- Kumar, V.; Prabhu, S.D.; Bansal, S.S. CD4(+) T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front. Cardiovasc. Med. 2022, 9, 992653. [Google Scholar] [CrossRef]
- Traub, J.; Schurmann, P.; Schmitt, D.; Gassenmaier, T.; Fette, G.; Frantz, S.; Stork, S.; Beyersdorf, N.; Boivin-Jahns, V.; Jahns, R.; et al. Features of metabolic syndrome and inflammation independently affect left ventricular function early after first myocardial infarction. Int. J. Cardiol. 2023, 370, 43–50. [Google Scholar] [CrossRef]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef]
- Iba, M.; Kim, C.; Sallin, M.; Kwon, S.; Verma, A.; Overk, C.; Rissman, R.A.; Sen, R.; Sen, J.M.; Masliah, E. Neuroinflammation is associated with infiltration of T cells in Lewy body disease and alpha-synuclein transgenic models. J. Neuroinflammation. 2020, 17, 214. [Google Scholar] [CrossRef]
- Hong, X.; Bu, L.; Wang, Y.; Xu, J.; Wu, J.; Huang, Y.; Liu, J.; Suo, H.; Yang, L.; Shi, Y.; et al. Increases in the risk of cognitive impairment and alterations of cerebral beta-amyloid metabolism in mouse model of heart failure. PLoS ONE 2013, 8, e63829. [Google Scholar] [CrossRef]
- Guggilam, A.; Patel, K.P.; Haque, M.; Ebenezer, P.J.; Kapusta, D.R.; Francis, J. Cytokine blockade attenuates sympathoexcitation in heart failure: Cross-talk between nNOS, AT-1R and cytokines in the hypothalamic paraventricular nucleus. Eur. J. Heart Fail. 2008, 10, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Diaz, H.S.; Toledo, C.; Andrade, D.C.; Marcus, N.J.; Del Rio, R. Neuroinflammation in heart failure: New insights for an old disease. J. Physiol. 2020, 598, 33–59. [Google Scholar] [CrossRef]
- Isegawa, K.; Hirooka, Y.; Katsuki, M.; Kishi, T.; Sunagawa, K. Angiotensin II type 1 receptor expression in astrocytes is upregulated leading to increased mortality in mice with myocardial infarction-induced heart failure. Am. J. Physiol. Heart Circ. Physiol 2014, 307, H1448–H1455. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, K.; Bhandare, A.M.; Nedoboy, P.E.; Mohammed, S.; Farnham, M.M.; Pilowsky, P.M. Dynamic changes in the relationship of microglia to cardiovascular neurons in response to increases and decreases in blood pressure. Neuroscience 2016, 329, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Rosenzweig, R.; Asalla, S.; Nehra, S.; Prabhu, S.D.; Bansal, S.S. TNFR1 Contributes to Activation-Induced Cell Death of Pathological CD4(+) T Lymphocytes During Ischemic Heart Failure. JACC Basic Transl. Sci. 2022, 7, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Westland, K.W.; Pollard, J.D.; Sander, S.; Bonner, J.G.; Linington, C.; McLeod, J.G. Activated non-neural specific T cells open the blood-brain barrier to circulating antibodies. Brain 1999, 122, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Du, L.; Jiang, J.; Yang, M.; Wang, Z.; Wang, Y.; Tang, T.; Fu, X.; Hao, J. Microglial TREM2 Mitigates Inflammatory Responses and Neuronal Apoptosis in Angiotensin II-Induced Hypertension in Middle-Aged Mice. Front. Aging Neurosci. 2021, 13, 716917. [Google Scholar] [CrossRef]
- Carnevale, D.; Mascio, G.; Ajmone-Cat, M.A.; D’Andrea, I.; Cifelli, G.; Madonna, M.; Cocozza, G.; Frati, A.; Carullo, P.; Carnevale, L.; et al. Role of neuroinflammation in hypertension-induced brain amyloid pathology. Neurobiol. Aging 2012, 33, 205.e19–205.e29. [Google Scholar] [CrossRef]
- Bajwa, E.; Klegeris, A. Neuroinflammation as a mechanism linking hypertension with the increased risk of Alzheimer’s disease. Neural. Regen. Res. 2022, 17, 2342–2346. [Google Scholar] [CrossRef]
- Mowry, F.E.; Biancardi, V.C. Neuroinflammation in hypertension: The renin-angiotensin system versus pro-resolution pathways. Pharmacol. Res. 2019, 144, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Masson, G.S.; Nair, A.R.; Silva Soares, P.P.; Michelini, L.C.; Francis, J. Aerobic training normalizes autonomic dysfunction, HMGB1 content, microglia activation and inflammation in hypothalamic paraventricular nucleus of SHR. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1115–H1122. [Google Scholar] [CrossRef] [PubMed]
- Lappegard, K.T.; Pop-Purceleanu, M.; van Heerde, W.; Sexton, J.; Tendolkar, I.; Pop, G. Improved neurocognitive functions correlate with reduced inflammatory burden in atrial fibrillation patients treated with intensive cholesterol lowering therapy. J. Neuroinflamm. 2013, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, J.; Lappegard, K.T.; Sexton, J.; Plesiewicz, I.; Lap, P.; Bouwels, L.; Sprong, T.; Mollnes, T.E.; Verheugt, F.; van Heerde, W.L.; et al. Persisting thrombin activity in elderly patients with atrial fibrillation on oral anticoagulation is decreased by anti-inflammatory therapy with intensive cholesterol-lowering treatment. J. Clin. Lipidol. 2011, 5, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Ousta, A.; Piao, L.; Fang, Y.H.; Vera, A.; Nallamothu, T.; Garcia, A.J., 3rd; Sharp, W.W. Microglial Activation and Neurological Outcomes in a Murine Model of Cardiac Arrest. Neurocrit. Care 2022, 36, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Zhu, J.; Wang, D.; Li, H.; He, Y.; Liu, K.; Wang, X.; Peng, Y.; Pan, S.; Huang, K. NLRP3 inflammasome-mediated microglial pyroptosis is critically involved in the development of post-cardiac arrest brain injury. J. Neuroinflamm. 2020, 17, 219. [Google Scholar] [CrossRef]
- Ostovaneh, M.R.; Moazzami, K.; Yoneyama, K.; Venkatesh, B.A.; Heckbert, S.R.; Wu, C.O.; Shea, S.; Post, W.S.; Fitzpatrick, A.L.; Burke, G.L.; et al. Change in NT-proBNP (N-Terminal Pro-B-Type Natriuretic Peptide) Level and Risk of Dementia in Multi-Ethnic Study of Atherosclerosis (MESA). Hypertension 2020, 75, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, P.; Sabayan, B.; Wijsman, L.W.; Poortvliet, R.K.; Mooijaart, S.P.; de Ruijter, W.; Gussekloo, J.; de Craen, A.J.; Westendorp, R.G. NT-proBNP, blood pressure, and cognitive decline in the oldest old: The Leiden 85-plus Study. Neurology 2014, 83, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.A.; Aimo, A.; Bernardini, S.; Clerico, A. Inflammageing and Cardiovascular System: Focus on Cardiokines and Cardiac-Specific Biomarkers. Int. J. Mol. Sci. 2023, 24, 844. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nistico, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhao, K.; Xia, H.; Xu, Y. Peripheral inflammatory biomarkers in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis. Psychogeriatrics 2019, 19, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Traub, J.; Otto, M.; Sell, R.; Gopfert, D.; Homola, G.; Steinacker, P.; Oeckl, P.; Morbach, C.; Frantz, S.; Pham, M.; et al. Serum phosphorylated tau protein 181 and neurofilament light chain in cognitively impaired heart failure patients. Alzheimers Res. Ther. 2022, 14, 149. [Google Scholar] [CrossRef] [PubMed]
- Traub, J.; Otto, M.; Sell, R.; Homola, G.A.; Steinacker, P.; Oeckl, P.; Morbach, C.; Frantz, S.; Pham, M.; Stork, S.; et al. Serum glial fibrillary acidic protein indicates memory impairment in patients with chronic heart failure. ESC Heart Fail. 2022, 9, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Traub, J.; Grondey, K.; Gassenmaier, T.; Schmitt, D.; Fette, G.; Frantz, S.; Boivin-Jahns, V.; Jahns, R.; Stork, S.; Stoll, G.; et al. Sustained Increase in Serum Glial Fibrillary Acidic Protein after First ST-Elevation Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 10304. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.Y.; Wang, X.; Ip, N.Y. Targeting Neuroinflammation as a Therapeutic Strategy for Alzheimer’s Disease: Mechanisms, Drug Candidates, and New Opportunities. ACS Chem. Neurosci. 2019, 10, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Budni, J.; Garcez, M.L.; de Medeiros, J.; Cassaro, E.; Bellettini-Santos, T.; Mina, F.; Quevedo, J. The Anti-Inflammatory Role of Minocycline in Alzheimer s Disease. Curr. Alzheimer Res. 2016, 13, 1319–1329. [Google Scholar] [CrossRef]
- Seabrook, T.J.; Jiang, L.; Maier, M.; Lemere, C.A. Minocycline affects microglia activation, Abeta deposition, and behavior in APP-tg mice. Glia 2006, 53, 776–782. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, H.S.; Shin, K.Y.; Kim, E.M.; Kim, M.; Kim, H.S.; Park, C.H.; Jeong, Y.H.; Yoo, J.; Lee, J.P.; et al. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology 2007, 32, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Feng, N.; Liu, Y.C.; Guo, Q.; Wang, J.K.; Bai, Y.Z.; Ye, X.M.; Yang, Z.; Yang, H.; Liu, Y.; et al. Neuroinflammation inhibition by small-molecule targeting USP7 noncatalytic domain for neurodegenerative disease therapy. Sci. Adv. 2022, 8, eabo0789. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cardiac Disease (Chapter in Article) | Evidence of (Chronic) Neuroinflammation | ||
|---|---|---|---|
| Preclinical Models | Human Studies | ||
| Myocardial infarction/coronary artery disease (5.1) | Microglial activation in the RVLM, NTS, PAG, and PVN [77,78,79] | TSPO on microglia in the cerebellum, temporal and frontobasal cortex, and hypothalamus [75,76] | |
| Phenotypic global changes to microglia [80,81,82] | |||
| Astroglial activation [76,84,85] | |||
| Monocyte and neutrophil abundance [87,88,89] | |||
| Heart failure (5.2) | Inflammatory genes in the cortex and hippocampus [94] | ||
| Astrocyte activation in the RVLM [97] | |||
| Microglia co-localization and activation in the PVN, RVLM, and NTS [98] | |||
| Hypertension (5.3) | Activation and TREM2 production by microglia in the cortex and hippocampus [102] | ||
| Neuroinflammation before amyloid-beta deposition [102,103] | |||
| Pro-inflammatory pathways involved in sympathetic control [105] | |||
| Atrial fibrillation (5.4) | Interleukin-1β and tumor necrosis factor-α levels [107] | ||
| Aortic valve stenosis (5.5) | |||
| Cardiac arrest (5.6) | Microglial activation and neurodegeneration in the cornu ammonis area 1 [109] | ||
| NLRP3 inflammasome [110] | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traub, J.; Frey, A.; Störk, S. Chronic Neuroinflammation and Cognitive Decline in Patients with Cardiac Disease: Evidence, Relevance, and Therapeutic Implications. Life 2023, 13, 329. https://doi.org/10.3390/life13020329
Traub J, Frey A, Störk S. Chronic Neuroinflammation and Cognitive Decline in Patients with Cardiac Disease: Evidence, Relevance, and Therapeutic Implications. Life. 2023; 13(2):329. https://doi.org/10.3390/life13020329
Chicago/Turabian StyleTraub, Jan, Anna Frey, and Stefan Störk. 2023. "Chronic Neuroinflammation and Cognitive Decline in Patients with Cardiac Disease: Evidence, Relevance, and Therapeutic Implications" Life 13, no. 2: 329. https://doi.org/10.3390/life13020329
APA StyleTraub, J., Frey, A., & Störk, S. (2023). Chronic Neuroinflammation and Cognitive Decline in Patients with Cardiac Disease: Evidence, Relevance, and Therapeutic Implications. Life, 13(2), 329. https://doi.org/10.3390/life13020329