
Modeling the Effect on a Novel Fungal Peptaibol Placed in an All-Atom Bacterial Membrane Mimicking System via Accelerated Molecular Dynamics Simulations

 ,
,  , , and
, , and 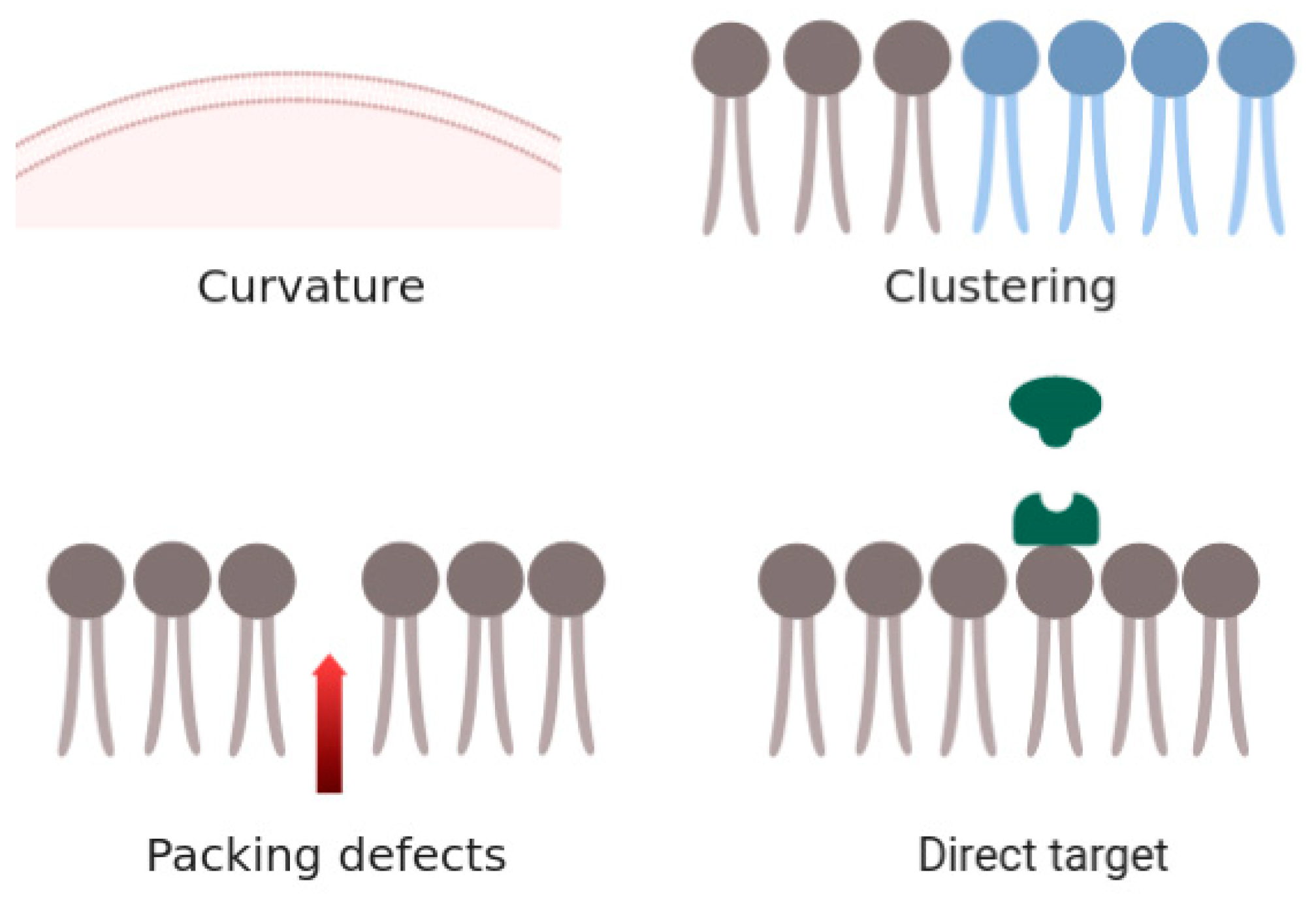{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
- AcAib1-Ser2-Ala3-Aib4-Vxx5-Gln6-Vxx7-Aib8-Vxx9-Ala10-Vxx11-Aib12-Pro13-Lxx14-Aib15-Vxx16-Gln17-Pheol18
3. Results
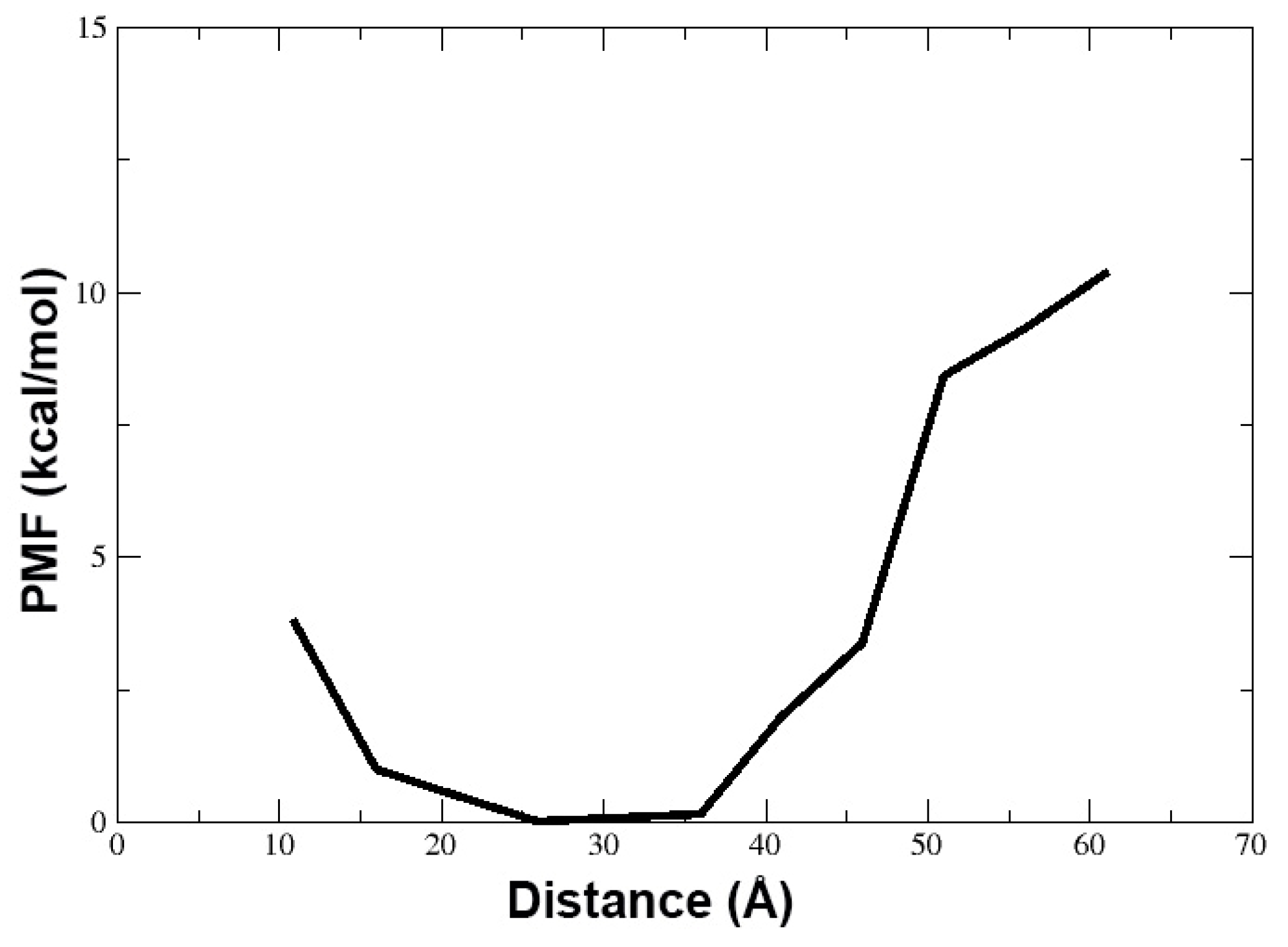
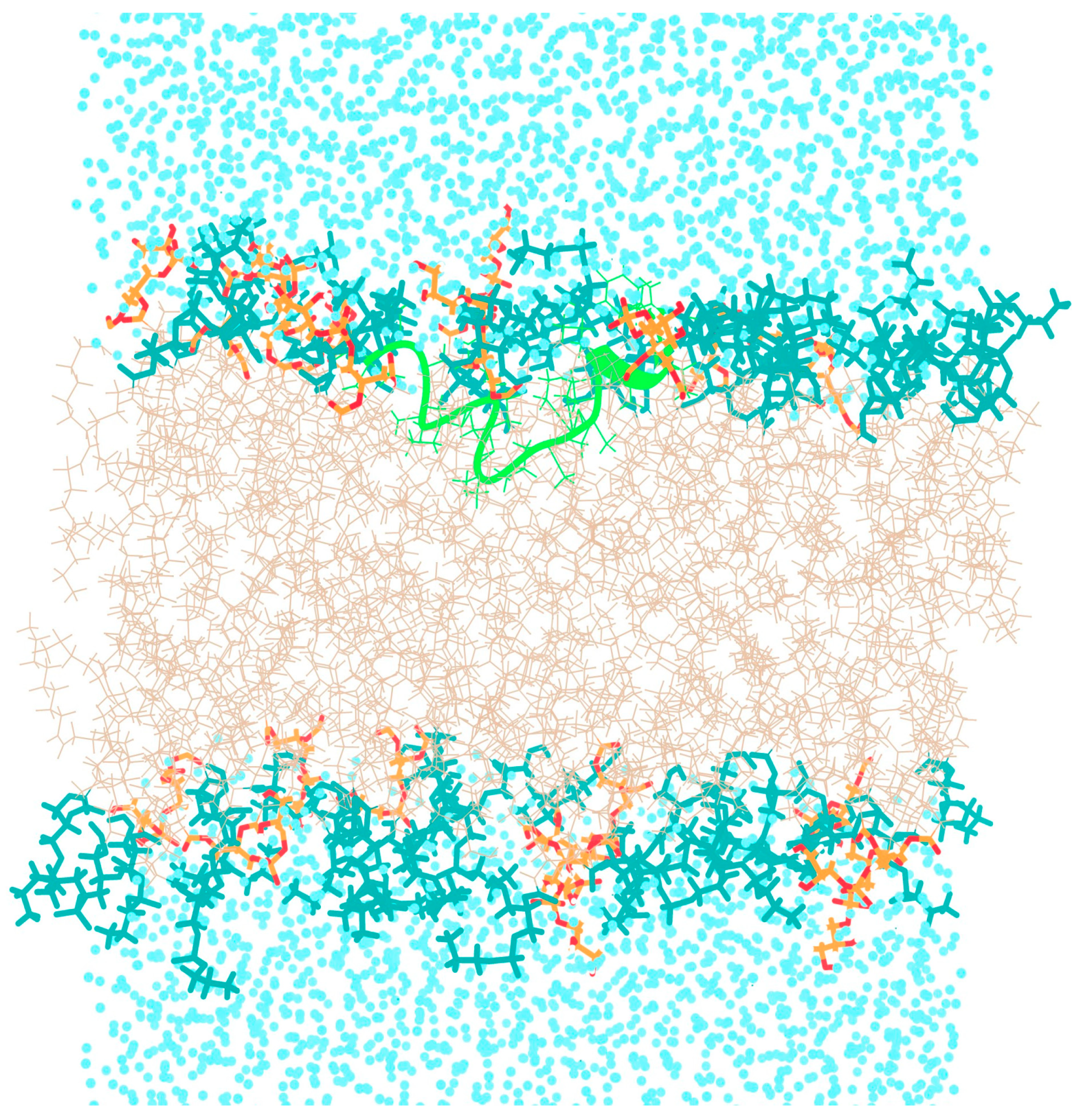
3.1. Behavior of TPN XIIc Peptide in DOPE:DOPG System with 3:1 Ratio
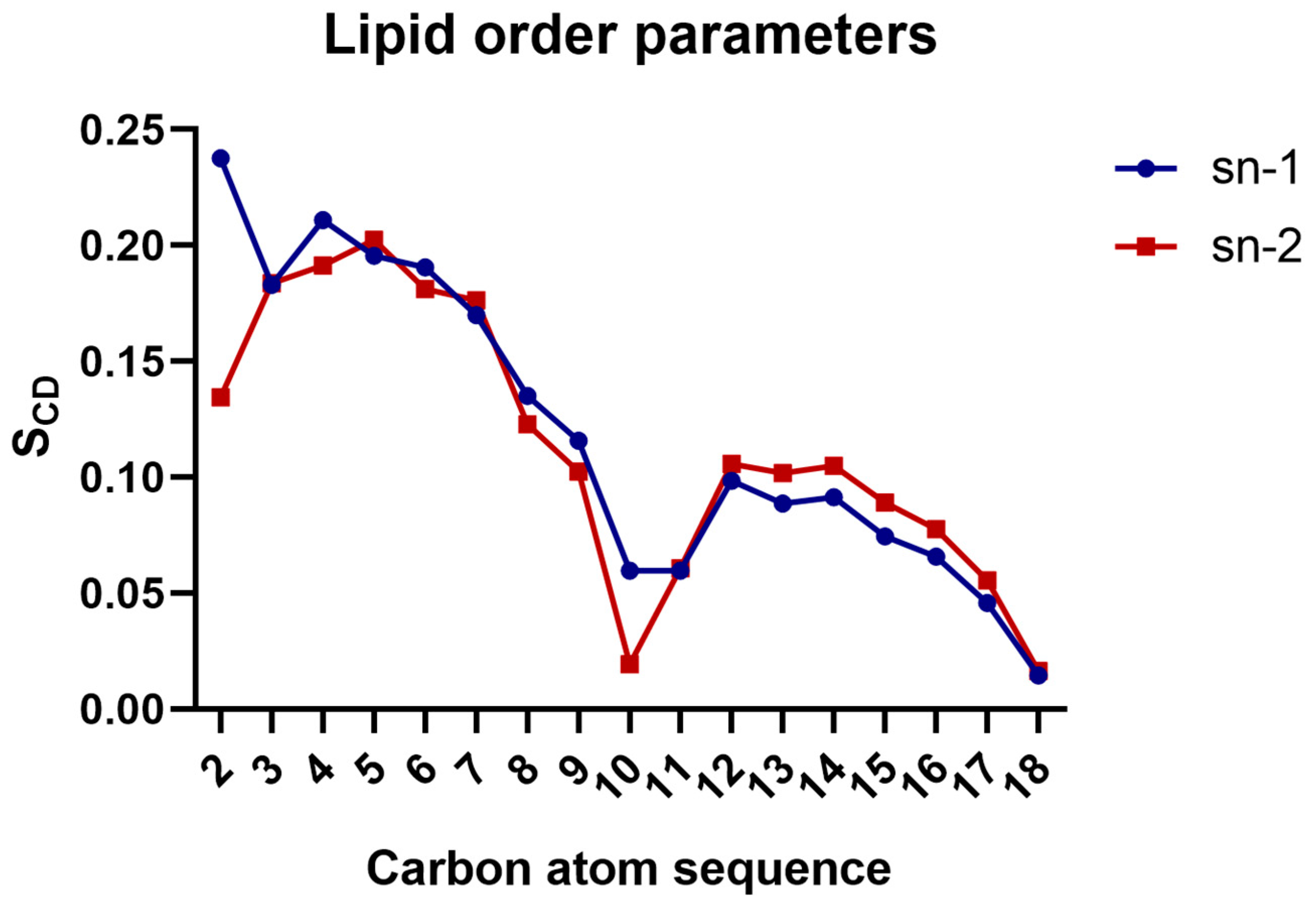
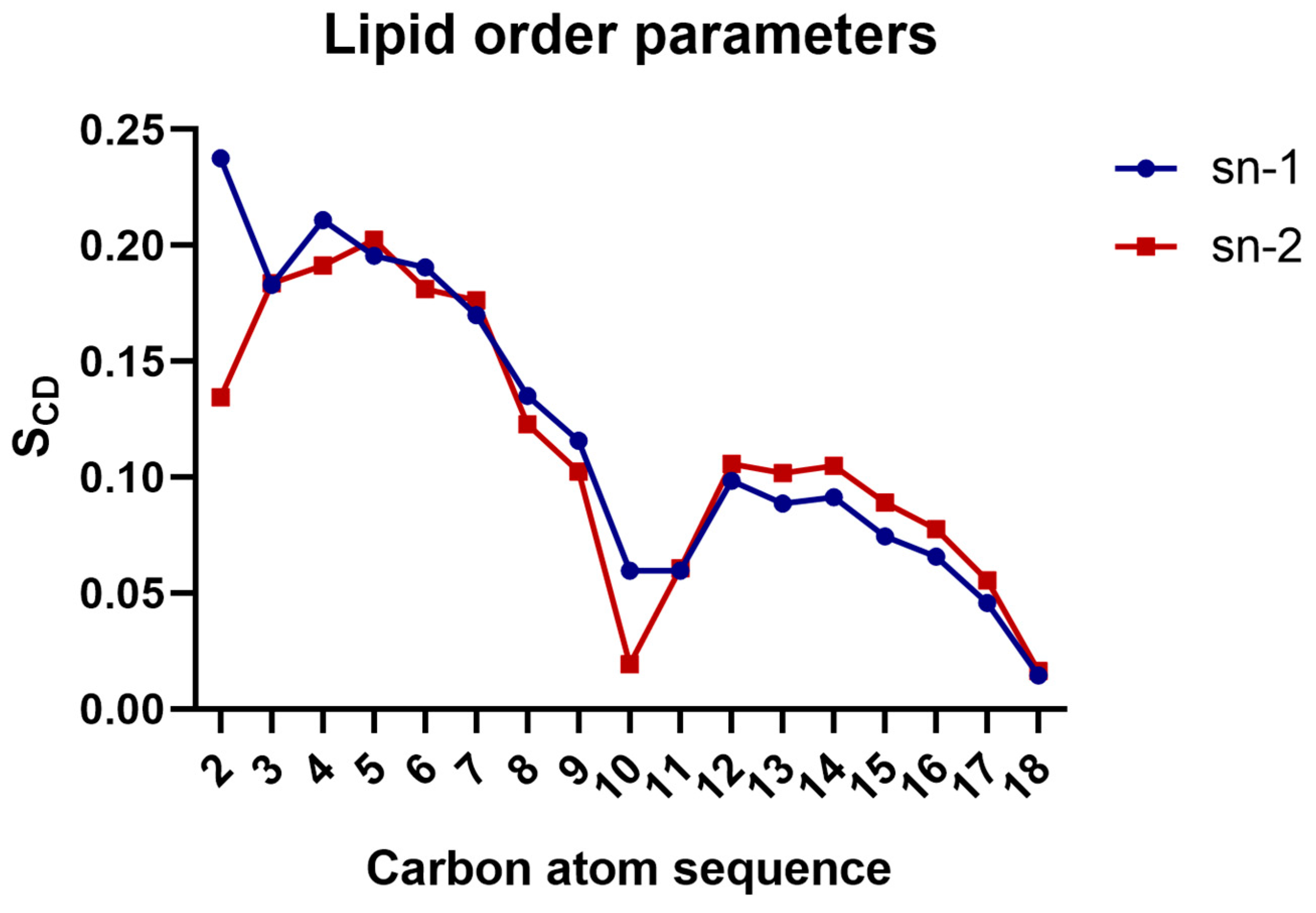
3.2. Calculation of Lipid Order Parameters
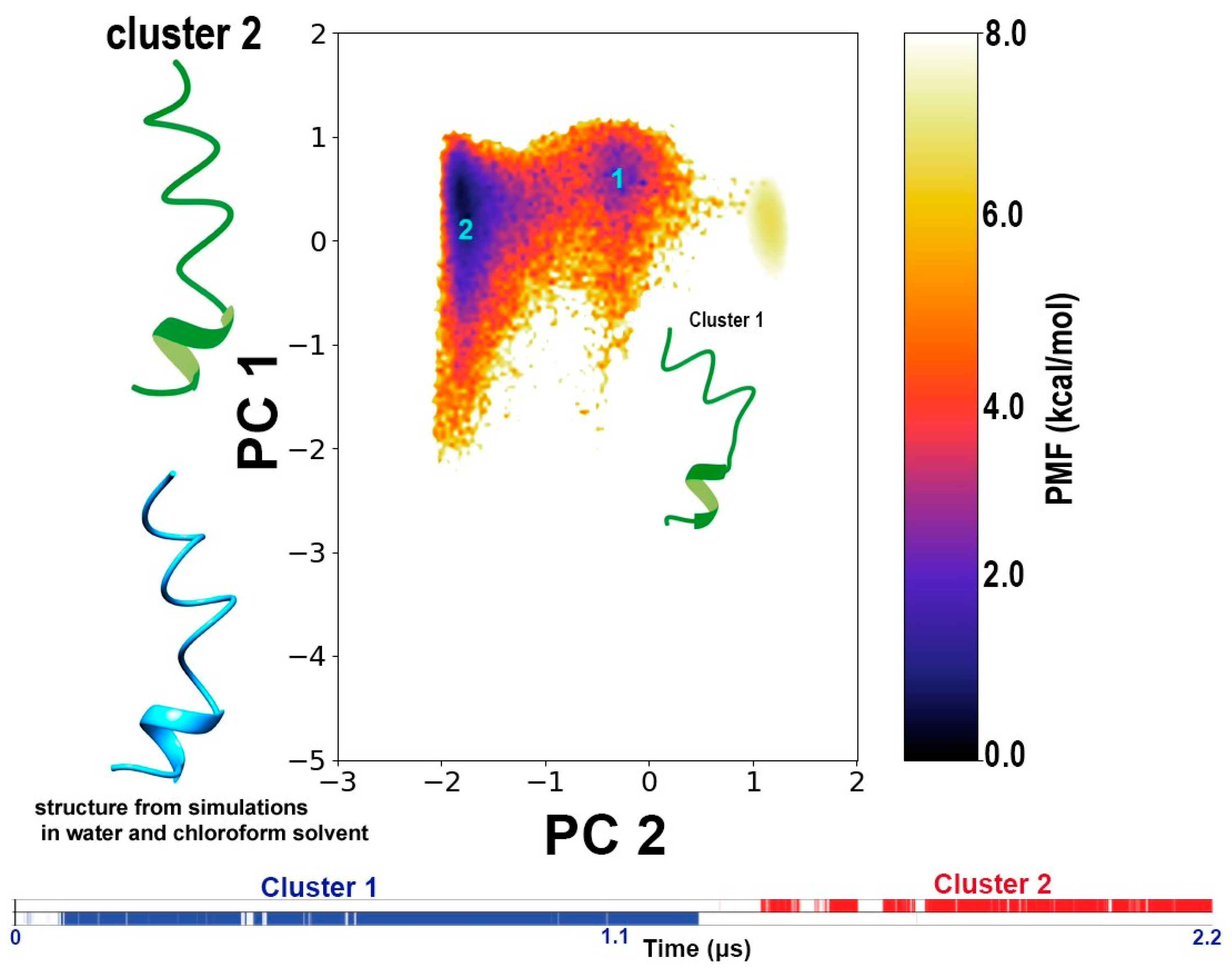
3.3. Conformational Dynamics of TPN during Interaction with a Bilayer Membrane
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benedetti, E.; Bavoso, A.; Di Blasio, B.; Pavone, V.; Pedone, C.; Toniolo, C.; Bonora, G.M. Peptaibol antibiotics: A study on the helical structure of the 2-9 sequence of emerimicins III and IV. Proc. Natl. Acad. Sci. USA 1982, 79, 7951–7954. [Google Scholar] [CrossRef] [PubMed]
- Brückner, H.; Graf, H. Paracelsin, a peptide antibiotic containing α-aminoisobutyric acid, isolated from Trichoderma reesei Simmons Part A. Experientia 1983, 39, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Degenkolb, T.; Brückner, H. Peptaibiomics: Towards a myriad of bioactive peptides containing Cα-dialkylamino acids? Chem. Biodivers. 2008, 5, 1817–1843. [Google Scholar] [CrossRef] [PubMed]
- Zocher, R.; Keller, U. Thiol template peptide synthesis systems in bacteria and fungi. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: Gainesville, FL, USA, 1996; Volume 38, pp. 85–131. [Google Scholar]
- Marahiel, M.A.; Stachelhaus, T.; Mootz, H.D. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 1997, 97, 2651–2674. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yang, F.; Straney, D.C. Multiple non-ribosomal peptide synthetase genes determine peptaibol synthesis in Trichoderma virens. Can. J. Microbiol. 2005, 51, 423–429. [Google Scholar] [CrossRef]
- Marik, T.; Urbán, P.; Tyagi, C.; Szekeres, A.; Leitgeb, B.; Vágvölgyi, M.; Manczinger, L.; Druzhinina, I.; Vágvölgyi, C.; Kredics, L. Diversity profile and dynamics of peptaibols produced by green mould Trichoderma species in interactions with their hosts Agaricus bisporus and Pleurotus ostreatus. Chem. Biodivers. 2017, 14, e1700033. [Google Scholar] [CrossRef]
- Tyagi, C.; Marik, T.; Vágvölgyi, C.; Kredics, L.; Szekeres, A.; Ötvös, F. Tripleurin XIIc: Peptide folding dynamics in aqueous and hydrophobic environment mimic using accelerated molecular dynamics. Molecules 2019, 24, 358. [Google Scholar] [CrossRef]
- Mularski, A.; Wilksch, J.J.; Wang, H.; Hossain, M.A.; Wade, J.D.; Separovic, F.; Strugnell, R.A.; Gee, M.L. Atomic force microscopy reveals the mechanobiology of lytic peptide action on bacteria. Langmuir 2015, 31, 6164–6171. [Google Scholar] [CrossRef]
- Hu, K.; Jiang, Y.; Xie, Y.; Liu, H.; Liu, R.; Zhao, Z.; Lai, R.; Yang, L. Small-anion selective transmembrane “holes” induced by an antimicrobial peptide too short to span membranes. J. Phys. Chem. B 2015, 119, 8553–8560. [Google Scholar] [CrossRef]
- Voievoda, N.; Schulthess, T.; Bechinger, B.; Seelig, J. Thermodynamic and biophysical analysis of the membrane-association of a histidine-rich peptide with efficient antimicrobial and transfection activities. J. Phys. Chem. B 2015, 119, 9678–9687. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.I.; Ishibe, N.; Ueha, M.; Nakata, S.; Miyajima, K.; Epand, R.M. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry 1998, 37, 11856–11863. [Google Scholar] [CrossRef]
- Bozelli, J.C., Jr.; Sasahara, E.T.; Pinto, M.R.; Nakaie, C.R.; Schreier, S. Effect of head group and curvature on binding of the antimicrobial peptide tritrpticin to lipid membranes. Chem. Phys. Lipids 2012, 165, 365–373. [Google Scholar] [CrossRef]
- Koller, D.; Lohner, K. The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Perrin, B.S.; Sodt, A.J.; Cotten, M.L.; Pastor, R.W. The curvature induction of surface-bound antimicrobial peptides piscidin 1 and piscidin 3 varies with lipid chain length. J. Membr. Biol. 2015, 248, 455–467. [Google Scholar] [CrossRef]
- Epand, R.M.; Rotem, S.; Mor, A.; Berno, B.; Epand, R.F. Bacterial membranes as predictors of antimicrobial potency. J. Am. Chem. Soc. 2008, 130, 14346–14352. [Google Scholar] [CrossRef]
- Epand, R.F.; Wang, G.; Berno, B.; Epand, R.M. Lipid segregation explains selective toxicity of a series of fragments derived from the human cathelicidin LL-37. Antimicrob. Agents Chemother. 2009, 53, 3705–3714. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular mechanisms of membrane targeting antibiotics. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Machaidze, G.; Ziegler, A.; Seelig, J. Specific binding of Ro 09-0198 (cinnamycin) to phosphatidylethanolamine: A thermodynamic analysis. Biochemistry 2002, 41, 1965–1971. [Google Scholar] [CrossRef]
- Pogliano, J.; Pogliano, N.; Silverman, J.A. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 2012, 194, 4494–4504. [Google Scholar] [CrossRef]
- Wada, S.I.; Iida, A.; Asami, K.; Fujita, T. Ion channel-forming property of trichorovin-XII, an 11-residue peptaibol from the fungus Trichoderma viride, in planar lipid bilayer membranes. Bioorg. Med. Chem. Lett. 1996, 6, 2275–2278. [Google Scholar] [CrossRef]
- Tyagi, C. Structural Investigation of Peptaibols Using Accelerated Molecular Dynamics Simulations. Ph.D. Thesis, University of Szeged, Szeged, Hungary, 2020. [Google Scholar]
- Juhl, D.W.; Glattard, E.; Aisenbrey, C.; Bechinger, B. Antimicrobial peptides: Mechanism of action and lipid-mediated synergistic interactions within membranes. Faraday Discuss. 2021, 232, 419–434. [Google Scholar] [CrossRef]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2016, 45, D555–D559. [Google Scholar] [CrossRef]
- Schott-Verdugo, S.; Gohlke, H. PACKMOL-Memgen: A simple-to-use, generalized workflow for membrane-protein–lipid-bilayer system building. J. Chem. Inf. Model. 2019, 59, 2522–2528. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Glykos, N.M. Software news and updates. Carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Koukos, P.I.; Glykos, N.M. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J. Comput. Chem. 2013, 34, 2310–2312. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; McCammon, J.A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. [Google Scholar] [CrossRef]
- Pastor, R.W.; Venable, R.M.; Karplus, M.; Szabo, A. A simulation-based model of NMR T 1 relaxation in lipid bilayer vesicles. J. Chem. Phys. 1988, 89, 1128–1140. [Google Scholar] [CrossRef]
- Pastor, R.W.; Venable, R.M.; Karplus, M. Model for the structure of the lipid bilayer. Proc. Natl. Acad. Sci. USA 1991, 88, 892–896. [Google Scholar] [CrossRef]
- Venable, R.M.; Brown, F.L.; Pastor, R.W. Mechanical properties of lipid bilayers from molecular dynamics simulation. Chem. Phys. Lipids 2015, 192, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, C.; Marik, T.; Vágvölgyi, C.; Kredics, L.; Ötvös, F. Accelerated molecular dynamics applied to the peptaibol folding problem. Int. J. Mol. Sci. 2019, 20, 4268. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Schertzer, J.W.; Yong, X. Molecular dynamics modeling of Pseudomonas aeruginosa outer membranes. Phys. Chem. Chem. Phys. 2018, 20, 23635–23648. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.F. In Search of a Molecular View of Peptide–Lipid Interactions in Membranes. Langmuir 2023, 39, 10289–10300. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C.; Ladokhin, A.S. Energetics of Peptide–Bilayer Interactions. Meth. Enzymol. 1998, 295, 62–87. [Google Scholar]
- Almeida, P.F.; Ladokhin, A.S.; White, S.H. Hydrogen-bond energetics drive helix formation in membrane interfaces. Biochim. Biophys. Acta (BBA)-Biomembr. 2012, 1818, 178–182. [Google Scholar] [CrossRef] [PubMed]
- De Zotti, M.; Sella, L.; Bolzonello, A.; Gabbatore, L.; Peggion, C.; Bortolotto, A.; Elmaghraby, I.; Tundo, S.; Favaron, F. Targeted amino acid substitutions in a Trichoderma peptaibol confer activity against fungal plant pathogens and protect host tissues from Botrytis cinerea infection. Int. J. Mol. Sci. 2020, 21, 7521. [Google Scholar] [CrossRef]
- Baccelli, I.; Luti, S.; Bernardi, R.; Favaron, F.; De Zotti, M.; Sella, L. Water-soluble Trichogin GA IV-derived peptaibols protect tomato plants from Botrytis cinerea infection with limited impact on plant defenses. Front. Plant. Sci. 2022, 13, 881961. [Google Scholar] [CrossRef]
- Afanasyeva, E.F.; Syryamina, V.N.; Dzuba, S.A. Communication: Alamethicin can capture lipid-like molecules in the membrane. J. Chem. Phys. 2017, 146, 011103. [Google Scholar] [CrossRef]
- Su, Z.; Shodiev, M.; Leitch, J.J.; Abbasi, F.; Lipkowski, J. In situ electrochemical and PM-IRRAS studies of alamethicin ion channel formation in model phospholipid bilayers. J. Electroanal. Chem. 2018, 819, 251–259. [Google Scholar] [CrossRef]
- Putzu, M.; Kara, S.; Afonin, S.; Grage, S.L.; Bordessa, A.; Chaume, G.; Brigaud, T.; Ulrich, A.S.; Kubař, T. Structural behavior of the peptaibol harzianin HK VI in a DMPC bilayer: Insights from MD simulations. Biophys. J. 2017, 112, 2602–2614. [Google Scholar] [CrossRef] [PubMed]
- Finger, S.; Kerth, A.M.; Dathe, M.; Blume, A. The impact of non-ideality of lipid mixing on peptide induced lipid clustering. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183248. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyagi, C.; Marik, T.; Szekeres, A.; Vágvölgyi, C.; Kredics, L.; Ötvös, F. Modeling the Effect on a Novel Fungal Peptaibol Placed in an All-Atom Bacterial Membrane Mimicking System via Accelerated Molecular Dynamics Simulations. Life 2023, 13, 2288. https://doi.org/10.3390/life13122288
Tyagi C, Marik T, Szekeres A, Vágvölgyi C, Kredics L, Ötvös F. Modeling the Effect on a Novel Fungal Peptaibol Placed in an All-Atom Bacterial Membrane Mimicking System via Accelerated Molecular Dynamics Simulations. Life. 2023; 13(12):2288. https://doi.org/10.3390/life13122288
Chicago/Turabian StyleTyagi, Chetna, Tamás Marik, András Szekeres, Csaba Vágvölgyi, László Kredics, and Ferenc Ötvös. 2023. "Modeling the Effect on a Novel Fungal Peptaibol Placed in an All-Atom Bacterial Membrane Mimicking System via Accelerated Molecular Dynamics Simulations" Life 13, no. 12: 2288. https://doi.org/10.3390/life13122288
APA StyleTyagi, C., Marik, T., Szekeres, A., Vágvölgyi, C., Kredics, L., & Ötvös, F. (2023). Modeling the Effect on a Novel Fungal Peptaibol Placed in an All-Atom Bacterial Membrane Mimicking System via Accelerated Molecular Dynamics Simulations. Life, 13(12), 2288. https://doi.org/10.3390/life13122288