

Virtual Insights into Natural Compounds as Potential 5α-Reductase Type II Inhibitors: A Structure-Based Screening and Molecular Dynamics Simulation Study
 , ,
, ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of 3D Structure of 5αR2
2.2. Retrieval and Preparation of Flavonoid Library
2.3. Structure-Based Virtual Screening
2.4. Physiochemical and Toxicity Prediction
2.5. Molecular Dynamics (MD) Simulations
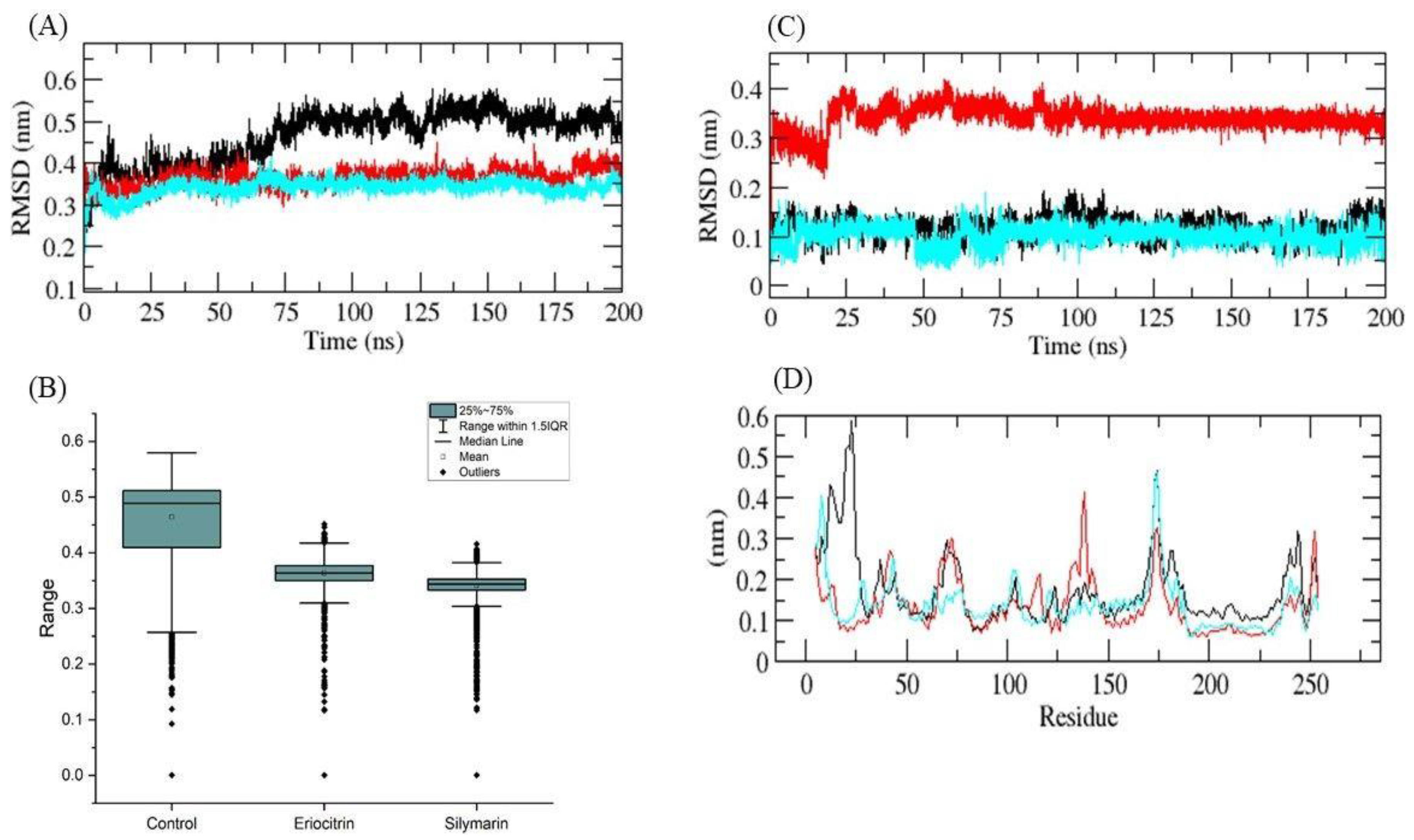
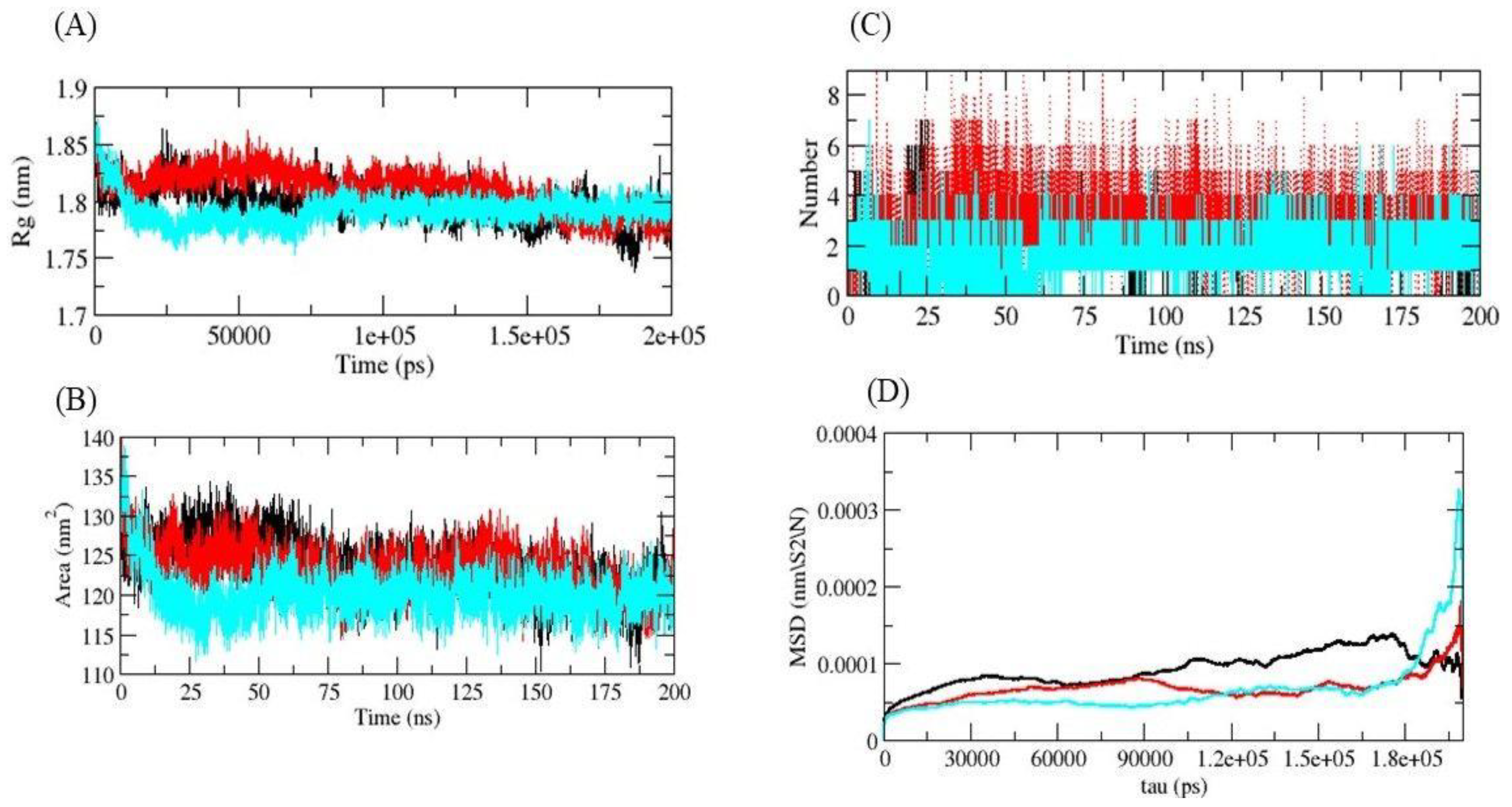
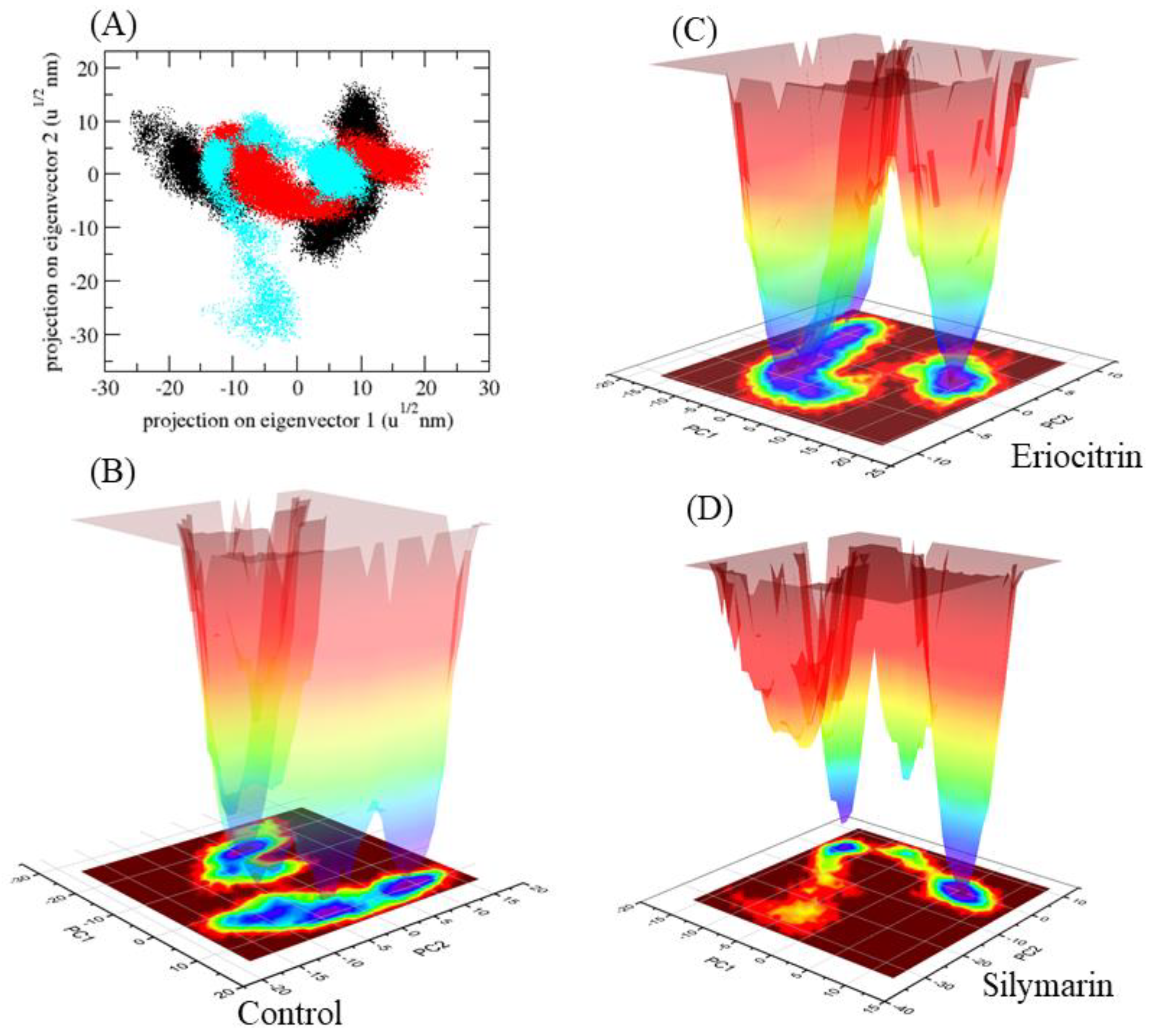
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koch, S.L.; Tridico, S.R.; Bernard, B.A.; Shriver, M.D.; Jablonski, N.G. The biology of human hair: A multidisciplinary review. Am. J. Hum. Biol. 2020, 32, e23316. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.B.; Yuan, X.; Geller, F.; Waterworth, D.; Bataille, V.; Glass, D.; Song, K.; Waeber, G.; Vollenweider, P.; Aben, K.K.; et al. Male-pattern baldness susceptibility locus at 20p11. Nat. Genet. 2008, 40, 1282–1284. [Google Scholar] [CrossRef] [PubMed]
- Adil, A.; Godwin, M. The effectiveness of treatments for androgenetic alopecia: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2017, 77, 136–141.e5. [Google Scholar] [CrossRef] [PubMed]
- Sultan, C.; Lumbroso, S.; Poujol, N.; Boudon, C.; Georget, V.; Terouanne, B.; Belon, C.; Lobaccaro, J.M. Genetics and endocrinology of male sex differentiation: Application to molecular study of male pseudohermaphroditism. C. R. Seances Soc. Biol. Fil. 1995, 189, 713–740. [Google Scholar]
- Trueb, R.M. Molecular mechanisms of androgenetic alopecia. Exp. Gerontol. 2002, 37, 981–990. [Google Scholar] [CrossRef]
- Asakawa, K.; Toyoshima, K.E.; Ishibashi, N.; Tobe, H.; Iwadate, A.; Kanayama, T.; Hasegawa, T.; Nakao, K.; Toki, H.; Noguchi, S.; et al. Hair organ regeneration via the bioengineered hair follicular unit transplantation. Sci. Rep. 2012, 2, 424. [Google Scholar] [CrossRef]
- Toyoshima, K.E.; Asakawa, K.; Ishibashi, N.; Toki, H.; Ogawa, M.; Hasegawa, T.; Irie, T.; Tachikawa, T.; Sato, A.; Takeda, A.; et al. Fully functional hair follicle regeneration through the rearrangement of stem cells and their niches. Nat. Commun. 2012, 3, 784. [Google Scholar] [CrossRef]
- Gupta, A.K.; Carviel, J.; MacLeod, M.A.; Shear, N. Assessing finasteride-associated sexual dysfunction using the FAERS database. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1069–1075. [Google Scholar] [CrossRef]
- Olsen, E.A.; Weiner, M.S.; Amara, I.A.; DeLong, E.R. Five-year follow-up of men with androgenetic alopecia treated with topical minoxidil. J. Am. Acad. Dermatol. 1990, 22, 643–646. [Google Scholar] [CrossRef]
- Barbareschi, M. The use of minoxidil in the treatment of male and female androgenetic alopecia: A story of more than 30 years. G. Ital. Dermatol. Venereol. 2018, 153, 102–106. [Google Scholar] [CrossRef]
- Mysore, V.; Shashikumar, B.M. Guidelines on the use of finasteride in androgenetic alopecia. Indian J. Dermatol. Venereol. Leprol. 2016, 82, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Yim, E.; Nole, K.L.; Tosti, A. 5alpha-Reductase inhibitors in androgenetic alopecia. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Nonell-Canals, A.; Avila, C.; Sanchez-Martinez, M. Computer-Aided Drug Design (CADD) to De-Orphanize Marine Molecules: Finding Potential Therapeutic Agents for Neurodegenerative and Cardiovascular Diseases. Mar. Drugs 2022, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Maghsoudi, S.; Taghavi Shahraki, B.; Rameh, F.; Nazarabi, M.; Fatahi, Y.; Akhavan, O.; Rabiee, M.; Mostafavi, E.; Lima, E.C.; Saeb, M.R.; et al. A review on computer-aided chemogenomics and drug repositioning for rational COVID-19 drug discovery. Chem. Biol. Drug Des. 2022, 100, 699–721. [Google Scholar] [CrossRef]
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 2020, 11, 733. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Romano, J.D.; Tatonetti, N.P. Informatics and Computational Methods in Natural Product Drug Discovery: A Review and Perspectives. Front. Genet. 2019, 10, 368. [Google Scholar] [CrossRef]
- Martins, B.T.; Correia da Silva, M.; Pinto, M.; Cidade, H.; Kijjoa, A. Marine natural flavonoids: Chemistry and biological activities. Nat. Prod. Res. 2019, 33, 3260–3272. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, L.; Supekar, S.; Shen, T.; Liu, H.; Ye, F.; Huang, J.; Fan, H.; Wei, Z.; Zhang, C. Structure of human steroid 5alpha-reductase 2 with the anti-androgen drug finasteride. Nat. Commun. 2020, 11, 5430. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Ballante, F.; Kooistra, A.J.; Kampen, S.; de Graaf, C.; Carlsson, J. Structure-Based Virtual Screening for Ligands of G Protein-Coupled Receptors: What Can Molecular Docking Do for You? Pharmacol. Rev. 2021, 73, 527–565. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Fernandes, C.L.; Verli, H. GROMOS96 43a1 performance on the characterization of glycoprotein conformational ensembles through molecular dynamics simulations. Carbohydr. Res. 2009, 344, 491–500. [Google Scholar] [CrossRef]
- Libecco, J.F.; Bergfeld, W.F. Finasteride in the treatment of alopecia. Expert Opin. Pharmacother. 2004, 5, 933–940. [Google Scholar] [CrossRef]
- Kaufman, K.D.; Dawber, R.P. Finasteride, a Type 2 5alpha-reductase inhibitor, in the treatment of men with androgenetic alopecia. Expert Opin. Investig. Drugs 1999, 8, 403–415. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Khan, M.B.; Ahmad, K.; Lim, J.H.; Shaikh, S.; Lee, E.J.; Choi, I. Biocomputational Screening of Natural Compounds against Acetylcholinesterase. Molecules 2021, 26, 2641. [Google Scholar] [CrossRef] [PubMed]
- Mojica, L.; de Mejia, E.G. Optimization of enzymatic production of anti-diabetic peptides from black bean (Phaseolus vulgaris L.) proteins, their characterization and biological potential. Food Funct. 2016, 7, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Ahmad, K.; Shaikh, S.; Lim, J.H.; Chun, H.J.; Ahmad, S.S.; Lee, E.J.; Choi, I. Identification and Evaluation of Traditional Chinese Medicine Natural Compounds as Potential Myostatin Inhibitors: An In Silico Approach. Molecules 2022, 27, 4303. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Liu, W.; Bashir, M.; Nisar, M.F.; Wan, C.C. Eriocitrin: A review of pharmacological effects. Biomed. Pharmacother. 2022, 154, 113563. [Google Scholar] [CrossRef]
- Karimi, G.; Vahabzadeh, M.; Lari, P.; Rashedinia, M.; Moshiri, M. “Silymarin”, a promising pharmacological agent for treatment of diseases. Iran. J. Basic Med. Sci. 2011, 14, 308–317. [Google Scholar]
- Aggarwal, S.; Thareja, S.; Verma, A.; Bhardwaj, T.R.; Kumar, M. An overview on 5alpha-reductase inhibitors. Steroids 2010, 75, 109–153. [Google Scholar] [CrossRef]
- Iamsumang, W.; Leerunyakul, K.; Suchonwanit, P. Finasteride and Its Potential for the Treatment of Female Pattern Hair Loss: Evidence to Date. Drug Des. Dev. Ther. 2020, 14, 951–959. [Google Scholar] [CrossRef]
- Dhurat, R.; Sharma, A.; Rudnicka, L.; Kroumpouzos, G.; Kassir, M.; Galadari, H.; Wollina, U.; Lotti, T.; Golubovic, M.; Binic, I.; et al. 5-Alpha reductase inhibitors in androgenetic alopecia: Shifting paradigms, current concepts, comparative efficacy, and safety. Dermatol. Ther. 2020, 33, e13379. [Google Scholar] [CrossRef]
- Erdemir, F.; Harbin, A.; Hellstrom, W.J. 5-alpha reductase inhibitors and erectile dysfunction: The connection. J. Sex. Med. 2008, 5, 2917–2924. [Google Scholar] [CrossRef]
- Mushtaq, S.; Abbasi, B.H.; Uzair, B.; Abbasi, R. Natural products as reservoirs of novel therapeutic agents. EXCLI J. 2018, 17, 420–451. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Ali, S.; Lim, J.H.; Chun, H.J.; Ahmad, K.; Ahmad, S.S.; Hwang, Y.C.; Han, K.S.; Kim, N.R.; Lee, E.J.; et al. Dipeptidyl peptidase-4 inhibitory potentials of Glycyrrhiza uralensis and its bioactive compounds licochalcone A and licochalcone B: An in silico and in vitro study. Front. Mol. Biosci. 2022, 9, 1024764. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Shaikh, S.; Ahmad, K.; Ahmad, S.S.; Lim, J.H.; Park, S.; Yang, H.J.; Cho, W.K.; Park, S.J.; Lee, Y.H.; et al. Isolation and Characterization of Compounds from Glycyrrhiza uralensis as Therapeutic Agents for the Muscle Disorders. Int. J. Mol. Sci. 2021, 22, 876. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Lee, E.J.; Ahmad, K.; Ahmad, S.S.; Lim, J.H.; Choi, I. A Comprehensive Review and Perspective on Natural Sources as Dipeptidyl Peptidase-4 Inhibitors for Management of Diabetes. Pharmaceuticals 2021, 14, 591. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compound Name | Binding Energy (kcal/mol) |
|---|---|---|
| 1. | Eriocitrin | −12.1 |
| 2. | Obacunone | −11.8 |
| 3. | Oroxin_B | −11.7 |
| 4. | Silymarin | −11.7 |
| 5. | Hesperidin | −11.7 |
| 6. | Baicalin | −11.6 |
| 7. | Diosmin | −11.6 |
| 8. | Scutellarin | −11.6 |
| 9. | Methyl-Hesperidin | −11.6 |
| 10. | Narirutin | −11.6 |
| 11. | Isosilybin | −11.6 |
| 12. | Finasteride (positive control) | −11.2 |
| Compounds | Structure | No. of H-Bond | H-Bonds Interacting Residues | Van der Waals Interactions |
|---|---|---|---|---|
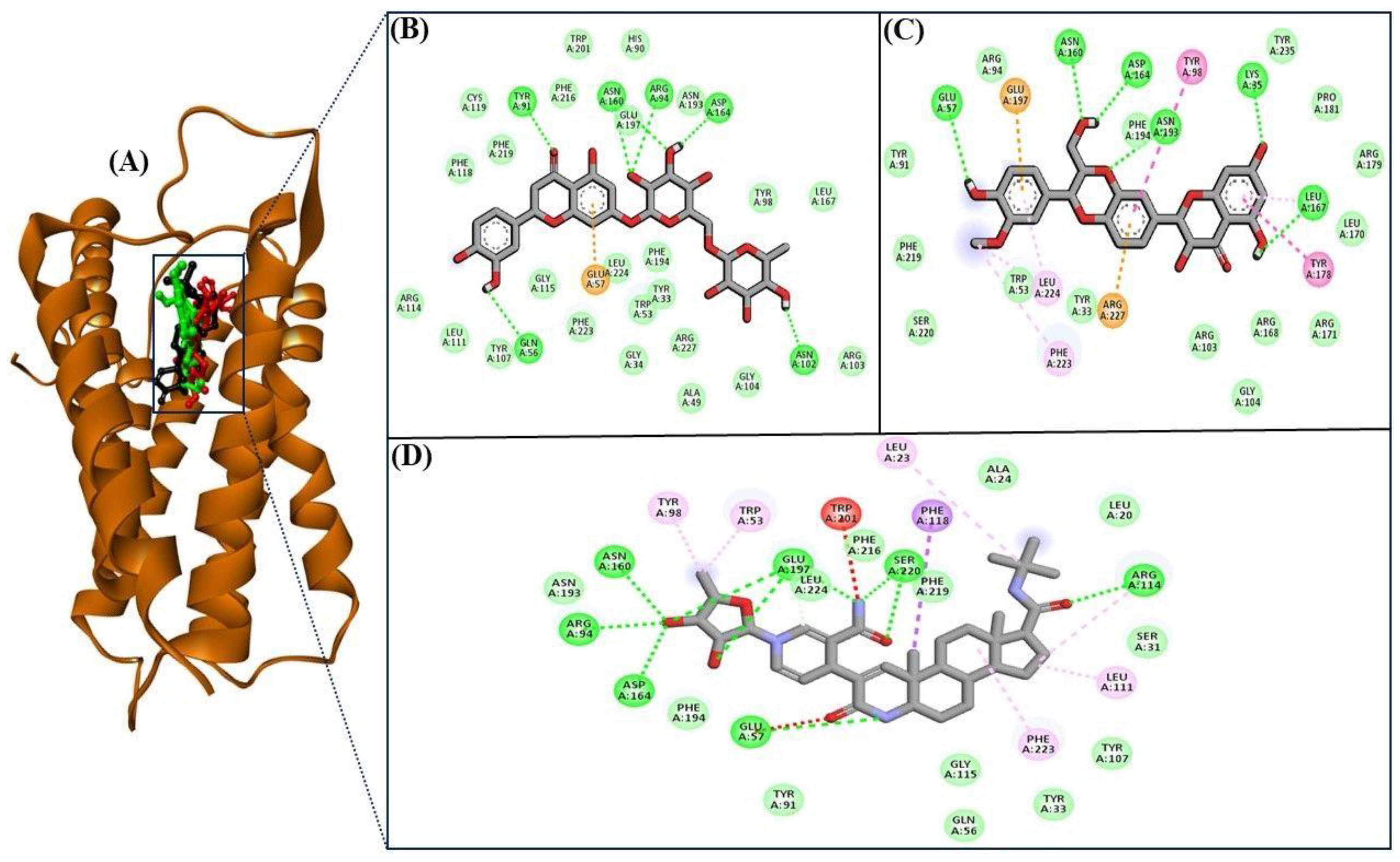
| Eriocitrin | 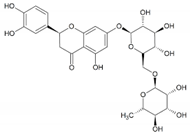 | 6 | Gln56, Tyr91, Arg94, Asn102, Asn160, and Asp164 | Tyr107, Leu111, Arg114, Phe118, Phe219, Cys119, Phe216, Trp201, His90, Glu197, Asn193, Tyr98, Leu167, Arg103, Gly104, Ala49, Arg227, Phe194, Tyr33, Trp53, Gly34, Leu224, Phe223, and Gly115 |
| Silymarin | 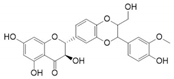 | 6 | Lys35, Glu57, Asn160, Asp164, Leu167, and Asn193 | Ser220, Phe219, Tyr91, Arg94, Phe194, Tyr235, Pro181, Arg179, Leu170, Arg171, Arg168, Gly104, Arg103, Tyr33, and Trp53 |
| Finasteride | 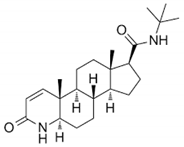 | 7 | Glu57, Arg94, Arg114, Asn160, Asp164, Glu197, and Ser220 | Asn193, Leu224, Tyr91, Phe194, Phe216, Phe219, Ala24, Leu20, Ser31, Tyr107, Tyr33, Gly115, and Gln56 |
| Properties | Compound Name | |||
|---|---|---|---|---|
| Physicochemical Properties | Eriocitrin | Silymarin | Finasteride (Control) | |
| MW | 596.53 | 482.44 | 372.54 | |
| Heavy atoms | 42 | 35 | 27 | |
| Aromatic heavy atoms | 12 | 18 | 0 | |
| Fraction Csp3 | 0.52 | 0.24 | 0.83 | |
| RB | 6 | 4 | 3 | |
| HBA | 15 | 10 | 2 | |
| HBD | 9 | 5 | 2 | |
| Molar Refractivity | 136.94 | 120.55 | 113.18 | |
| TPSA | 245.29 | 155.14 | 58.20 | |
| Lipophilicity | ||||
| iLOGP | 1.95 | 2.79 | 3.32 | |
| XLOGP3 | −1.35 | 1.9 | 3.03 | |
| WLOGP | −1.78 | 1.71 | 3.43 | |
| MLOGP | −3.24 | −0.4 | 3.46 | |
| Silicos-IT Log P | −2.1 | 1.92 | 3.20 | |
| Consensus Log P | −1.3 | 1.59 | 3.29 | |
| Water Solubility | ||||
| ESOL | Log S | −2.5 | −4.14 | −3.86 |
| Solubility (mg/mL) | 1.87 × 100 | 3.46 × 10−2 | 5.13 × 10−2 | |
| Class | Soluble | Moderately soluble | Soluble | |
| Ali | Log S | −3.3 | −4.78 | −3.92 |
| Solubility (mg/mL) | 2.98 × 10−1 | 7.99 × 10−3 | 4.50 × 10−2 | |
| Class | Soluble | Moderately soluble | Soluble | |
| Silicos-IT | LogSw | 0.1 | −4.5 | −4.54 |
| Solubility (mg/mL) | 7.56 × 102 | 1.53 × 10−2 | 1.07 × 10−2 | |
| class | Soluble | Moderately soluble | Moderately soluble | |
| Pharmacokinetics | ||||
| GI absorption | Low | Low | High | |
| BBB permeant | No | No | Yes | |
| Pgp substrate | Yes | No | Yes | |
| inhibitor | CYP1A2 | No | No | No |
| CYP2C19 | No | No | No | |
| CYP2C9 | No | No | No | |
| CYP2D6 | No | No | No | |
| CYP3A4 | No | Yes | No | |
| log Kp (cm/s) | −10.9 | −7.89 | −6.42 | |
| Classification | Target | Prediction | Probability | ||||
|---|---|---|---|---|---|---|---|
| Silymarin | Eriocitrin | Finasteride (Control) | Silymarin | Eriocitrin | Finasteride (Control) | ||
| Organ toxicity | Hepatotoxicity | IA | IA | IA | 0.78 | 0.8 | 0.98 |
| Toxicity endpoints | Carcinogenicity | IA | IA | IA | 0.72 | 0.91 | 0.61 |
| Immunotoxicity | A | A | A | 0.97 | 0.99 | 0.99 | |
| Mutagenicity | IA | IA | IA | 0.69 | 0.88 | 0.81 | |
| Cytotoxicity | IA | IA | IA | 0.77 | 0.64 | 0.79 | |
| Tox21-Nuclear receptor signaling pathways | AhR | A | IA | IA | 0.99 | 0.83 | 0.99 |
| Androgen Receptor (AR) | IA | IA | IA | 0.95 | 0.98 | 0.87 | |
| AR-LBD | IA | IA | IA | 0.99 | 0.99 | 0.99 | |
| Aromatase | IA | IA | IA | 0.8 | 0.99 | 0.97 | |
| Estrogen Receptor Alpha (ER) | IA | IA | IA | 0.71 | 0.95 | 0.93 | |
| ER-LBD | IA | IA | IA | 0.96 | 0.99 | 0.98 | |
| PPAR-Gamma | IA | IA | IA | 0.97 | 0.98 | 0.98 | |
| Tox21-stress response pathways | nrf2/ARE | IA | IA | IA | 0.92 | 0.99 | 0.97 |
| HSE | IA | IA | IA | 0.92 | 0.99 | 0.97 | |
| MMP | IA | IA | IA | 0.73 | 0.97 | 0.93 | |
| p53 | IA | IA | IA | 0.91 | 0.9 | 0.97 | |
| ATAD5 | IA | IA | IA | 0.94 | 0.99 | 0.99 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaikh, S.; Ali, S.; Lim, J.H.; Ahmad, K.; Han, K.S.; Lee, E.J.; Choi, I. Virtual Insights into Natural Compounds as Potential 5α-Reductase Type II Inhibitors: A Structure-Based Screening and Molecular Dynamics Simulation Study. Life 2023, 13, 2152. https://doi.org/10.3390/life13112152
Shaikh S, Ali S, Lim JH, Ahmad K, Han KS, Lee EJ, Choi I. Virtual Insights into Natural Compounds as Potential 5α-Reductase Type II Inhibitors: A Structure-Based Screening and Molecular Dynamics Simulation Study. Life. 2023; 13(11):2152. https://doi.org/10.3390/life13112152
Chicago/Turabian StyleShaikh, Sibhghatulla, Shahid Ali, Jeong Ho Lim, Khurshid Ahmad, Ki Soo Han, Eun Ju Lee, and Inho Choi. 2023. "Virtual Insights into Natural Compounds as Potential 5α-Reductase Type II Inhibitors: A Structure-Based Screening and Molecular Dynamics Simulation Study" Life 13, no. 11: 2152. https://doi.org/10.3390/life13112152
APA StyleShaikh, S., Ali, S., Lim, J. H., Ahmad, K., Han, K. S., Lee, E. J., & Choi, I. (2023). Virtual Insights into Natural Compounds as Potential 5α-Reductase Type II Inhibitors: A Structure-Based Screening and Molecular Dynamics Simulation Study. Life, 13(11), 2152. https://doi.org/10.3390/life13112152