
L-DOPA and Droxidopa: From Force Field Development to Molecular Docking into Human β2-Adrenergic Receptor



Abstract
1. Introduction
2. Materials and Methods
2.1. Force Field Development Protocol
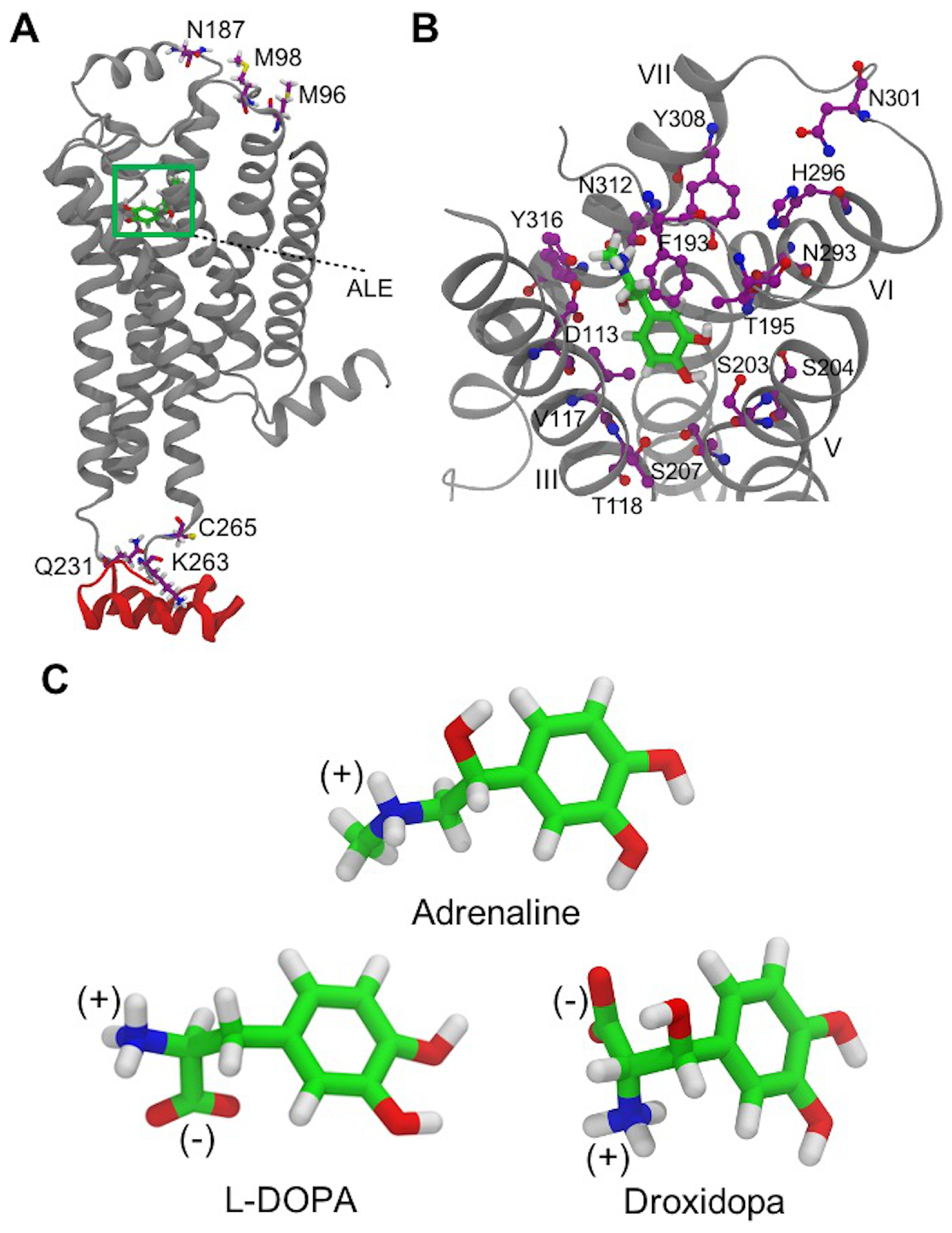
2.2. 2AR Molecular Modeling
2.3. Molecular Docking Protocol
2.4. MD Simulations of 2AR-Catecholamine Complexes
3. Results
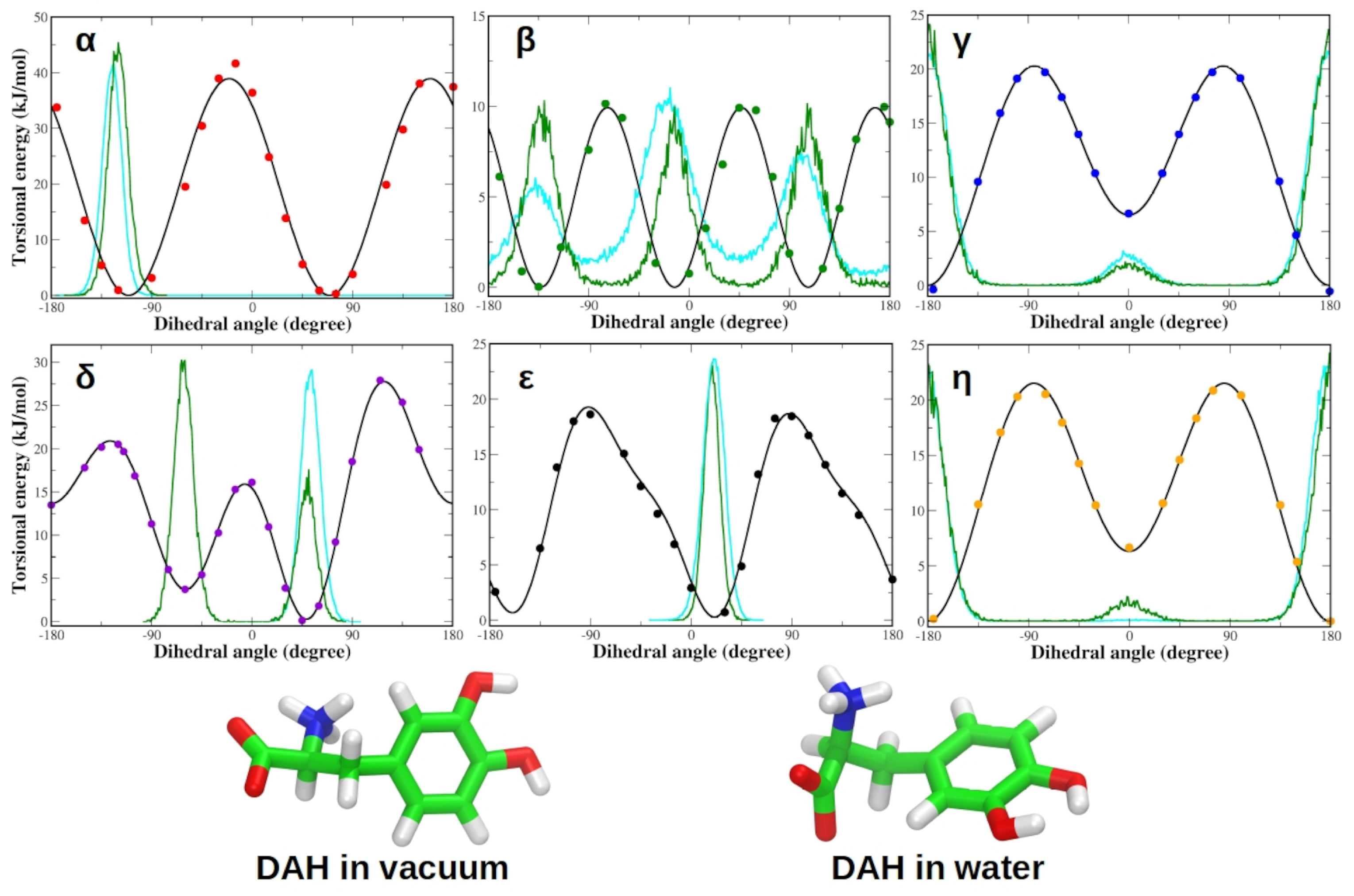
3.1. Force Field Parameterization and Md Simulations of L-Dopa and Droxidopa
3.2. Validation of the 2AR Model: Redocking of Adrenaline
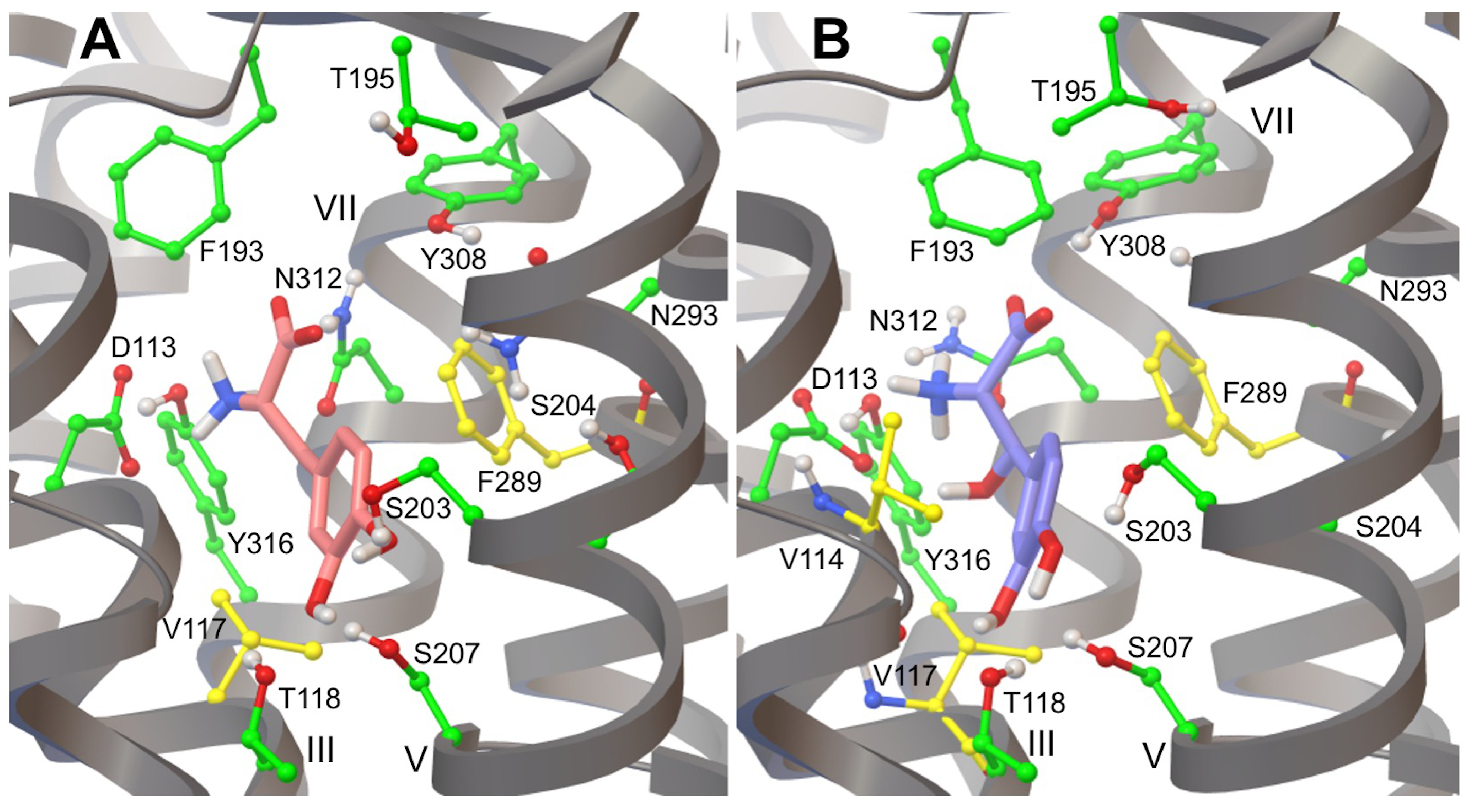
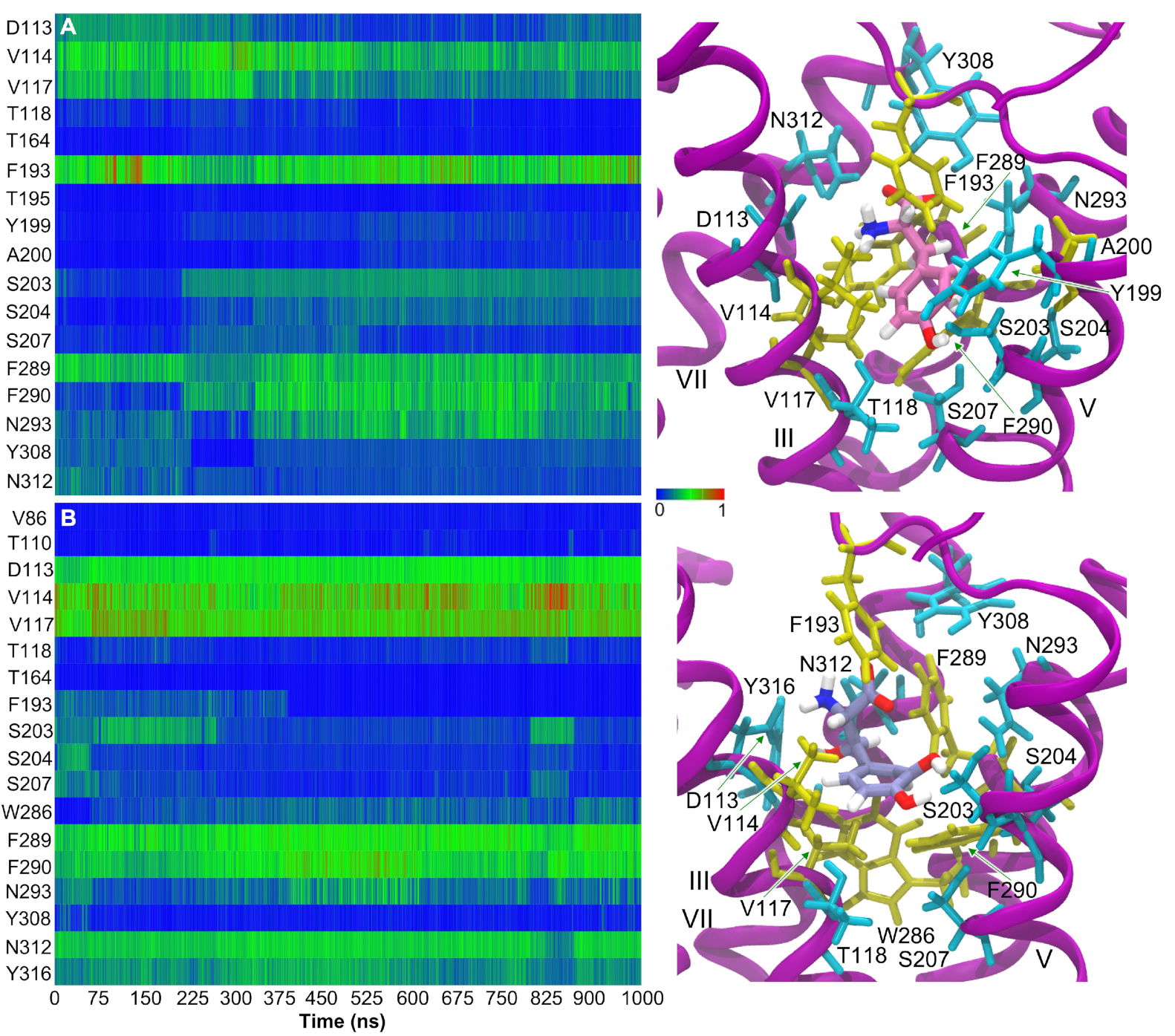
3.3. L-DOPA Binding to the 2AR Receptor
3.4. Droxidopa Binding to the 2AR Receptor
3.5. Hydrogen Bonds in 2AR-Adrenaline Complexes
3.6. Hydrogen Bonds in 2AR in Complex with L-DOPA and Droxidopa
3.7. Hydrophobic Contacts of 2AR Agonists
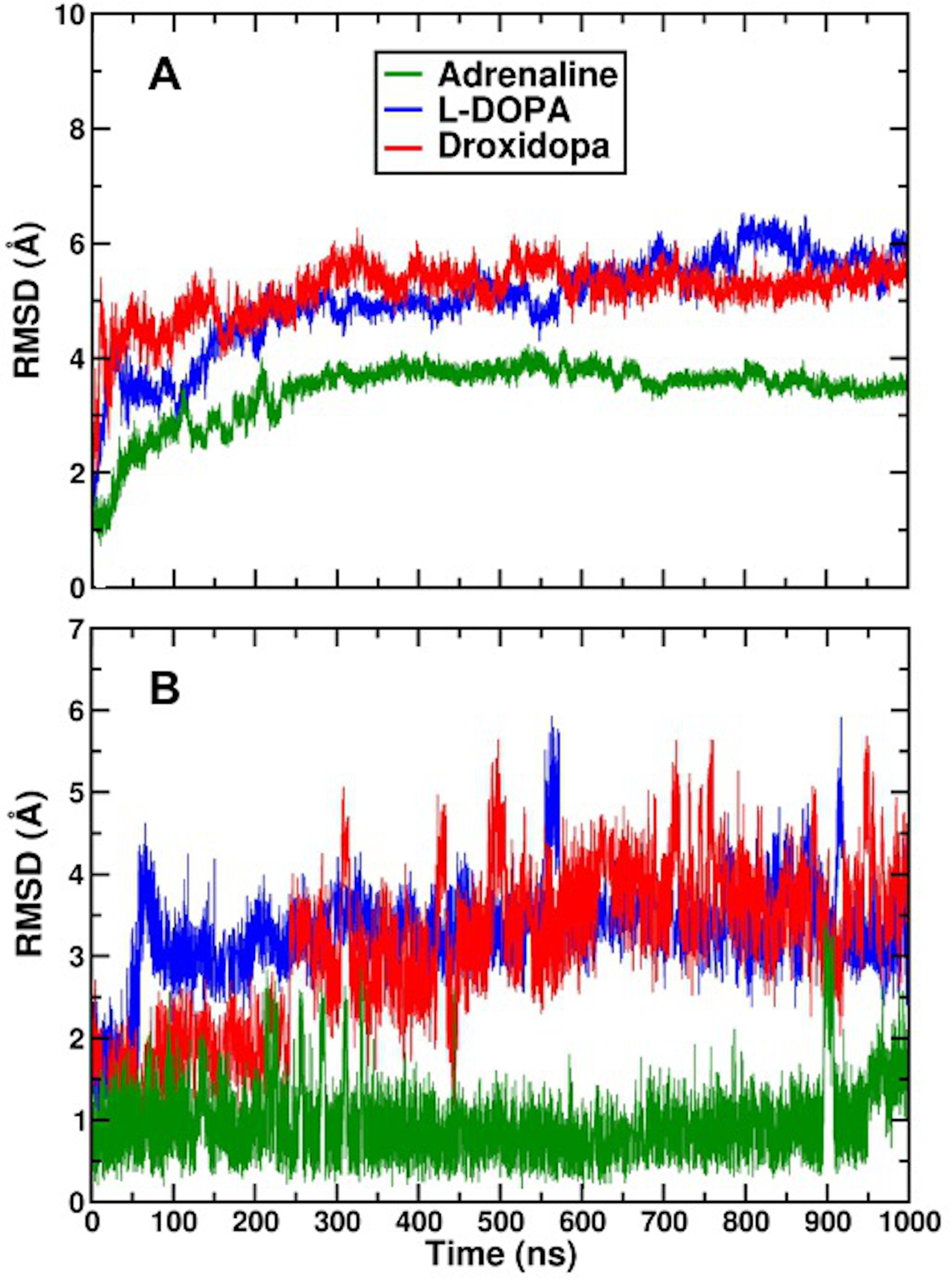
3.8. Protein–Ligand Interactions in MD Simulations of 2AR-Catecholamine Complexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | All Atom |
| 2AR | Beta2 Adrenergic Receptor |
| pKd | Binding Affinity |
| BFE | Binding Free Energy |
| GPCR | Guanine Nucleotide-Binding Protein Coupled Receptor |
| DFT | Density-Functional Theory |
| FF | Force Field |
| L-DOPA | Levodopa |
| MD | Molecular Dynamics |
| PD | Parkinson’s Disease |
| QM | Quantum Mechanical |
References
- Lee, Y.; Lazim, R.; Macalino, S.J.Y.; Choi, S. Importance of protein dynamics in the structure-based drug discovery of class A G protein-coupled receptors (GPCRs). Curr. Opin. Struct. Biol. 2019, 55, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.; Dror, R.O. GPCR dynamics: Structures in motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Gacasan, S.B.; Baker, D.L.; Parrill, A.L. G protein-coupled receptors: The evolution of structural insight. AIMS Biophys. 2017, 4, 491–527. [Google Scholar] [CrossRef] [PubMed]
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef]
- Yang, J.; Villar, V.A.M.; Armando, I.; Jose, P.A.; Zeng, C. G protein–coupled receptor kinases: Crucial regulators of blood pressure. J. Am. Heart Assoc. 2016, 5, e003519. [Google Scholar] [CrossRef]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR drug targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Tyndall, J.D.; Sandilya, R. GPCR agonists and antagonists in the clinic. Med. Chem. 2005, 1, 405–421. [Google Scholar] [CrossRef]
- Chattopadhyay, A. GPCRs: Lipid-dependent membrane receptors that act as drug targets. Adv. Biol. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Paila, Y.D. Lipid–protein interactions, regulation and dysfunction of brain cholesterol. Biochem. Biophys. Res. Commun. 2007, 354, 627–633. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein–coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Nakliang, P.; Lazim, R.; Chang, H.; Choi, S. Multiscale Molecular Modeling in G Protein-Coupled Receptor (GPCR)-Ligand Studies. Biomolecules 2020, 10, 631. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Macalino, S.J.; Park, J.; Clavio, N.A.; Kang, S.; Choi, S. Exploring G protein-coupled receptors (GPCRs) ligand space via cheminformatics approaches: Impact on rational drug design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef]
- Heifetz, A.; James, T.; Morao, I.; Bodkin, M.J.; Biggin, P.C. Guiding lead optimization with GPCR structure modeling and molecular dynamics. Curr. Opin. Pharmacol. 2016, 30, 14–21. [Google Scholar] [CrossRef]
- Yuan, X.; Xu, Y. Recent trends and applications of molecular modeling in GPCR–ligand recognition and structure-based drug design. Int. J. Mol. Sci. 2018, 19, 2105. [Google Scholar] [CrossRef]
- Lee, Y.; Basith, S.; Choi, S. Recent advances in structure-based drug design targeting class AG protein-coupled receptors utilizing crystal structures and computational simulations. J. Med. Chem. 2018, 61, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Espigares, I.; Torrens-Fontanals, M.; Tiemann, J.K.S.; Aranda-García, D.; Ramírez-Anguita, J.M.; Stepniewski, T.M.; Worp, N.; Varela-Rial, A.; Morales-Pastor, A.; Medel-Lacruz, B.; et al. GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat. Methods 2020, 17, 777–787. [Google Scholar] [CrossRef]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein–coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Yao, X.J.; Weis, W.I.; Stevens, R.C.; et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; DeVree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef]
- Swaminath, G.; Xiang, Y.; Lee, T.W.; Steenhuis, J.; Parnot, C.; Kobilka, B.K. Sequential binding of agonists to the β2 adrenoceptor: Kinetic evidence for intermediate conformational states. J. Biol. Chem. 2004, 279, 686–691. [Google Scholar] [CrossRef]
- Swaminath, G.; Deupi, X.; Lee, T.W.; Zhu, W.; Thian, F.S.; Kobilka, T.S.; Kobilka, B. Probing the β2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J. Biol. Chem. 2005, 280, 22165–22171. [Google Scholar] [CrossRef]
- Kobilka, B.K.; Deupi, X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 397–406. [Google Scholar] [CrossRef]
- Kobilka, B. The structural basis of G-protein-coupled receptor signaling (Nobel Lecture). Angew. Chem. Int. Edit. 2013, 52, 6380–6388. [Google Scholar] [CrossRef]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of β2-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef]
- Katsube, J.; Narabayashi, H.; Hayashi, A.; Tanaka, C.; Suzuki, T. Development of L-threo-DOPS, a norepinephrine precursor amino acid. Yakugaku Zasshi 1994, 114, 823–846. [Google Scholar] [CrossRef][Green Version]
- Goole, J.; Amighi, K. Levodopa delivery systems for the treatment of Parkinson’s disease: An overview. Int. J. Pharm. 2009, 380, 1–15. [Google Scholar] [CrossRef]
- Rosebraugh, M.; Voight, E.A.; Moussa, E.M.; Jameel, F.; Lou, X.; Zhang, G.G.Z.; Mayer, P.T.; Stolarik, D.; Carr, R.A.; Enright, B.P.; et al. Foslevodopa/Foscarbidopa: A New Subcutaneous Treatment for Parkinson’s Disease. Ann. Neurol. 2021, 90, 52–61. [Google Scholar] [CrossRef]
- Rosebraugh, M.; Liu, W.; Neenan, M.; Facheris, M.F. Foslevodopa/Foscarbidopa Is Well Tolerated and Maintains Stable Levodopa and Carbidopa Exposure Following Subcutaneous Infusion. J. Park. Dis. 2021, 11, 1695–1702. [Google Scholar] [CrossRef]
- Facheris, M.; Robieson, W.; Fisseha, N.; Standaert, D. Efficacy and safety of foslevodopa/foscarbidopa versus oral carbidopa/levodopa in advanced Parkinson’s disease patients: Design of a phase 3, randomized, double-blind, double-dummy, active controlled 12-week trial. J. Neurol. Sci. 2021, 429, 331. [Google Scholar] [CrossRef]
- Barone, V.; Cacelli, I.; De Mitri, N.; Licari, D.; Monti, S.; Prampolini, G. J oyce and U lysses: Integrated and user-friendly tools for the parameterization of intramolecular force fields from quantum mechanical data. Phys. Chem. Chem. Phys. 2013, 15, 3736–3751. [Google Scholar] [CrossRef]
- Cacelli, I.; Prampolini, G. Parametrization and Validation of Intramolecular Force Fields Derived from DFT Calculations. J. Chem. Theory Comput. 2007, 3, 1803–1817. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Marenich, A.V.; Jerome, S.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Vilseck, J.Z.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation of CM5 charges for condensed-phase modeling. J. Chem. Theory Comput. 2014, 10, 2802–2812. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Damm, W.; Frontera, A.; Tirado–Rives, J.; Jorgensen, W.L. OPLS all-atom force field for carbohydrates. J. Comput. Chem. 1997, 18, 1955–1970. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Comm. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.F.; Choi, H.J.; DeVree, B.T.; Sunahara, R.K.; et al. Structure and Function of an Irreversible Agonist-β2 Adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.Ø.; Piana, S.; Shaw, D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef]
- Pieper, U.; Webb, B.M.; Dong, G.Q.; Schneidman-Duhovny, D.; Fan, H.; Kim, S.J.; Khuri, N.; Spill, Y.G.; Weinkam, P.; Hammel, M.; et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014, 42, D336–D346. [Google Scholar] [CrossRef]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef]
- Dilcan, G.; Doruker, P.; Akten, E.D. Ligand-binding affinity of alternative conformers of human β2-adrenergic receptor in the presence of intracellular loop 3 (ICL3) and their potential use in virtual screening studies. Chem. Biol. Drug Des. 2019, 93, 883–899. [Google Scholar] [CrossRef]
- Vanni, S.; Neri, M.; Tavernelli, I.; Rothlisberger, U. A Conserved Protonation-Induced Switch can Trigger “Ionic-Lock” Formation in Adrenergic Receptors. J. Mol. Biol. 2010, 397, 1339–1349. [Google Scholar] [CrossRef]
- Ranganathan, A.; Dror, R.O.; Carlsson, J. Insights into the Role of Asp792.50 in β2 Adrenergic Receptor Activation from Molecular Dynamics Simulations. Biochemistry 2014, 53, 7283–7296. [Google Scholar] [CrossRef] [PubMed]
- Bang, I.; Choi, H.J. Structural Features of β2 Adrenergic Receptor: Crystal Structures and Beyond. Mol. Cells 2015, 38, 105–111. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tosso, R.D.; Parravicini, O.; Zarycz, M.N.C.; Angelina, E.; Vettorazzi, M.; Peruchena, N.; Andujar, S.; Enriz, R.D. Conformational and electronic study of dopamine interacting with the D2 dopamine receptor. J. Comput. Chem. 2020, 41, 1898–1911. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Feinstein, W.P.; Brylinski, M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminform. 2015, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual Screening with AutoDock: Theory and Practice. Expert. Opin. Drug. Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef]
- Cosconati, S.; Marinelli, L.; Di Leva, F.S.; La Pietra, V.; De Simone, A.; Mancini, F.; Andrisano, V.; Novellino, E.; Goodsell, D.S.; Olson, A.J. Protein Flexibility in Virtual Screening: The BACE-1 Case Study. J. Chem. Inf. Model. 2012, 52, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Del Carmine, R.; Ambrosio, C.; Sbraccia, M.; Cotecchia, S.; Ijzerman, A.P.; Costa, T. Mutations inducing divergent shifts of constitutive activity reveal different modes of binding among catecholamine analogues to the β2-adrenergic receptor. Br. J. Pharmacol. 2002, 135, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.D.; Catte, A.; Mancini, G.; Barone, V. Analysis of L-DOPA and Droxidopa binding to Human β2-Adrenergic Receptor. Biophys. J. 2021, 7, 1–18. [Google Scholar] [CrossRef]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel Parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 12. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Maciejewski, A.; Pasenkiewicz-Gierula, M.; Cramariuc, O.; Vattulainen, I.; Róg, T. Refined OPLS All-Atom Force Field for Saturated Phosphatidylcholine Bilayers at Full Hydration. J. Phys. Chem. B 2014, 118, 4571–4581. [Google Scholar] [CrossRef]
- Kulig, W.; Pasenkiewicz-Gierula, M.; Róg, T. Topologies, structures and parameter files for lipid simulations in GROMACS with the OPLS-aa force field: DPPC, POPC, DOPC, PEPC, and cholesterol. Data Brief 2015, 5, 333–336. [Google Scholar] [CrossRef]
- Kulig, W.; Pasenkiewicz-Gierula, M.; Róg, T. Cis and trans unsaturated phosphatidylcholine bilayers: A molecular dynamics simulation study. Chem. Phys. Lipids 2016, 195, 12–20. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A.; Open Source Drug Discovery Consortium. g_mmpbsa–A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Del Galdo, S.; Mancini, G.; Daidone, I.; Polzi Zanetti, L.; Amadei, A.; Barone, V. Tyrosine absorption spectroscopy: Backbone protonation effects on the side chain electronic properties. J. Comput. Chem. 2018, 39, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Reynolds, K.A.; Cherezov, V.; Hanson, M.A.; Roth, C.B.; Yeager, M.; Abagyan, R. Analysis of Full and Partial Agonists Binding to β2-Adrenergic Receptor Suggests a Role of Transmembrane Helix V in Agonist-Specific Conformational Changes. J. Mol. Recognit. 2009, 22, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Álvarez Diduk, R.; Galano, A. Adrenaline and Noradrenaline: Protectors against Oxidative Stress or Molecular Targets? J. Phys. Chem. B 2015, 119, 3479–3491. [Google Scholar] [CrossRef]
- Del Carmine, R.; Molinari, P.; Sbraccia, M.; Ambrosio, C.; Costa, T. “Induced-Fit” Mechanism for Catecholamine Binding to the β2-Adrenergic Receptor. Mol. Pharmacol. 2004, 66, 356–363. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Strader, C.D.; Sigal, I.S.; Candelore, M.R.; Rands, E.; Hill, W.S.; Dixon, R.A. Conserved aspartic acid residues 79 and 113 of the beta-adrenergic receptor have different roles in receptor function. J. Biol. Chem. 1988, 263, 10267–10271. [Google Scholar] [CrossRef]
- Suryanarayana, S.; Kobilka, B.K. Amino acid substitutions at position 312 in the seventh hydrophobic segment of the beta 2-adrenergic receptor modify ligand-binding specificity. Mol. Pharmacol. 1993, 44, 111–114. [Google Scholar]
- Hannawacker, A.; Krasel, C.; Lohse, M. Mutation of Asn293 to Asp in transmembrane helix VI abolishes agonist-induced but not constitutive activity of the beta(2)-adrenergic receptor. Mol. Pharmacol. 2002, 62, 1431–1437. [Google Scholar] [CrossRef]
- Wieland, K.; Zuurmond, H.M.; Krasel, C.; Ijzerman, A.P.; Lohse, M.J. Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta 2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 9276–9281. [Google Scholar] [CrossRef]
- Ambrosio, C.; Molinari, P.; Cotecchia, S.; Costa, T. Catechol-binding serines of β2-adrenergic receptors control the equilibrium between active and inactive receptor states. Mol. Pharmacol. 2000, 57, 198–210. [Google Scholar]
- Liapakis, G.; Ballesteros, J.A.; Papachristou, S.; Chan, W.C.; Chen, X.; Javitch, J.A. The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the beta 2-adrenergic receptor. J. Biol. Chem. 2000, 275, 37779–37788. [Google Scholar] [CrossRef]
- Liapakis, G.; Chan, W.C.; Papadokostaki, M.; Javitch, J.A. Synergistic contributions of the functional groups of epinephrine to its affinity and efficacy at the beta2 adrenergic receptor. Mol. Pharmacol. 2004, 65, 1181–1190. [Google Scholar] [CrossRef]
- Strader, C.D.; Candelore, M.R.; Hill, W.S.; Sigal, I.S.; Dixon, R.A. Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. J. Biol. Chem. 1989, 264, 13572–13578. [Google Scholar] [CrossRef]
- Bandaru, S.; Alvala, M.; Nayarisseri, A.; Sharda, S.; Goud, H.; Mundluru, H.P.; Singh, S.K. Molecular dynamic simulations reveal suboptimal binding of salbutamol in T164I variant of β2 adrenergic receptor. PLoS ONE 2017, 12, e0186666. [Google Scholar] [CrossRef]
- Isin, B.; Estiu, G.; Wiest, O.; Oltvai, Z.N. Identifying Ligand Binding Conformations of the β2-Adrenergic Receptor by Using Its Agonists as Computational Probes. PLoS ONE 2012, 7, e50186. [Google Scholar] [CrossRef]
- Plazinska, A.; Kolinski, M.; Wainer, I.W.; Jozwiak, K. Molecular interactions between fenoterol stereoisomers and derivatives and the β2-adrenergic receptor binding site studied by docking and molecular dynamics simulations. J. Mol. Model. 2013, 19, 4919–4930. [Google Scholar] [CrossRef]
- Dickson, C.J.; Hornak, V.; Velez-Vega, C.; McKay, D.J.J.; Reilly, J.; Sandham, D.A.; Shaw, D.; Fairhurst, R.A.; Charlton, S.J.; Sykes, D.A.; et al. Uncoupling the Structure–Activity Relationships of β2 Adrenergic Receptor Ligands from Membrane Binding. J. Med. Chem. 2016, 59, 5780–5789. [Google Scholar] [CrossRef]
- Manna, M.; Niemelä, M.; Tynkkynen, J.; Javanainen, M.; Kulig, W.; Müller, D.J.; Rog, T.; Vattulainen, I. Mechanism of allosteric regulation of β2-adrenergic receptor by cholesterol. Elife 2016, 5, e18432. [Google Scholar] [CrossRef]
- Zocher, M.; Fung, J.J.; Kobilka, B.K.; Müller, D.J. Ligand-specific interactions modulate kinetic, energetic, and mechanical properties of the human β2 adrenergic receptor. Structure 2012, 20, 1391–1402. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Calculated Binding Affinity pKd a | Experimental Binding Affinity pKd | |
|---|---|---|---|
| Rigid Model | Flexible Model | ||
| Adrenaline | 5.9 | 9.2 | 6.1 b–6.5 c (3.3) d |
| L-DOPA | 4.7 | 9.4 | NA (3.9) |
| Droxidopa | 5.4 | 10.3 | NA (9.2) |
| Hydrogen Bonds (2AR-Ligand) | Donor-Acceptor Distance (Å) | |||||
|---|---|---|---|---|---|---|
| Rigid Model | Flexible Model | |||||
| ALE | DAH | DRO | ALE | DAH | DRO | |
| OD1 (D113)-N (amino) | 3.7 (4.1) a | 3.0 | 3.4 | 2.8 | 2.8 | 2.8 |
| OD2 (D113)-N (amino) | 2.7 (2.8) | 2.5 | 2.4 | 2.6 | 2.5 | 2.3 |
| OD1 (D113)-O (-OH) | 3.0 (2.8) | - c | 2.7 | 3.1 | - | 2.9 |
| OG1 (T118)-O (para) | 3.3 (4.4) | 6.2 | 3.6 | 3.4 | 3.1 | 3.1 |
| OG1 (T118)-O (meta) | 3.2 (7.0) | 3.7 | 3.3 | 2.6 | 2.7 | 3.1 |
| OG (S203)-O (para) | 4.7 (3.7) | 2.8 | 4.3 | 2.7 | 2.6 | 4.7 |
| OG (S203)-O (meta) | 7.1 (3.2) | 7.1 | 5.1 | 5.1 | 4.9 | 7.5 |
| OG (S204)-O (para) | 6.5 (5.7) | 3.5 | 6.3 | 6.2 | 5.5 | 6.9 |
| OG (S204)-O (meta) | 8.7 (4.8) | 6.2 | 8.7 | 8.0 | 7.8 | 8.8 |
| OG (S207)-O (para) | 2.6 (3.5) | 4.5 | 2.9 | 3.1 | 2.5 | 2.7 |
| OG (S207)-O (meta) | 4.0 (6.0) | 2.9 | 4.1 | 2.8 | 3.1 | 3.9 |
| OD1 (N312)-N (amino) | 2.8 (2.8) | 2.9 | 2.7 | 3.0 | 3.0 | 2.9 |
| ND2 (N312)-O (-OH) | 2.9 (2.8) | - | 2.8 | 4.9 | - | 3.0 |
| ND2 (N312)-O1 (-COO−) | - b | 2.7 | 4.6 | - | 2.5 | 3.5 |
| ND2 (N312)-O2 (-COO−) | - | 4.0 | 5.3 | - | 4.0 | 5.0 |
| OH (Y316)-N (amino) | 3.4 (3.5) | 3.0 | 2.9 | 3.3 | 2.4 | 2.4 |
| OH (Y316)-O (-OH) | 3.9 (3.5) | - | 3.5 | 4.1 | - | 3.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catte, A.; Biswas, A.D.; Mancini, G.; Barone, V. L-DOPA and Droxidopa: From Force Field Development to Molecular Docking into Human β2-Adrenergic Receptor. Life 2022, 12, 1393. https://doi.org/10.3390/life12091393
Catte A, Biswas AD, Mancini G, Barone V. L-DOPA and Droxidopa: From Force Field Development to Molecular Docking into Human β2-Adrenergic Receptor. Life. 2022; 12(9):1393. https://doi.org/10.3390/life12091393
Chicago/Turabian StyleCatte, Andrea, Akash Deep Biswas, Giordano Mancini, and Vincenzo Barone. 2022. "L-DOPA and Droxidopa: From Force Field Development to Molecular Docking into Human β2-Adrenergic Receptor" Life 12, no. 9: 1393. https://doi.org/10.3390/life12091393
APA StyleCatte, A., Biswas, A. D., Mancini, G., & Barone, V. (2022). L-DOPA and Droxidopa: From Force Field Development to Molecular Docking into Human β2-Adrenergic Receptor. Life, 12(9), 1393. https://doi.org/10.3390/life12091393