
Up-Date on Diabetic Nephropathy

 ,
,  ,
,
Abstract
1. Introduction
2. Diabetic Nephropathy: State of Art
3. DN and Risk for Progression to ESKD—Novel Biomarkers
4. Old and New Drugs Capable of Ameliorating Risk in Patients with DN
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end-products |
| ADA | American Diabetes Association |
| Ang-II | Angiotensin –II |
| ACE-i | Angiotensin-converting enzyme inhibitors |
| ARBs | Angiotensin Receptor Blockers |
| ADMA | Asymmetric dimethylarginine |
| BNP | Brain-Natriuretic-Peptide |
| CV | Cardiovascular |
| CKD | Chronic Kidney Disease |
| DM | Diabetes Mellitus |
| DN | Diabetic nephropathy |
| DKD | Diabetic kidney disease |
| ESKD | End-stage-kidney-disease |
| ERA | Endothelin-1 receptor antagonists |
| ETAr | Endothelin-1 binding to ET receptors type A |
| eGFR | Estimated glomerular filtration rate |
| ECM | Extracellular matrix |
| KDIGO | Global Outcomes Work Group |
| GLP1-RA | Glucagon-like peptide-1 receptor agonists |
| GDF-15 | Growth differentiation factor-15 |
| hs-cTnT and hs-cTnI | High-sensitivity cardiac troponins |
| IGF-1 | Insulin-like growth factor 1 |
| JAK-STAT | Janus kinase/signal transducers and activators of transcription |
| KIM-1 | Kidney Injury Molecule -1 |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MACE | Major cardiovascular events |
| MMP-10 | Matrix metalloproteinases—10 |
| MRA | Mineralocorticoid receptor antagonist |
| NGAL | Neutrophil Gelatinase-Associated Lipocalin |
| NO | Nitric oxide |
| NP-DN | Non-proteinuric diabetic nephropathy |
| NF-κB | Nuclear factor-κB |
| PKC | Protein kinase C |
| uPAR | Protein urokinase receptor |
| ROS | Reactive oxygen species |
| RAASi | Renin-angiotensin-system inhibitors |
| RBP-4 | Retinol-binding protein-4 |
| SGLT2is | Sodium-glucose co-transporter inhibitors |
| suPAR | Soluble urokinase-type plasminogen activator receptor |
| SDMA | Symmetric Dimethylarginine |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TGF-β1 | Transforming growth factor—β1 |
| TNFR-1 and TNFR-2 | Tumor Necrosis Factors receptors |
| ULK1 | Unc-51-like kinase 1 |
| VEGF | Vascular endothelial growth factor |
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E. How to protect the kidney in diabetic patients: With special reference to IDDM. Diabetes 1997, 46, S104–S111. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Pharmacotherapy to delay the progression of diabetic kidney disease in people with type 2 diabetes: Past, present and future. Ther. Adv. Endocrinol. Metab. 2022, 13, 20420188221081601. [Google Scholar] [CrossRef] [PubMed]
- KDOQI. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am. J. Kidney Dis. 2007, 49, S12–S154. [Google Scholar] [CrossRef]
- American Diabetes Association. 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44, S151–S167. [Google Scholar] [CrossRef]
- Rossing, P.; Hougaard, P.; Parving, H.H. Progression of microalbuminuria in type 1 diabetes: Ten-year prospective observational study. Kidney Int. 2005, 68, 1446–1450. [Google Scholar] [CrossRef]
- Parving, H.H.; Lewis, J.B.; Ravid, M.; Remuzzi, G.; Hunsicker, L.G.; DEMAND Investigators. Prevalence and risk factors for microalbuminuria in a referred cohort of type II diabetic patients: A global perspective. Kidney Int. 2006, 69, 2057–2063. [Google Scholar] [CrossRef]
- Ali, M.K.; Bullard, K.M.; Saydah, S.; Imperatore, G.; Gregg, E.W. Cardiovascular and renal burdens of prediabetes in the USA: Analysis of data from serial cross-sectional surveys, 1988–2014. Lancet Diabetes Endocrinol. 2018, 6, 392–403. [Google Scholar] [CrossRef]
- He, F.; Xia, X.; Wu, X.F.; Yu, X.Q.; Huang, F.X. Diabetic retinopathy in predicting diabetic nephropathy in patients with type 2 diabetes and renal disease: A meta-analysis. Diabetologia 2013, 56, 457–466. [Google Scholar] [CrossRef]
- Parving, H.H.; Gall, M.A.; Skøtt, P.; Jørgensen, H.E.; Løkkegaard, H.; Jørgensen, F.; Nielsen, B.; Larsen, S. Prevalence and causes of albuminuria in non-insulin-dependent diabetic patients. Kidney Int. 1992, 41, 758–762. [Google Scholar] [CrossRef] [PubMed]
- De Boer, I.H.; Rue, T.C.; Cleary, P.A.; Lachin, J.M.; Molitch, M.E.; Steffes, M.W.; Sun, W.; Zinman, B.; Brunzell, J.D.; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Group; et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: An analysis of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort. Arch. Intern. Med. 2011, 171, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Böttinger, E.P. Diabetic nephropathy: A frontier for personalized medicine. J. Am. Soc. Nephrol. 2006, 17, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Krishan, P.; Chakkarwar, V.A. Diabetic nephropathy: Aggressive involvement of oxidative stress. J. Pharm. Educ. Res. 2011, 2, 35–41. [Google Scholar]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef]
- Campbell, K.; Yacoub, R. Inhibition of RAS in diabetic nephropathy. Int. J. Nephrol. Renov. Dis. 2015, 8, 29–40. [Google Scholar] [CrossRef]
- Gilbert, R.E.; Krum, H.; Wilkinson-Berka, J.; Kelly, D.J. The renin-angiotensin system and the long-term complications of diabetes: Pathophysiological and therapeutic considerations. Diabet. Med. 2003, 20, 607–621. [Google Scholar] [CrossRef]
- Navar, L.G.; Inscho, E.W.; Majid, D.S.A.; Imig, J.D.; Harrison-Bernard, L.M.; Mitchell, K.D. Paracrine regulation of the renal microcirculation. Physiol. Rev. 1996, 76, 425–536. [Google Scholar] [CrossRef]
- Wennmann, D.O.; Hsu, H.-H.; Pavenstädt, H. The renin-angiotensin-aldosterone system in podocytes. Semin. Nephrol. 2012, 32, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Hong, H.C.; Cho, M.J.; Kim, Y.J.; Choi, H.Y.; Eun, C.R.; Yang, S.J.; Yoo, H.J.; Kim, H.Y.; Seo, J.A.; et al. Effect of eplerenone, a selective aldosterone blocker, on the development of diabetic nephropathy in type 2 diabetic rats. Diabetes Metab. J. 2012, 36, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Siragy, H.M. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrinol. Metab. 2003, 14, 274–281. [Google Scholar] [CrossRef]
- Nguyen, G.; Delarue, F.; Burcklé, C.; Bouzhir, L.; Giller, T.; Sraer, J.D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef]
- Siragy, H.M.; Carey, R.M. Protective role of the angiotensin AT2 receptor in a renal wrap hypertension model. Hypertension 1999, 33, 1237–1242. [Google Scholar] [CrossRef]
- Price, D.A.; Porter, L.E.; Gordon, M.; Fisher, N.D.; De’Oliveira, J.M.; Laffel, L.M.; Passan, D.R.; Williams, G.H.; Hollenberg, N.K. The paradox of the low-renin state in diabetic nephropathy. J. Am. Soc. Nephrol. 1999, 10, 2382–2391. [Google Scholar] [CrossRef]
- Choi, K.C.; Kim, N.H.; An, M.R.; Kang, D.G.; Kim, S.W.; Lee, J. Alterations of intrarenal renin-angiotensin and nitric oxide systems in streptozotocin-induced diabetic rats. Kidney Int. Suppl. 1997, 60, S23–S27. [Google Scholar]
- Hsieh, T.J.; Zhang, S.L.; Filep, J.G.; Tang, S.S.; Ingelfinger, J.R.; Chan, J.S. High glucose stimulates angiotensinogen gene expression via reactive oxygen species generation in rat kidney proximal tubular cells. Endocrinology 2002, 143, 2975–2985. [Google Scholar] [CrossRef]
- Bonnet, F.; Candido, R.; Carey, R.M.; Casley, D.; Russo, L.M.; Osicka, T.M.; Cooper, M.E.; Cao, Z. Renal expression of angiotensin receptors in long-term diabetes and the effects of angiotensin type 1 receptor blockade. J. Hyperten. 2002, 20, 1615–1624. [Google Scholar] [CrossRef]
- Provenzano, M.; Andreucci, M.; Garofalo, C.; Faga, T.; Michael, A.; Ielapi, N.; Grande, R.; Sapienza, P.; Franciscis, S.; Mastroroberto, P.; et al. The Association of Matrix Metalloproteinases with Chronic Kidney Disease and Peripheral Vascular Disease: A Light at the End of the Tunnel? Biomolecules 2020, 10, 154. [Google Scholar] [CrossRef]
- Llorens-Cebrià, C.; Molina-Van den Bosch, M.; Vergara, A.; Jacobs-Cachá, C.; Soler, M.J. Antioxidant Roles of SGLT2 Inhibitors in the Kidney. Biomolecules 2022, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [PubMed]
- Osorio, H.; Bautista, R.; Rios, A.; Franco, M.; Santamaría, J.; Escalante, B. Effect of treatment with losartan on salt sensitivity and SGLT2 expression in hypertensive diabetic rats. Diabetes Res. Clin. Pract. 2009, 86, e46–e49. [Google Scholar] [CrossRef]
- Freitas, H.S.; Schaan, B.D.; David-Silva, A.; Sabino-Silva, R.; Okamoto, M.M.; Alves-Wagner, A.B.; Mori, R.C.; Machado, U.F. SLC2A2 gene expression in kidney of diabetic rats is regulated by HNF-1alpha and HNF-3beta. Mol. Cell Endocrinol. 2009, 305, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Chen, J.; Hao, C.M.; Lin, S.; Gu, Y. Aldosterone promotes fibronectin production through a Smad2-dependent TGF-beta1 pathway in mesangial cells. Biochem. Res. Commun. 2006, 348, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Miyata, K.; Sun, G.P.; Rahman, M.; Kimura, S.; Miyatake, A.; Kiyomoto, H.; Kohno, M.; Abe, Y.; Yoshizumi, M.; et al. Aldosterone stimulates collagen gene expression and synthesis via activation of ERK1/2 in rat renal fibroblasts. Hypertension 2005, 46, 1039–1045. [Google Scholar] [CrossRef]
- Vidotti, D.B.; Casarini, D.E.; Cristovam, P.C.; Leite, C.A.; Schor, N.; Boim, M.A. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am. J. Physiol. 2004, 286, F1039–F1045. [Google Scholar] [CrossRef]
- Navarro, J.F.; Mora, C. Role of inflammation in diabetic complications. Nephrol. Dial. Transplant. 2005, 20, 2601–2604. [Google Scholar] [CrossRef]
- Awad, A.S.; Kinsey, G.R.; Khutsishvili, K.; Gao, T.; Bolton, W.K.; Okusa, M.D. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am. J. Physiol Ren. Physiol. 2011, 301, F1358–F1366. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros de Fuentes, M.; García-Pérez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef]
- Pérez-Morales, R.E.; del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Inflammation in diabetic kidney disease. Nephron 2019, 143, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Rivero, A.; Mora, C.; Muros, M.; Garcia, J.; Herrera, H.; Navarro-Gonzalez, J.F. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin. Sci. 2009, 116, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; van Raalte, D.H. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: A promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef]
- Mori, H.; Inoki, K.; Masutani, K.; Wakabayashi, Y.; Komai, K.; Nakagawa, R.; Guan, K.L.; Yoshimura, A. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem. Biophys. Res. Commun. 2009, 384, 471–475. [Google Scholar] [CrossRef]
- Zobel, E.H.; von Scholten, B.J.; Reinhard, H.; Persson, F.; Teerlink, T.; Hansen, T.W.; Parving, H.; Jacobsen, P.K.; Rossing, P. Symmetric and asymmetric dimethylarginine as risk markers of cardiovascular disease, all-cause mortality and deterioration in kidney function in persons with type 2 diabetes and microalbuminuria. Cardiovasc. Diabetol. 2017, 16, 888–896. [Google Scholar] [CrossRef]
- Looker, H.C.; Colombo, M.; Hess, S.; Brosnan, M.J.; Farran, B.; Dalton, R.N.; Wong, M.C.; Turner, C.; Palmer, C.N.; Nogoceke, E.; et al. Biomarkers of rapid chronic kidney disease progression in type 2 diabetes. Kidney Int. 2015, 88, 888–896. [Google Scholar] [CrossRef]
- Marcovecchio, M.L.; Dalton, R.N.; Turner, C.; Prevost, A.T.; Widmer, B.; Amin, R.; Dunger, D.B. Symmetric dimethylarginine, an endogenous marker of glomerular filtration rate, and the risk for microalbuminuria in young people with type 1 diabetes. Arch. Dis. Child. 2010, 95, 119–124. [Google Scholar] [CrossRef]
- El-Khoury, J.M.; Bunch, D.R.; Hu, B.; Payto, D.; Reineks, E.Z.; Wang, S. Comparison of symmetric dimethylarginine with creatinine, cystatin C and their eGFR equations as markers of kidney function. Clin. Biochem. 2016, 49, 1140–1143. [Google Scholar] [CrossRef]
- Stevens, L.A.; Schmid, C.H.; Greene, T.; Li, L.; Beck, G.J.; Joffe, M.M.; Froissart, M.; Kusek, J.W.; Zhang, Y.L.; Coresh, J.; et al. Factors other than glomerular filtration rate affect serum cystatin C levels. Kidney Int. 2009, 75, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Wiesli, P.; Brändle, M.; Schwegler, B.; Schmid, C. Impact of thyroid dysfunction on serum cystatin C. Kidney Int. 2003, 63, 1944–1947. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Song, S.H.; Kim, I.J.; Jeon, Y.K.; Kim, B.H.; Kwak, I.S.; Lee, E.K.; Kim, Y.K. Urinary cystatin C and tubular proteinuria predict progression of diabetic nephropathy. Diabetes Care 2013, 36, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Domingos, M.A.; Moreira, S.R.; Gomez, L.; Goulart, A.; Lotufo, P.A.; Benseñor, I.; Titan, S. Urinary Retinol-Binding Protein: Relationship to Renal Function and Cardiovascular Risk Factors in Chronic Kidney Disease. PLoS ONE 2016, 11, e0162782. [Google Scholar] [CrossRef]
- Zeni, L.; Norden, A.; Cancarini, G.; Unwin, R.J. A more tubulocentric view of diabetic kidney disease. J. Nephrol. 2017, 30, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Donadio, C.; Lucchesi, A.; Ardini, M.; Giordani, R. Cystatin C, beta 2-microglobulin, and retinol-binding protein as indicators of glomerular filtration rate: Comparison with plasma creatinine. J. Pharm Anal. 2001, 24, 835–842. [Google Scholar] [CrossRef]
- Park, S.E.; Lee, N.S.; Park, J.W.; Rhee, E.J.; Lee, W.Y.; Oh, K.W.; Park, S.W.; Park, C.Y.; Youn, B.S. Association of urinary RBP4 with insulin resistance, inflammation, and microalbuminuria. Eur. J. Endocrinol. 2014, 171, 443–449. [Google Scholar] [CrossRef]
- Cabré, A.; Lázaro, I.; Girona, J.; Manzanares, J.; Marimón, F.; Plana, N.; Heras, M.; Masana, L. Retinol-binding protein 4 as a plasma biomarker of renal dysfunction and cardiovascular disease in type 2 diabetes. J. Intern Med. 2007, 262, 496–503. [Google Scholar] [CrossRef]
- Mohapatra, J.; Sharma, M.; Acharya, A.; Pandya, G.; Chatterjee, A.; Balaraman, R.; Jain, M.R. Retinol-binding protein 4: A possible role in cardiovascular complications. Br. J. Pharmacol. 2011, 164, 1939–1948. [Google Scholar] [CrossRef][Green Version]
- Singer, E.; Markó, L.; Paragas, N.; Barasch, J.; Dragun, D.; Müller, D.N.; Budde, K.; Schmidt-Ott, K.M. Neutrophil gelatinase-associated lipocalin: Pathophysiology and clinical applications. Acta Physiol. 2013, 207, 663–672. [Google Scholar] [CrossRef]
- Kern, E.F.; Erhard, P.; Sun, W.; Genuth, S.; Weiss, M.F. Early urinary markers of diabetic kidney disease: A nested case-control study from the Diabetes Control and Complications Trial (DCCT). Am. J. Kidney Dis. 2010, 55, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.J.; Xiong, S.L.; Fang, Y.G.; Wen, S.; Chen, M.L.; Deng, R.T.; Zheng, L.; Wang, S.B.; Pen, L.F.; Wang, Q. Urinary tubular biomarkers in short-term type 2 diabetes mellitus patients: A cross-sectional study. Endocrine 2012, 41, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Vijay, S.; Hamide, A.; Senthilkumar, G.P.; Mehalingam, V. Utility of urinary biomarkers as a diagnostic tool for early diabetic nephropathy in patients with type 2 diabetes mellitus. Diabetes Met. Syndr. 2018, 12, 649–652. [Google Scholar] [CrossRef]
- Fu, W.J.; Li, B.L.; Wang, S.B.; Chen, M.L.; Deng, R.T.; Ye, C.Q.; Liu, L.; Fang, A.J.; Xiong, S.L.; Wen, S.; et al. Changes of the tubular markers in type 2 diabetes mellitus with glomerular hyperfiltration. Diabetes Res. Clin. Pract. 2012, 95, 105–109. [Google Scholar] [CrossRef]
- Giasson, J.; Li, G.H.; Chen, Y. Neutrophil gelatinase-associated lipocalin (NGAL) as a new biomarker for non-acute kidney injury (AKI) diseases. Inflamma Allergy Drug Targets 2011, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, J.A.M.; Tatsch, E.; Hausen, B.S.; Bollick, Y.; Moretto, M.B.; Duarte, T.; Duarte, M.M.; Londero, S.W.; Premaor, M.O.; Comin, F.V.; et al. Urinary kidney injury molecule-1 and neutrophil gelatinase-associated lipocalin as indicators of tubular damage in normoalbuminuric patients with type 2 diabetes. Clin. Biochem. 2015, 49, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Kammer, M.; Heinzel, A.; Willency, J.A.; Duffin, K.L.; Mayer, G.; Simons, K.; Gerl, M.J.; Klose, C.; Heinze, G.; Reindl-Schwaighofer, R.; et al. Integrative analysis of prognostic biomarkers derived from multiomics panels helps discrimination of chronic kidney disease trajectories in people with type 2 diabetes. Kidney Int. 2019, 96, 1381–1388. [Google Scholar] [CrossRef]
- Nauta, F.L.; Boertien, W.E.; Bakker, S.J.; van Goor, H.; van Oeveren, W.; de Jong, P.E.; Bilo, H.; Gansevoort, R.T. Glomerular and tubular damage markers are elevated in patients with diabetes. Diabetes Care 2011, 34, 975–981. [Google Scholar] [CrossRef]
- Colombo, M.; Valo, E.; McGurnaghan, S.J.; Sandholm, N.; Blackbourn, L.; Dalton, R.N.; Dunger, D.; Groop, P.H.; McKeigue, P.M.; Forsblom, C.; et al. Biomarker panels associated with progression of renal disease in type 1 diabetes. Diabetologia 2019, 62, 1616–1627. [Google Scholar] [CrossRef]
- Moresco, R.N.; Bochi, G.V.; Stein, C.S.; De Carvalho, J.; Cembranel, B.M.; Bollick, Y.S. Urinarykidneyinjury molecule-1 in renaldisease. Clin. Chim. Acta 2018, 487, 15–21. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-βsignaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. Ren. Physiol. 2015, 310, F596–F606. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kasama, Y.; Lee, J.S.; Jim, B.; Marin, M.; Ziyadeh, F.N. Podocyte-derived vascular endothelial growth factor mediates the stimulation of alpha3 (IV) collagen production by transforming growth factor-beta1 in mouse podocytes. Diabetes 2004, 53, 2939–2949. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Yu, M.R.; Yang, Y.; Jiang, Z.; Ha, H. Transforming growth factor β1-induced apoptosis inpodocytes via the extracellular signal-regulatedkinase-mammalian target of rapamycin complex1-NADPH oxidase 4 Axis. J. Am. Soc. Nephrol. 2005, 14, S241–S245. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh, F.N. Mediators of diabetic renal disease: The case for tgf-Beta as the major mediator. J. Am. Soc Nephrol. 2004, 15, S55–S57. [Google Scholar] [CrossRef]
- Hoffman, B.B.; Sharma, K.; Zhu, Y.; Ziyadeh, F.N. Transcriptional activation of transforming growth factor-beta1 in mesangial cell culture by high glucose concentration. Kidney Int. 1998, 54, 1107–1116. [Google Scholar] [CrossRef]
- Qiao, Y.C.; Chen, Y.L.; Pan, Y.H.; Ling, W.; Tian, F.; Zhang, X.X.; Zhao, H.L. Changes of transforming growth factor beta 1 in patients with type 2 diabetes and diabetic nephropathy: A PRISMA-compliant systematic review and meta-analysis. Medicine 2017, 96, e6583. [Google Scholar] [CrossRef]
- Andrésdóttir, G.; Jensen, M.L.; Carstensen, B.; Parving, H.H.; Rossing, K.; Hansen, T.W.; Rossing, P. Improved survival and renal prognosis of patients with type 2 diabetes and nephropathy with improved control of risk factors. Diabetes Care 2014, 37, 1660–1667. [Google Scholar] [CrossRef]
- Khamaisi, M.; Schrijvers, B.F.; De Vriese, A.S.; Raz, I.; Flyvbjerg, A. The emerging role of VEGF in diabetic kidney disease. Nephrol. Dial. Transpl. 2018, 18, 1427–1430. [Google Scholar] [CrossRef][Green Version]
- Mayer, G. Capillary rarefaction, hypoxia, VEGF and angiogenesis in chronic renal disease. Nephrol. Dial. Transplant. 2011, 26, 1132–1137. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Wong, J.S. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int. Suppl. 2000, 77, S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Baelde, H.J.; Eikmans, M.; Lappin, D.W.; Doran, P.P.; Hohenadel, D.; Brinkkoetter, P.T.; van der Woude, F.J.; Waldherr, R.; Rabelink, T.J.; de Heer, E.; et al. Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int. 2007, 71, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, B.F.; Flyvbjerg, A.; Tilton, R.G.; Rasch, R.; Lameire, N.H.; De Vriese, A.S. Pathophysiological role of vascular endothelial growth factor in the remnant kidney. Nephron. Exp. Nephrol. 2005, 101, e9–e15. [Google Scholar] [CrossRef]
- Hayek, S.S.; Quyyumi, A.A.; Reiser, J. Soluble Urokinase Receptor and Chronic Kidney Disease. N. Engl. J. Med. 2016, 374, 891. [Google Scholar] [CrossRef]
- RotbainCurovic, V.; Theilade, S.; Winther, S.A.; Tofte, N.; Eugen-Olsen, J.; Persson, F.; Hansen, T.W.; Jeppesen, J.; Rossing, P. Soluble Urokinase Plasminogen Activator Receptor Predicts Cardiovascular Events, Kidney Function Decline, and Mortality in Patients with Type 1 Diabetes. Diabetes Care 2019, 42, 1112–1119. [Google Scholar] [CrossRef]
- Theilade, S.; Lyngbaek, S.; Hansen, T.W.; Eugen-Olsen, J.; Fenger, M.; Rossing, P.; Jeppesen, J.L. Soluble urokinase plasminogen activator receptor levels are elevated and associated with complications in patients with type 1 diabetes. J. Intern. Med. 2015, 277, 362–371. [Google Scholar] [CrossRef]
- Guthoff, M.; Wagner, R.; Randrianarisoa, E.; Hatziagelaki, E.; Peter, A.; Häring, H.U.; Fritsche, A.; Heyne, N. Soluble urokinase receptor (suPAR) predicts microalbuminuria in patients at risk for type 2 diabetes mellitus. Sci. Rep. 2017, 7, 40627. [Google Scholar] [CrossRef]
- Adela, R.; Banerjee, S.K. GDF-15 as a Target and Biomarker for Diabetes and Cardiovascular Diseases: A Translational Prospective. J. Diabetes Res. 2015, 2015, 490842. [Google Scholar] [CrossRef]
- Hellemons, M.E.; Mazagova, M.; Gansevoort, R.T.; Henning, R.H.; de Zeeuw, D.; Bakker, S.J.; Lambers-Heerspink, H.J.; Deelman, L.E. Growth-differentiation factor 15 predicts worsening of albuminuria in patients with type 2 diabetes. Diabetes Care 2012, 35, 2340–2346. [Google Scholar] [CrossRef]
- Lajer, M.; Jorsal, A.; Tarnow, L.; Parving, H.H.; Rossing, P. Plasma growth differentiation factor-15 independently predicts all-cause and cardiovascular mortality as well as deterioration of kidney function in type 1 diabetic patients with nephropathy. Diabetes Care 2010, 33, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, H.; Çelik, H.T.; Gurel, O.M.; Bilgic, M.A.; Namuslu, M.; Bozkurt, H.; Ayyildiz, A.; Inan, O.; Bavbek, N.; Akcay, A. Increased serum levels of GDF-15 associated with mortality and subclinical atherosclerosis in patients on maintenance hemodialysis. Herz 2015, 40, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Wollert, K.C.; Larson, M.G.; Coglianese, E.; McCabe, E.L.; Cheng, S.; Ho, J.E.; Fradley, M.G.; Ghorbani, A.; Xanthakis, V.; et al. Prognostic utility of novel biomarkers of cardiovascular stress: The Framingham Heart Study. Circulation 2012, 126, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Kousios, A.; Kouis, P.; Panayiotou, A.G. Matrix Metalloproteinases and Subclinical Atherosclerosis in Chronic Kidney Disease: A Systematic Review. Int. J. Nephrol. 2016, 2016, 9498013. [Google Scholar] [CrossRef]
- Coll, B.; Rodríguez, J.A.; Craver, L.; Orbe, J.; Martínez-Alonso, M.; Ortiz, A.; Díez, J.; Beloqui, O.; Borras, M.; Valdivielso, J.M.; et al. Serum levels of matrix metalloproteinase-10 are associated with the severity of atherosclerosis in patients with chronic kidney disease. Kidney Int. 2010, 78, 1275–1280. [Google Scholar] [CrossRef]
- Toni, M.; Hermida, J.; Goñi, M.J.; Fernández, P.; Parks, W.C.; Toledo, E.; Montes, R.; Díez, N. Matrix metalloproteinase-10 plays an active role in microvascular complications in type 1 diabetic patients. Diabetologia 2013, 56, 2743–2752. [Google Scholar] [CrossRef]
- Carlsson, A.C.; Ingelsson, E.; Sundström, J.; Carrero, J.J.; Gustafsson, S.; Feldreich, T.; Stenemo, M.; Larsson, A.; Lind, L.; Ärnlöv, J. Use of Proteomics to Investigate Kidney Function Decline over 5 Years. Clin. J. Soc. Nephrol. 2017, 12, 1226–1235. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Heerspink, H.J.L. Time for clinical decision support systems tailoring individual patient therapy to improve renal and cardiovascular outcomes in diabetes and nephropathy. Nephrol. Dial Transplant. 2020, 35, ii38–ii42. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropthy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef]
- Petrykiv, S.I.; de Zeeuw, D.; Persson, F.; Rossing, P.; Gansevoort, R.T.; Laverman, G.D.; Heerspink, H.J.L. Variability in response to albuminuria-lowering drugs: True or random? Br. J. Clin. Pharmacol. 2017, 83, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Schievink, B.; de Zeeuw, D.; Parving, H.H.; Rossing, P.; Lambers Heerspink, H.J. The renal protective effect of angiotensin receptor blockers depends on intra-individual response variation in multiple risk markers. Br. J. Clin. Pharmacol. 2015, 80, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Garofalo, C.; Chiodini, P.; Mancuso, C.; Barbato, E.; De Nicola, L.; Andreucci, M. Ruolo della proteinuria nella ricerca clinica: Per ogni vecchia risposta, una nuova domanda [Role of proteinuria in clinicalresearch: For eachold-answer, a new key-question.]. Recenti. Prog. Med. 2020, 111, 74–81. (In Italian) [Google Scholar] [CrossRef] [PubMed]
- Laverman, G.D.; de Zeeuw, D.; Navis, G. Between-patient differences in the renal response to renin-angiotensin system intervention: Clue to optimisingrenoprotective therapy? J. Renin. Angiotensin Aldosterone Syst. 2002, 3, 205–213. [Google Scholar] [CrossRef]
- Parving, H.H.; Brenner, B.M.; McMurray, J.J.; de Zeuww, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Persson, F.; Desai, A.S.; Nicolaides, M.; et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N. Engl. J. Med. 2012, 367, 2204–2213. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Heerspink, H.J.L.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Eng. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Fried, L.F.; Duckworth, W.; Zhang, J.H.; O’Connor, T.; Brophy, M.; Emanuele, N.; Huang, G.D.; McCullough, P.A.; Palevsky, P.M.; Seliger, S.; et al. Design of combination angiotensin receptor blocker and angiotensin-converting enzyme inhibitor for treatment of diabetic nephropathy (VA NEPHRON-D). Clin. J. Am. Soc. Nephrol. 2009, 4, 361–368. [Google Scholar] [CrossRef]
- Mann, J.F.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G.; ASCEND Study Group. Avosentan for overt diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 527–535. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.-F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improvesendothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: Apilot study. Cardiovasc. Diabetol. 2017, 16, 138. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Rivoli, L.; Garofalo, C.; Faga, T.; Pelagi, E.; Perticone, M.; Serra, R.; Michael, A.; Comi, N.; Andreucci, M. Renal resistive index in chronickidneydiseasepatients: Possibledeterminants and riskprofile. PLoS ONE 2020, 15, e0230020. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Andreucci, M.; Garofalo, C.; Minutolo, R.; Serra, R.; De Nicola, L. Selective endothelin A receptor antagonism in patients with proteinuric chronic kidney disease. Expert Opin. Investig. Drugs 2021, 30, 253–262. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef]
- Sawaf, H.; Thomas, G.; Taliercio, J.J.; Nakhoul, G.; Vachharajani, T.J.; Mehdi, A. Therapeutic Advances in Diabetic Nephropathy. J. Clin. Med. 2022, 11, 378. [Google Scholar] [CrossRef]
- Tanaka, T.; Higashijima, Y.; Wada, T.; Nangaku, M. The potential for renoprotection with incretin-based drugs. Kidney Int. 2014, 86, 701–711. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Lakshmanan, M.C.; Rayner, B.; Busch, R.S.; Zimmermann, A.G.; Woodward, D.B.; Botros, F.T. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): A multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol. 2018, 6, 605–617. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Sattar, N.; Rosenstock, J.; Ramasundarahettige, C.; Pratley, R.; Lopes, R.D.; Lam, C.S.P.; Khurmi, N.S.; Heenan, L.; Del Prato, S.; et al. Cardiovascular and Renal Outcomes with Efpeglenatide in Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 896–907. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Remuzzi, G.; Parving, H.H.; Keane, W.F.; Zhang, Z.; Shahinfar, S.; Snappin, S.; Cooper, M.E.; Mitch, W.E.; Brenner, B.M. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: Lessons from RENAAL. Kidney Int. 2004, 65, 2309–2320. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Ninomiya, T.; Persson, F.; Brenner, B.M.; Brunel, P.C.; Chaturverdi, N.; Desai, A.; Haffner, S.M.; McMurray, J.J.; Solomon, S.; et al. Is a reduction in albuminuria associated with renal and cardiovascular protection? A post hoc analysis of the ALTITUDE trial. Diabetes Obes. Metab. 2016, 18, 169–177. [Google Scholar] [CrossRef] [PubMed]
- MacIsaac, R.J.; Ekinci, E.I.; Jerums, G. ‘Progressive diabetic nephropathy. How useful is microalbuminuria?: Contra’. Kidney Int. 2014, 86, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Perkins, B.A.; Ficociello, L.H.; Roshan, B.; Warram, J.H.; Krolewski, A.S. In patients with type 1 diabetes and new-onset microalbuminuria the development of advanced chronic kidney disease may not require progression to proteinuria. Kidney Int. 2010, 77, 57–64. [Google Scholar] [CrossRef]


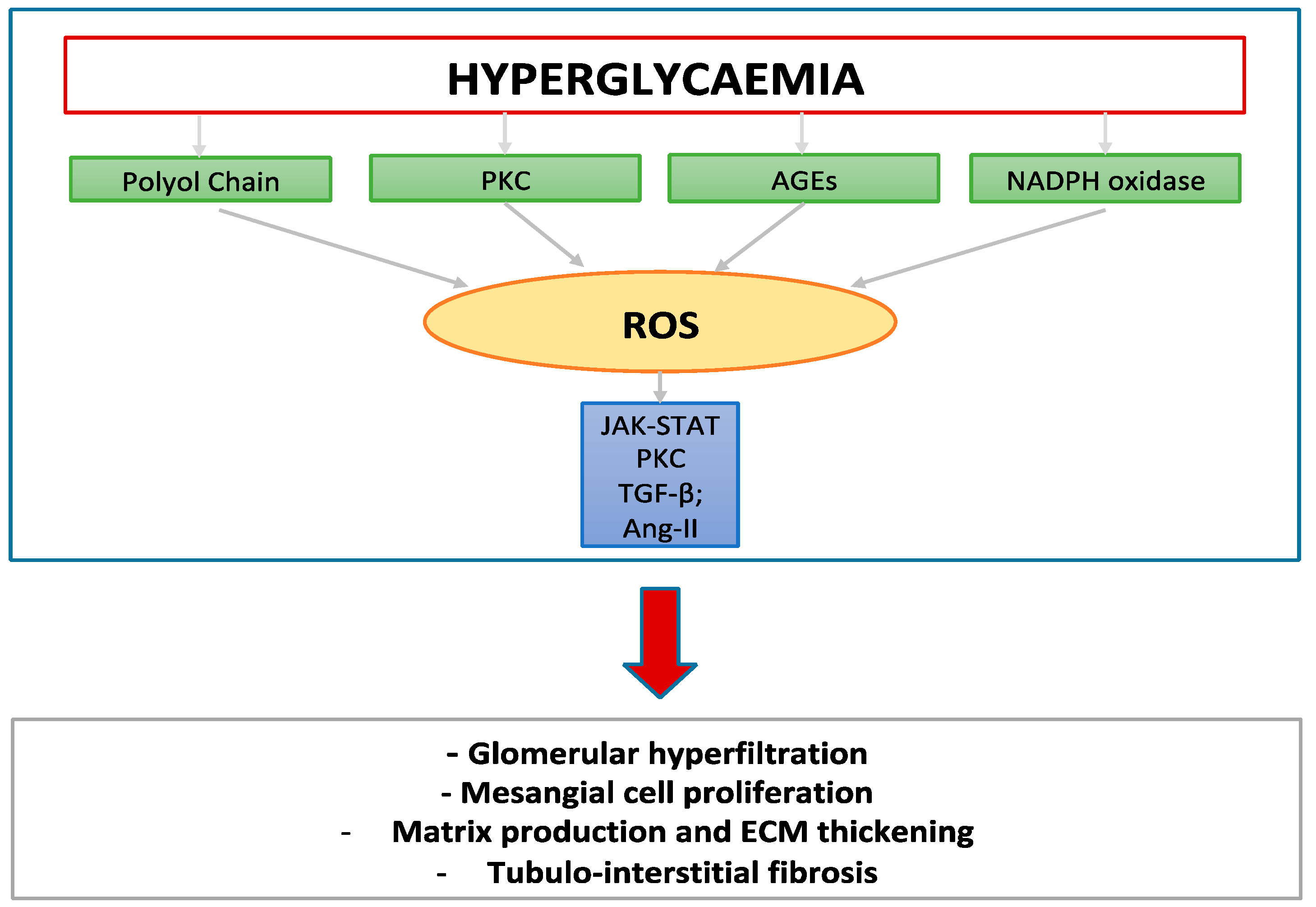{kind=link}
| Biomarkers | Characteristics | Prognostic/Predictive Values |
|---|---|---|
| SDMA | It is a catabolic product of arginine methylated proteins, excreted through the kidneys | Increased in patients with T2DM and microalbuminuria and is associated with impaired renal function and cardiovascular disease [34]. Is associated lower eGFR in patients with T2DM and DN; lower values have been observed in proteinuric patients perhaps due to hyperfiltration [35,36] |
| Cystatin C | It is a low molecular weight protein produced by all types of nucleated cells. It acts as inhibitor of cysteine protease and is freely filtered by the renal glomeruli, then 99% reabsorbed and metabolized in the renal proximal tube. It is not secreted. It is also a marker of tubular damage [38] | The concentration of cystatin C in T2DM patients is independently associated with eGFR, and its increasing is observed in patients with normoalbuminuria and decreased GFR [40] |
| RBP-4 | It is a carrier of retinol in plasma, which is not reabsorbed by tubuli when they are damaged | It is a marker of tubular damage, and its urinary concentration can be considered a predictive marker of DN in diabetic and macroalbuminuric patients [41,42]. Although the plasma concentration of RBP-4 is not a better marker of cystitis C or creatinine [43], in humans and animal models it is a marker of insulin resistance and cardiovascular risk factors [44,45,46] |
| TNFR-1 and TNFR-2 | They are membrane receptors that bind TNF alpha. It has been hypothesized that they have a direct toxic effect on the kidney, activating pathways of inflammation and apoptosis | Plasma levels of TNFR-1 and TNFR-2 are linked with an enhanced risk of CKD progression and ESKD. Moreover, they may help to ameliorate risk stratification of DKD patients [57]. They predict ESKD in absence of proteinuria, so they may play a possible predictive role in the earlier stages of DN and in non-proteinuric phenotypes of DN |
| NGAL | It is a protein produced in the renal tubule due to inflammation injury [47] | In T2DM patients, plasma levels of NGAL are inversely related to eGFR and positively related to albuminuria [48,49]. In normoalbuminuric patients with DM compared to non-diabetic control subjects, NGAL is higher [50]. Furthermore, NGAL urinary levels are higher in T2DM diabetic patients with hyperfiltration compared to T2DM with normal eGFR [51] |
| KIM-1 | It is a type 1 transmembrane glycoprotein located in the proximal tubules, and it is proposed as a marker of acute kidney injury | In T2DM patients with normoalbuminuria or mild albuminuria, its plasma concentrations are high [53]. The use of KIM-1 associated with pro b-type natriuretic peptide (pro-BNP) or beta 2 microglobulin, eGFR, and albuminuria in T2DM seemed to improve prediction of kidney function decline [54,55]. It is not associated with albuminuria. Moreover, in T1DM patients, it does not seem like it is a predictor for progression of DN [56] |
| Cardiac troponins (hs-cTnT and hs-cTnI) | They are enzymes present in both skeletal and cardiac muscles. They regulated muscle contraction by controlling the calcium-mediated interaction of actin and myosin | Raises in their values are related to acute myocardial damage. In patients with kidney disease, the dosage of both hs-cTnT and hs-cTnI improves CV risk stratification |
| NT-proBNP | Amino terminal fragment of the natriuretic type B peptide, normally produced in the heart and released in the case of cardiac stresses consequent to water overload conditions | NT-proBNP has shown to predict CV and kidney outcomes in subjects with kidney disease |
| TGF β 1 | TGF β 1 is a cytokine and a mediator of kidney damage, leading interstitial fibrosis, mesangial matrix expansion, and glomerular membrane thickening | TGFβ1 seems to cause oxidative stress in podocytes by itself, and podocyte injury leads proteinuria [60,61]. Moreover, in vitro studies showed that high glucose concentration stimulates TGFβ1 secretion and activation, so it is proposed as a mediator of DN in animal models [62,63]. A positive correlation of high levels of serum and urinary TGFβ1with albuminuria has been reported in a large metanalysis [64] |
| VEGF | VEGF is an important angiogenic factor [66]. In kidney, reduction of oxygen delivery is a stimulus of expression of VEGF [67,68] | Studies in vitro demonstrated that chronic hyperglycemia can increase the production of the VEGF protein [69], in diabetic humans biopsies, it is found a down regulation of VEGF-A expression that is correlated with loss of podocyte [70]. Moreover, in people with T1DM and T2DM and advanced DN, higher levels of urinary VEGF have been found. Although, there is contradictory evidence in intervention studies; in fact, both inhibition and administration of VEGF has been shown to improve kidney function [71] |
| suPAR | It is the circulating form of membrane protein urokinase receptor (uPAR), regulates both cell adhesion and migration [72] | Its increased levels are independent risk factors of cardiovascular diseases and kidney disease. Some evidence indicated suPAR as a marker of early kidney disease. In a cohort study that included patients with T1DM, suPAR is correlated with decline in eGFR and cardiovascular risk, but not with albuminuria [73]. Conversely, in another studies, it demonstrated a positive correlation in T1DM and T2DM patients between levels of suPAR and albuminuria [74,75] |
| GDF-15 | It is a member of TGF-cytokine family, released in response to cellular stress. It seems to have a role in regulating inflammatory processes, apoptosis, cell repair, and cell growth [76] | Higher levels are correlated with an increased risk for several adverse outcomes, particularly to a progression of albuminuria in T2DM patients [77], eGFR decline and cardiovascular risk in T1DM patients [78], early death in patients in haemodialysis [79], and in the general population it is associated with incident heart failure [80] |
| MMP-10 | It is a calcium-dependent endopeptidases that contains zinc, involved in the various processes of tissue development and cellular homeostasis. MMP-10 remodels matrix and its degradation products promote mesangium expansion [85] | It is involved in kidney and cardiovascular disease [81]. In patients with CKD, elevated concentrations of MMP-10 are independently associated with atherosclerosis and in T1DM patients are associated with DN [82,83]. Conversely, it is not associated with eGFR decline [84] |
| Key Points | |
|---|---|
| |
| DiabeticNephropaty |
|
| |
| |
| |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelle, M.C.; Provenzano, M.; Busutti, M.; Porcu, C.V.; Zaffina, I.; Stanga, L.; Arturi, F. Up-Date on Diabetic Nephropathy. Life 2022, 12, 1202. https://doi.org/10.3390/life12081202
Pelle MC, Provenzano M, Busutti M, Porcu CV, Zaffina I, Stanga L, Arturi F. Up-Date on Diabetic Nephropathy. Life. 2022; 12(8):1202. https://doi.org/10.3390/life12081202
Chicago/Turabian StylePelle, Maria Chiara, Michele Provenzano, Marco Busutti, Clara Valentina Porcu, Isabella Zaffina, Lucia Stanga, and Franco Arturi. 2022. "Up-Date on Diabetic Nephropathy" Life 12, no. 8: 1202. https://doi.org/10.3390/life12081202
APA StylePelle, M. C., Provenzano, M., Busutti, M., Porcu, C. V., Zaffina, I., Stanga, L., & Arturi, F. (2022). Up-Date on Diabetic Nephropathy. Life, 12(8), 1202. https://doi.org/10.3390/life12081202