Abstract
Heart failure management has been repeatedly reviewed over time. This strategy has resulted in improved quality of life, especially in patients with heart failure with reduced ejection fraction (HFrEF). It is for this reason that new mechanisms involved in the development and progression of heart failure, along with specific therapies, have been identified. This review focuses on the most recent guidelines of therapeutic interventions, trials that explore novel therapies, and also new molecules that could improve prognosis of different HFrEF phenotypes.
1. Introduction
Optimal medical therapy directed by current heart failure (HF) guidelines is the most important pillar of treatment. Despite clear recommendations, the prognosis for these patients is still marked by high morbidity and mortality rates. Given its significant increase in prevalence worldwide, HF remains an important public health problem. Rapid diagnosis and adequate treatment are essential for these patients. Special attention must be focused on the mechanisms of HF development and targeted therapies [1]. Distinct etiologies, clinical characteristics and comorbidities determine different mechanisms of HF development and progression. Neurohormonal modulation and hemodynamic control proved to be of great benefit, but the analysis of intercellular signaling pathway modulation is believed to have more precise therapeutic potential. Different phenotype-based subgroups related to aging and frequent comorbidities cause myocardial impairment through inflammation and microvascular coronary endothelial dysfunction. Given the heterogeneity of HF patients, several trials have been conducted to evaluate the safety and efficiency of some new molecules. Favorable results will lead to the optimal implementation of novel individualized therapies. Furthermore, present, new, promising and revolutionary research data seem to provide cutting-edge guideline recommendations for HF treatment. Additional targets, such as anatomical and physiological structures (cardiomyocytes and myocardial interstitium, microcirculation), must be considered for novel drug development. Novel therapies addressed to extracellular environment correction, angiogenesis, cellular viability, contractile function or microRNA are also evolving [2,3].
2. Materials and Methods
Based on most recent data, this review provides an overview of the current guideline-directed medical therapy of HFrEF and novel treatments tested in clinical trials. Our goal was to also present new therapeutic targets based on the progress in our understanding of the molecular and cellular mechanisms leading to HF. A systematic search of MEDLINE, Embase and the Cochrane Database of Systemic Reviews was performed using the following keywords—pharmacological targets, heart failure with reduced ejection fraction, trials. We searched for relevant English-language articles published between 1 January 2000 and June 2022, with a focus on randomized clinical trials, meta-analyses, systematic reviews and clinical practice guidelines. Additional publications were identified during a systematic review of the literature (Figure 1).
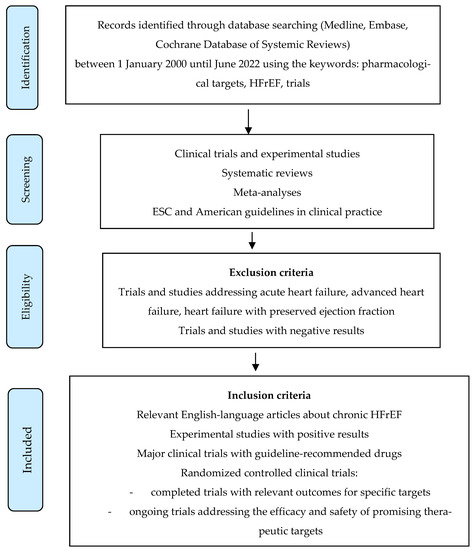
Figure 1.
Search strategy flow chart. HFrEF—heart failure with reduced ejection fraction.
3. Pathophysiological Mechanisms in HF
The sustained effort to develop a unifying hypothesis to explain the pathophysiology of HFrEF failed to generate a single conceptual model. The development of HFrEF represents the complex interaction between structural and functional biological changes that occur in the heart, autonomic nervous system, kidney, peripheral vascular system and skeletal muscle (Table 1) [4].

Table 1.
Pathophysiological mechanisms in HF.
HF is a continuously evolving paradigm. In the 1960s to early 1980s, HF was viewed as a hemodynamic model; it was then generally abandoned, except in patients admitted for decompensated HF. Until the late 1960s, congestion and edema were considered a consequence of heart–kidney interaction. In the 1970s, the concept had changed, with HR being viewed as a consequence of pump failure. The concept was based on the Frank–Starling mechanism and the vascular response, known as preload and afterload. From this moment on, the importance of vasodilation as a compensatory mechanism of pump dysfunction was intuited and developed [5]. A more recent hypothesis suggests that HF is a progressive impairment of the left heart, with secondary remodeling due to an index event evolving towards a clinical syndrome consisting of circulatory congestion and loss of cardiac function. This hypothesis suggests the importance of neurohumoral changes represented by sympathetic-adrenergic overactivity, the renin-angiotensin-aldosteron system (RAAS), vasopressin, cytokine activation, increased endothelin levels and natriuretic peptide dysfunction. The role of catecholamines was first suggested by Starling, who described tachycardia and vasoconstriction as mechanisms for increasing cardiac contractility, much later supported by Eugene Braunwald [6]. At the end of the 20th century, RAAS was still in the spotlight and well known for the cascade of long-term adverse cardiovascular effects. Short-term neurohumoral activation is beneficial for increasing pump function, stabilizing blood pressure and maintaining organ perfusion. However, chronic activation disrupts the physiological balance between vasoconstrictor and vasodilator hormones [7]. Therefore, a crucial moment in understanding the mechanisms of HF was the use of the beneficial effects of B-type natriuretic peptides (BNP), with a counter-regulatory effect on RAAS and catecholamines.
Inflammation plays an important role in the pathogenesis of HF and is associated with the up-regulation of proinflammatory cytokines tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), interleukin 1α (IL-1α), interleukin 1ß (IL-1ß) and interferon-γ (INF-γ) to the detriment of anti-inflammatory cytokines—interleukin 10 (IL-10) and transforming growth factor beta (TGFß). They initiate the process of apoptosis or necrosis and cause the loss of functional cardiomyocytes. The consequences are LV dysfunction, remodeling and increased collagen turnover of the extracellular matrix, all current targets in HF therapy. Particular attention is paid to TNF-α, considered to be the master regulator of the detrimental inflammatory effects on the heart. This cytokine increases catabolism and is possibly responsible for cardiac cachexia, which may accompany severely symptomatic HF [8]. In turn, inflammation causes microvascular endothelial dysfunction, deficiency in endogenous vasodilator molecules (nitric oxide–NO, prostaglandins) and excess endogenous vasoconstrictor products such as endothelin, a contributor to increased afterload. There is also an enhanced production of reactive oxygen species. Low NO bioavailability causes decreased protein kinase G, cyclic guanosine monophosphate (cGMP) activity and protein kinase C hyperfunction. The consequence is cardiomyocyte hypertrophy and fibrosis, with increased myocardial stiffness and diastolic dysfunction.
A remarkable point of progress in understanding the pathophysiological mechanisms of HF is its relationship with energy metabolism. In HF, there are dramatic changes in energy expenditure and energy supply, with an impact on electrophysiological functions, contractile protein interaction, abnormalities in intracellular calcium modulation and cAMP production. Under normal circumstances, oxidative metabolism in mitochondria provides 95% of cardiac energy, 5% deriving from anaerobic glycolysis. Approximately 70% of cardiac ATP is produced by the oxidation of fatty acids, the remaining 30% coming from the oxidation of glucose and lactate as well as small amounts of ketone bodies and certain amino acids. Most of the energy is consumed to maintain the excitation–contraction coupling, ion flow included. Efficient turnover of metabolic substrates is therefore a prerequisite for normal contractile and energetic function. Deficiencies are closely linked to both cellular oxidative stress and contractile dysfunction. In HF, the energy reserve is depleted and the use of the substrate is deranged, increasing the dependence on glucose metabolism and reducing fatty acid oxidation. The result is a decrease in stored ATP and phosphocreatine (PCr) and a reduction in PCr/ATP ratio, phenomena predictive of adverse events in HF. Given the paradigm of energy depletion in HF, improving cardiac energy metabolism is likely to be an essential target. Hence the undeniable benefit of SGTL2 inhibitors (SGTL2i). They make available an additional source of energy for the heart, namely the circulating ketones resulting from fatty acid mobilization in adipose tissue which are used by the liver for ketogenesis; this is beneficial for increasing functional efficiency of the heart [9].
Finally, intracellular calcium overload is not only caused by decreased myocardial glucose oxidation, but also by impaired function of the sarcoplasmic/endoplasmic reticulum Ca2+ -ATPase (SERCA2a), which regulates cardiac myocyte contraction and relaxation by transporting Ca2+ from the cytosol into the sarcoplasmic reticulum during diastole. Altered SERCA2a and abnormal handling of Ca2+ are associated with HF progression. Contractility and relaxation are both energy consuming processes and depend on ATP hydrolysis [10].
Overall, all these exhaustive structural changes involve loss of myofilaments, apoptosis, cytoskeleton disorganization, calcium homeostasis disturbance, receptor density changes, signal transduction and collagen synthesis, with devastating functional consequences. The loss of cardiac pump efficiency remains asymptomatic for variable periods of time because of compensatory mechanisms. Once the symptoms occur, these compensatory mechanisms are overwhelmed, determining myocardial damage and inducing disease progression irrespective of neurohormonal status. For all these reasons, the current research focuses on new pathophysiological targets, such as inflammation, cytokine inhibition, cardiac metabolism and cardiomyocytes, without minimizing the role of neurohormonal activation (Figure 2).
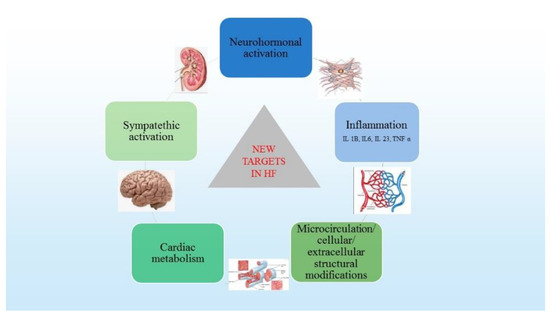
Figure 2.
New therapeutic targets in HF (adapted from Corealle et al. [2]).
4. Guideline Recommendations for Therapeutic Targets
During the last three decades, substantial progress has been made in chronic HF management. The main goals of HF treatment are better quality of life, improved symptoms and greater functional capacity in order to prevent disease recurrence and to prolong survival. The newest concept is phenotype-specific HF therapy. The 2021 European guidelines present HF classification according to left ventricular ejection fraction (LVEF) as: HF with reduced LVEF (<40%), HF with preserved LVEF (>50%) and HF with mildly reduced LVEF (40–49%). These three HF classes are characterized by distinct etiologies, comorbidities and demographic features as well as very different responses to treatment. Clinical trials assessing the efficacy of therapeutic agents demonstrated some benefit for HFrEF patients but no improvement in patients with HF with preserved EF. Two more staging systems are presented, one by the American College of Cardiology and American Heart Association (ACC/AHA) and one by the New York Heart Association (NYHA). The ACC/AHA classification is based on structural damage and clinical symptoms of HF, while the NYHA classification follows functional capacity associated with physical activity. Therefore, HF should be considered a multiple stage continuum, with each stage receiving enhanced therapy focused on risk factor modification, structural disease intervention and morbidity and mortality reduction [11,12,13]. The cornerstone treatments for chronic HF are angiotensin-converting enzyme inhibitors (ACE-I) or angiotensin receptor blockers (ARB), beta blockers (BB), mineralocorticoid receptor antagonists (MRA) and diuretics.
4.1. Novel Guideline Therapeutic Targets
After nearly two decades without new viable drugs, new classes of drugs for use in HFrEF were approved (Figure 3).
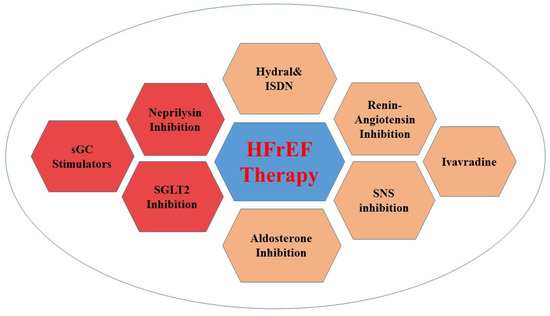
Figure 3.
Guideline recommendations for HFrEF therapy.
The most popular guideline recommendation is certainly the class of drug referred to as angiotensin receptor–neprilysin inhibitor (ARNI). Their mechanism of action consists of RAAS inhibition and activation of natriuretic peptides. The system includes structurally similar peptides (B and C type) with diuretic, natriuretic and vasodilator effects (mediated by cGMP receptors) as well as antifibrotic and antisympathetic actions. Combining sacubitril, a neprilysin inhibitor, with valsartan has been associated with improvement in HFrEF prognosis. PARADIGM-HF (Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure), a randomized double-blind trial of 8448 patients with HFrEF (EF < 40%) NYHA class II or III, demonstrated that sacubitril/valsartan was superior to enalapril in reducing the risk of CV death (13.3% vs. 16.5%, p < 0.001), HF hospitalizations (p < 0.001), prevention of worsening of symptoms (16.7% vs. 14.9%) and quality of life improvement (Table 2) [14].
Table 2.
Major clinical trials with guideline-recommended drugs (except ACE-I/ARB, beta blockers and spironolactone) in HFrEF.
Based on these results, the 2021 European guidelines recommend ARNI to replace ACE-I in patients with chronic symptomatic HFrEF as a standard therapy (class I, level B), while the 2022 American HF guidelines recommend ARNI as a first-line therapy (class I, level of evidence A) in patients with HFrEF NYHA class II or III so as to reduce mortality and morbidity [11,12]. Sacubtril/valsartan has been approved in symptomatic HF patients. The PIONEER-HF trial (Comparison of Sacubitril/Valsartan versus Enalapril on Effect on NT-proBNP in Patients Stabilized from an Acute Heart Failure Episode) demonstrated that ARNI reduced NT-proBNP levels in patients hospitalized for decompensated acute HF, without an increased rate of adverse events [15]. In the open-label TRANSITION trial (Comparison of Pre- and Post-Discharge Initiation of Sacubitril/Valsartan Therapy in HFrEF Patients After an Acute Decompensation Event), patients with reduced LVEF admitted for HF decompensation were randomized to start ARNI before or after discharge. Safety results were similar in both arms, suggesting that early initiation may simplify management (compared to ACE-I initiation and uptitration and subsequent replacement with ARNI) in the absence of contraindications. ARNI can also be initiated in symptomatic patients with chronic HF and preserved LVEF, but data are limited [16]. Adverse effects, such as hypotension, renal impairment and hyperkalemia, must not be ignored. Pregnancy is a contraindication to ARNI therapy because of its theratogenic potential. Additionally, some hypotheses suggest amiloid cerebral storage and cognitive dysfunction caused by neprilysin inhibition. A PARADIGM-HF trial subanalysis refuted this effect, but long-term studies are needed to confirm it. In this regard, the ongoing PERSPECTIVE trial (Prospective Evaluation of Cognitive Function in Heart Failure: Efficacy and Safety of Entresto compared to Valsartan on Cognitive Function in Patients with Chronic Heart Failure and Preserved Ejection Fraction—NCT02884206) is assessing cognitive decline in 592 patients with chronic heart failure and LVEF above 40% after three years of ARNI treatment compared to valsartan [17].
Another important drug class, newly introduced by guidelines, is sodium-glucose cotransporter 2 inhibitors (SGLT2i), also known as “gliflozins”. They reduce glucose reabsorption by inhibiting sodium–glucose cotransporter 2 found in the proximal nephron tubule. This determines the enhancement of chlorine (NaCl) concentration in the distal tubule and resets the tubuloglomerular feedback mechanism. Consequently, plasma volume contraction is achieved without sympathetic nervous system activation. Empagliflozin, Canagliflozin and Dapagliflozin reduced HF hospitalization rates in diabetic patients at high cardiovascular risk in three main trials: EMPA-REG outcome (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients), CANVAS program (CANagliflozin cardioVascular Assessment Study—Renal) and DECLARE TIMI 58 trial (Dapagliflozin Effect on CardiovascuLAR Events (Thrombolysis in Myocardial Infarction)) (Table 2). DAPA-HF (Dapagliflozin and Prevention of Adverse-outcomes in Heart Failure) is the first trial to investigate the effects of SGLT2i in HFrEF patients independent of diabetic status. This trial demonstrated a reduction in HF progression of 30% and in cardiovascular death risk of 18%. Moreover, Dapagliflozin reduced all-cause death risk by 17% and improved HF symptoms (hazard ratio, 0.74; 95% confidence interval [CI] 0.65 to 0.85; p < 0.001) [18]. Subsequently, EMPEROR-Reduced (Empagliflozin outcome trial in Patients with Chronic Heart Failure with Reduced Ejection Fraction) found that Empagliflozin reduced the combined risk of cardiovascular death and HF hospitalization by 25% in patients with NYHA class II-IV HF, LVEF 40% or less, and elevated natriuretic peptides despite optimal medical therapy (hazard ratio for cardiovascular death or hospitalization for heart failure 0.75; 95% CI 0.65 to 0.86; p < 0.001). Exclusion criteria were eGFR < 20 mL/min/1.73 m2 in EMPEROR-Reduced or <30 mL/min/1.73 m2 in DAPA-HF, type 1 diabetes, or systolic blood pressure < 95 to 100 mmHg [19]. Therefore, Dapagliflozin and Empagliflozin are recommended for all patients with HFrEF, regardless of the presence of diabetes (class I, level of evidence A), according to the European and American guidelines for HF [11,12]. The paradigm shift in HF therapy also includes adapting the medication to patient phenotype. From this point of view, Dapagliflozin works in most subgroups of HFrEF patients, namely patients with atrial fibrillation, systolic blood pressure > 95–100 mmHg or hypertension, high/low heart rates, hyperkalemia, chronic kidney disease and type 2 diabetes, respectively. Dapagliflozin and Empagliflozin have been shown to be effective and safe in improving cardiovascular and renal targets in patients with eGFR > 25 mL/min/1.73 m2 and 20 mL/min/1.73 m2, respectively. However, there is evidence of benefits in relation to the use of Dapagliflozin in patients with eGFR < 20 mL/min/1.73 m2 [20]. It is worth mentioning the modest and reversible decrease in eGFR in the first days after the initiation of SGLT2i, which does not require discontinuing medication because of the documented long-term beneficial effect on renal function. On the other hand, SGTL2i is not the perfect drug. Increased attention should be paid to maintaining euglycemic status, the risk of ketoacidosis, genital and soft tissue infections and, if necessary, adjusting diuretic treatment to prevent severe volume depletion [19]. In the SOLOIST-WHF (Effect of Sotagliflozin on Cardiovascular Events in Patients with Type 2 Diabetes and Worsening Heart Failure), a multicenter double-blind trial was conducted in 1222 patients with diabetes and recent HF hospitalization who were enrolled before discharge or within 3 days after discharge. The median follow-up time was 9.0 months. Sotagliflozin, a dual inhibitor of sodium–glucose cotransporters 1 and 2, reduced the combined endpoint of cardiovascular death, HF hospitalization or urgent HF visits by 33% (hazard ratio, 0.67; 95% CI 0.52 to 0.85; p < 0.001) but has not been approved by the US Food and Drug Administration (FDA). An essential aspect of HF management is the firm recommendation of both guidelines to associate ACE-I/ARNI, SGLT2i, beta blockers and aldosterone antagonists as early as possible, and to not administer them separately in a sequential manner [21].
Vericiguat, an oral soluble guanylate cyclase (GCs) stimulator that raises the production of cyclic guanylate monophosphate (cGMP), is also a new drug introduced in current guidelines. Phase 2 studies showed that Vericiguat is well tolerated by patients with HFrEF. The VICTORIA trial (Vericiguat Global Study in Subjects with Heart Failure with Reduced Ejection Fraction), a phase 3 randomized double-blind study, has evaluated the effects of Vericiguat on 4500 patients with chronic HFrEF and demonstrated a reduction in cardiovascular death (16.4% in Vericiguat group vs. 17.5% in placebo group; hazard ratio, 0.93; 95% CI, 0.81 to 1.06) and HF hospitalizations (37.9% in Vericiguat group vs. 40.9% in placebo group; hazard ratio, 0.90; 95% CI, 0.83 to 0.98; p = 0.02) (Table 2). Therefore, this drug is also recommended in the current HF guidelines (class II, level of evidence B) [22]. It has been demonstrated that deficiency in sGC-derived cyclic guanosine monophosphate (cGMP) induced by low NO bioavailability leads to myocardial dysfunction and endothelial dysfunction in coronary microcirculation. Nitric oxide activates guanylyl cyclases (GCs), followed by a rise in cGMP level in the vascular and nonvascular tissues, such as myocardium and kidney tissues. In HF, NO production is low, resulting in high arteriolar, pulmonary venous and systemic tone and leading to increased cardiac preload and afterload. In the myocardium, NO modulates the calcium channel activity, SERCA pump, sarcoplasmic reticulum and ryanodine receptor and has complex effects on mitochondrial metabolism. Different NO-synthase isoforms are also involved in the ventricular remodeling process. An increase in oxygen free radicals and a decrease in NO production are detected in HF, determining disease progression and reduction in beneficial vasodilator effects [10]. Thus, restoration of adequate nitric oxide (NO)-sGC-cGMP signaling is an important therapeutic target.
Another mechanism of HF progression is fast heart rate (HR). This reflects the imbalance between sympathetic hyperstimulation and parasympathetic inhibition, both components of neurohormonal activation. New studies demonstrate the contribution of this mechanism to high cardiovascular death and HF hospitalization rates, thus making it a new potential therapeutic target. Multiple drugs, such as BB, Digoxin, Amiodarone and Ivabradine, can modulate HR. Ivabradine is an If channel inhibitor in the sinoatrial node which controls spontaneous diastolic depolarization. It is recommended in patients with HFrEF and a LVEF less than 35% in sinus rhythm with HR over 70 beats per minute and who remain symptomatic despite maximum tolerated doses of BB therapy. The SHIFT trial (Systolic Heart Failure Treatment with the If Inhibitor Ivabradine) demonstrated a reduction in HF hospitalizations and death rates of 18% (HR 0.82; 95% CI, 0.75–0.90, p < 0.0001) (Table 2) [23]. Hence, Ivabradine is recommended by both the European and American HF guidelines (class I, level of evidence A) [11,12].
Aldosterone plays an important role in the pathophysiology of HF by inflammation, vascular rigidity, collagen synthesis and myocardial necrosis. At the same time, chronically high aldosterone levels are associated with coronary and renovascular remodeling, endothelial and baroreceptor dysfunction, and myocardial hypertrophy. Adding an MRA drug to standard HF therapy improves survival and reduces mortality in symptomatic chronic HF patients and also in subgroups with systolic dysfunction following myocardial infarction. Spironolactone was first studied in the RALES trial (Randomized Aldactone Evaluation Study). A blockade of aldosterone receptors by spironolactone, in addition to standard therapy, substantially reduced the risk of both morbidity and death among patients with severe HF (relative risk of death 0.70; 95% CI, 0.60 to 0.82; p < 0.001). Because of its antiandrogenic adverse effects (gynecomastia and breast sensitivity in males), eplerenone, a selective MRA with fewer endocrine side effects, was developed. It was investigated in EPHESUS (Eplerenone Post–Acute Myocardial Infarction Heart Failure Efficacy and Survival Study), where the rate of the primary endpoints—death from cardiovascular causes or hospitalization for cardiovascular events—was reduced (relative risk 0.87; 95% CI, 0.79 to 0.95; p = 0.002), as was the secondary endpoint—death from any cause or any hospitalization (relative risk, 0.92; 95% CI, 0.86 to 0.98; p = 0.02) (Table 2). It was also studied in the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial which demonstrated similar beneficial effects and less adverse effects compared to spironolactone (hazard ratio, 0.63; 95% CI, 0.54 to 0.74; p < 0.001) (Table 2). Based on these data, MRA treatment is recommended in all HF patients who remain symptomatic, despite ACE-I, ARB or ARNI treatment with a BB, to reduce HF hospitalizations and death (class I, level of evidence A) [11,12].
4.2. New Approaches for Studying the Additional Benefits of Some Previous Therapeutic Targets
Digoxin is the oldest, but also the most controversial, drug prescribed in HF therapy (class II, level of evidence B) according to the European HF guidelines. In the DIG study, it did not reduce mortality compared to placebo, though it did reduce HF hospitalizations (26.8 percent vs. 34.7 percent; risk ratio, 0.72; 95% CI, 0.66 to 0.79; p < 0.001). A retrospective analysis suggested that patients with a digoxin blood level between 0.5 and 0.9 mg/mL have some benefits [24,25]. An ongoing prospective placebo-controlled trial (DECISION trial—NCT03783429) is currently testing if lower digoxin doses guided by blood levels will reduce HF hospitalizations and cardiovascular death rate in approximately 1000 patients (https://clinicaltrials.gov/ct2/show/NCT03783429; accessed on 3 July 2022). The results will be published in 2025 (Table 3).
Table 3.
Clinical trials targeting old drugs, new drugs and potential targets in HFrEF.
Furthermore, a combination of hydralazine and isosorbide dinitrate may be considered (class II, level of evidence B) to reduce mortality in symptomatic HFrEF patients who do not tolerate ACE-I, ARNI or ARB or who are contraindicated [11,12].
Congestion, an important cause of the signs and symptoms of HF, causes atrial and ventricular remodeling, arrhythmias and renal impairment and is a predictor of poor prognosis. Congestion treatment is an essential part of HF management. However, its diagnosis remains a challenge. In some patients without clinical evidence of congestion, subclinical signs have been demonstrated using pleural and cardiac ultrasound, either in the interstitial space (lung B lines) or in the intravascular space (inferior vena cava distention). Loop diuretics are the mainstay of decongestion therapy. Although the most widely used loop diuretic is Furosemide, Bumetanide and Torsemide are better absorbed and released in the renal tubule. A meta-analysis of some small randomized and observational studies suggested the possible superiority of Torsemide compared to Furosemide; however, there are no randomized studies to verify this [26].
TRANSFORM-HF (ToRsemide compArisoN With furoSemide FOR Management of Heart Failure), an ongoing multicenter study, will randomize 6000 patients with decompensated HF before discharge to compare the effectiveness of Torsemide versus Furosemide and its effects on mortality and morbidity (Table 3) [27]. Resistant congestion can be managed by combining different classes of diuretics, although the efficacy of this strategy has not been tested in clinical trials.
Most of the sodium is reabsorbed in the proximal tubule of the nephron. Acetazolamide, a carbonic anhydrase inhibitor, decreases proximal tubular sodium reabsorption, therefore enhancing the effect of loop diuretics. ADVOR (Acetazolamide in Decompensated Heart Failure With Volume OveRload), a randomized double-blind placebo-controlled study which will enroll 500 HF patients, is planning to test the efficiency of this association (Table 3) [28]. Tolvaptan is an oral vasopressin type 2 receptor antagonist that enhances water excretion through the collecting tubules. Urine volume is high, but without enhanced electrolyte excretion. The EVEREST study (The Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan) compared the safety and efficacy of tolvaptan versus placebo in the treatment of patients with worsening congestive HF and showed relief of dyspnea. The composite endpoint of cardiovascular death or hospitalization for HF occurred in 871 tolvaptan group patients (42%) and 829 placebo group patients (40.2%) (hazard ratio, 1.04; 95% CI, 0.95–1.14; p = 0.55) (Table 3). Likewise, in the QUEST study (Efficacy and Safety of Tolvaptan in Heart Failure Patients with Volume Overload Despite the Standard Treatment with Conventional Diuretics), tolvaptan was associated with weight loss, relief of HF symptoms and signs and increased diuresis. Over a 3-year period in the tolvaptan group, the increase in total kidney volume was 2.8% per year (95% CI, 2.5 to 3.1) versus 5.5% per year in the placebo group (95% CI, 5.1 to 6.0; p < 0.001). The composite endpoint favored tolvaptan over placebo (44 vs. 50 events per 100 person-years, p = 0.01), with lower rates of worsening kidney function (2 vs. 5 events per 100 person-years, p < 0.001) and kidney pain (5 vs. 7 events per 100 person-years, p = 0.007). Nevertheless, some patients developed hypernatremia at high tolvaptan doses (Table 3) [29].
5. Evolving Therapeutic Targets
Despite the encouraging results obtained with the new molecules already introduced in HF guidelines, the battle with this disease is not fully won. The discovery of some new mechanisms involved in HF development offers attractive perspectives for the research of some new molecules with additional benefits. On the other side, most hospitalizations are related to worsening of symptoms and signs of chronic HF (acutely decompensated HF), a highly heterogeneous pathophysiologic condition. There is a large amount of information regarding the new strategies in chronic HF. Interesting therapeutic molecules targeting the causative mechanisms of HR decompensation are also intensively studied (Figure 4).
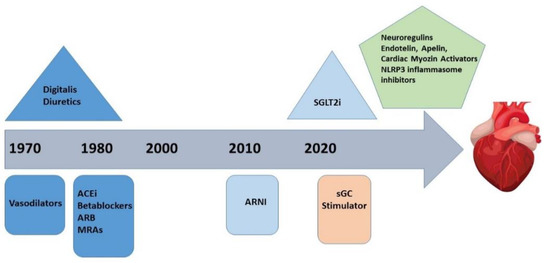
Figure 4.
Progression of therapeutic targets over time.
Finerenone, a novel non-steroidal MRA, combines spironolactone efficiency with eplerenone selectivity resulting in a lower level of hyperkalemia and a more significant decrease in natriuretic peptide levels. The ARTS-HF trial (Miner Alocorticoid Receptor antagonist Tolerability Study-Heart Failure) was a randomized, double-blind, phase 2b, multicenter study enrolling 1066 patients who received oral, once-daily finerenone or eplerenone. The study demonstrated a decrease of >30% in plasma N-terminal pro-B-type natriuretic peptide over 90 days in 37.2% of patients as a primary endpoint. The proportion of patients was similar between the subgroups. The composite endpoint included cardiovascular hospitalizations, acute worsening HF or 90-day all-cause mortality. The composite endpoint was statistically significant only in the 10 to 20 mg group (hazard ratio, 0.56; 95% CI, 0.35; 0.90; p = 0.02); however, further research is needed to confirm these findings (Table 3) [33].
Achieving optimal heart pump performance requires the use of significant amounts of ATP and therefore relies on such substrates as carbohydrates and fatty acids for energy requirements. In HF, insufficient ATP is generated by defects in glycolysis, oxidation of glucose and fatty acids and oxidative phosphorylation. Perhexilline is a metabolic modulator whose main action appears to be the inhibition of fatty acid oxidation by inhibiting carnitine palmitoyltransferases 1 and 2 (CPT-1/2). These are specific proteins that carry fatty acids from the cytoplasm into the mitochondria. After CPT-1/2 inhibition, beta-oxidation is reduced, shifting the metabolism to glycolysis and increasing mechanical efficiency, thus improving the efficiency of ATP generation. In a phase 2 randomized, double-blinded, placebo-controlled study using direct measures of energy metabolism in the left ventricle, cardiac magnetic resonance spectroscopy showed a clear biological effect on human cardiac metabolism but no changes in noninvasively assessed contractile function or invasive assessment of myocardial substrate utilization after 1 month of therapy (Table 3) [38]. Despite these results, the study is a strong model for a new phenotyping approach in HF patients.
Cardiotrophin 1 cytokine (CT1) is a new molecule that belongs to the IL-6 family. Initially discovered as a factor inducing cardiomyocyte hypertrophy, it has a variety of different effects, including the ability to stimulate the survival of cardiac and neuronal cells. Interestingly, although activation of the p42/p44 MAP kinase pathway is necessary for promoting the survival effects of CT1 in the cardiac cells, it is not necessary for its hypertrophic effect that is probably secondary to the activation of the Jak/STAT-3 pathway. Thus, CT-1 may be used as a new cardioprotective agent, especially if its hypertrophic effect can be specifically inhibited. In addition, CT1 has been shown to enhance the production of heat shock proteins hsp70 and hsp90, which protect cardiomyocytes from thermic and ischemic stress. Moreover, it seems that CT1 reduces the tumor necrosis factor, with beneficial effects in myocardial infarction and ischemic HF. In HF, CT1 promotes beneficial cardiac remodeling, reduces pathological cardiac structural and ischemic changes and also regulates obesity secondary to increased food intake and insulin resistance [39,40].
Interleukin 1 (IL-1) beta is also known to depress cardiac function. IL-1 inhibition has beneficial effects in HF because the acute inflammatory response is suppressed, thrombotic cardiovascular events are prevented, and cardiac function and patient quality of life are improved [41]. The extension phase of the CANTOS study [Cardiovascular Risk Reduction Study (Reduction in Recurrent Major CV Disease Events)], a randomized double-blind trial, suggests Il-1 as a potential therapeutic target in HF. Canakinumab, an IL-1β antibody, at a dose of 150 mg every 3 months significantly reduced the composite endpoint of HF hospitalization or HF–related mortality in 10,061 patients with a history of acute myocardial infarction (hazard ratio vs. placebo, 0.83; 95% CI, 0.73 to 0.95; p = 0.005) (Table 3) [35]. In the REDHART trial (the REcently Decompensated Heart failure Anakinra Response Trial), Anakinra, a recombinant human Il-1 receptor antagonist, improved peak aerobic exercise capacity after 12 weeks of treatment in patients with LVEF < 50% (from 14.5 mL/kg/minute to 16.1 mL/kg/minute; p = 0.009) (Table 3) [36]. A currently underway single center, randomized, placebo-controlled, double-blind, phase II, randomized clinical trial is evaluating the 24-week effect of anakinra on cardiorespiratory capacity in patients with recent hospitalization for acute HFrEF decompensation [30].
Dapansutrile (OLT1177) is a selective oral NLRP3 inflammasome inhibitor (intracellular sensor that detects endogenous danger signals, environmental irritants) aimed at inhibiting IL-1β and IL-18 activity. Recently, a randomized, double-blind, phase IB study on dapansutrile in 30 patients with stable HFrEF, NYHA class II or III reported that it is safe and well-tolerated over the 14-day treatment regardless of the administered doses. For example, in the group receiving 2000 mg of dapansutrile, LVEF and exercise time were improved (from 31.5% to 36.5%, p = 0.039 and from 570 to 616 s, p = 0.039, respectively) [42]. Endothelin antagonists seem to be a promising therapeutic target for HF, considering their role in pathological fibrosis, hypertrophy, arterial hypertension and overregulation. Despite encouraging preclinical data, some trials did not report significant benefits, while others reported adverse effects. This is probably secondary to the competitive effects in different cell types or receptor subtype selectivity [43].
Neuregulin-1 proteins are important in the development and function of cardiomyocytes. Some small phase 2 studies using recombinant human neuregulin-1 reported improved hemodynamics and reverse remodeling in HFrEF patients [44]. A phase 3 study (NCT03388593—Survival Study of the Recombinant Human Neuregulin-1β in Subjects With Chronic Heart Failure) is ongoing and seeks to assess the efficiency and safety of daily intravenous neuregulin-1 perfusion, followed by weekly bolus in HFrEF, on all-cause mortality (https://clinicaltrials.gov/ct2/show/NCT03388593; accessed on 3 July 2022) (Table 3).
Patiromer and sodium zirconium cyclosilicate are new oral treatments that bind potassium into the gastrointestinal tract and rapidly normalize serum potassium levels. Their use is still discussed, and they are not a guideline recommendation. Patiromer and sodium zirconium cyclosilicate could be useful in association with ACE-I, ARB or ARNI for reaching the maximum recommended doses, considering that these drugs should not be initiated if serum potassium level is over 5 mmol/L. Doses should be reduced, or therapy interrupted, if potassium serum level reaches 5.5 mmol/L. Hyperkalemia is a frequent finding in HF-related diabetes mellitus, chronic kidney disease or elderly patients. BIOSTAT-CHF (BIOlogy Study to TAilored Treatment in Chronic Heart Failure), an international, multicenter, prospective, observational study was specifically designed to assess the effects of ACE-I/ARB uptitration and its association with outcome. The study concluded that higher potassium levels are an independent predictor of enduring lower dosages, without attenuating the beneficial effects of uptitration [45]. The DIAMOND (Patiromer for the Management of Hyperkalemia in Subjects Receiving RAASi Medications for the Treatment of Heart Failure) trial, a prospective phase 3b multicenter, double-blind, placebo-controlled, randomized study, enrolled 878 eligible patients with HFrEF. The follow-up time was 13.0 weeks. Subjects with hyperkalemia (serum potassium level over 5.5 mmol/L) were randomized to patiromer (n = 439) versus control group (n = 439). The trial included a run-in phase during which the patients received patiromer and optimized doses of RAAS inhibitors and a randomized withdrawal blinded treatment phase. The primary endpoint, adjusted mean change in serum potassium level, was 0.03 mEq/L in the patiromer group versus 0.13 mEq/L in the control group (p < 0.001). Patiromer seems to be a beneficial strategy for managing patients at risk of hyperkalemia. Its administration also permits 85% of patients to receive appropriate doses of RAAS inhibitors (Table 3) [32].
Iron deficiency is a common comorbidity in HF patients and is associated with reduced quality of life and functional capacity, higher rates of hospitalization and mortality, regardless of the presence of anemia. Intravenous iron supplementation is a promising therapeutic target in HF patients given its essential role in mitochondrial aerobic respiration and cellular immune response, including cardiomyocytes. The European guidelines recommend that HFrEF patients should be tested for anemia and iron deficiency using serum ferritin and transferrin saturations. The European guidelines also recommend intravenous ferric carboxymaltose administration in symptomatic HF patients with documented iron deficiency to improve symptoms and quality of life (class IIa, Level of Evidence A recommendation). American guidelines support intravenous iron treatment as a class IIb, level of evidence B recommendation [11,12]. The FAIR-HF (Ferric Carboxymaltose Assessment in Patients With Iron Deficiency and Chronic Heart Failure) trial included 459 patients with chronic HF who received weekly intravenous ferric carboxymaltose until iron repletion. The results sustained significant improvements in NYHA functional class at week 24 (odds ratio for improvement by one class, 2.40; 95% CI, 1.55 to 3.71; p < 0.001), in distance on the 6-min walk test and in quality of life (evaluated by the European Quality of Life–5 Dimensions Visual Analog Scale and Kansas City Cardiomyopathy questionnaire) at weeks 4, 12 and 24 (p < 0.001 for all comparisons) [31] (Table 3). CONFIRM-HF (Ferric CarboxymaltOse evaluatioN on perFormance in patients with IRon deficiency in coMbination with chronic Heart Failure) was a double-blind, multi-center, prospective, randomized, placebo-controlled trial which enrolled 304 ambulatory patients with symptomatic HFrEF, iron deficiency (defined as ferritin < 100 ng/mL, or ferritin 100–300 ng/mL if transferrin saturation < 20%) and haemoglobin < 15 g/dL. The trial provided evidence that treatment with intravenous feric carboximaltose over one year improved exercise capacity, symptoms and quality of life, and is thus associated with a reduced risk of hospitalizations due to worsening HF (hazard ratio: 0.39, p = 0.009) [37] (Table 3). A more recent meta-analysis included data from four randomized trials using the same intravenous iron preparation (ferric carboxymaltose). FAIR-HF and CONFIRM-HF contributed approximately 90% of the total number of subjects included. This meta-analysis concluded that intravenous ferric carboxymaltose in patients with iron deficiency may decrease recurrent hospitalizations in HFrEF [46]. Three large randomized trials studying the benefits of intravenous iron on mortality and hospitalizations rates in chronic HFrEF are still active (FAIR-HF2: https://clinicaltrials.gov/ct2/show/NCT03036462; HEART-FID: https://clinicaltrials.gov/ct2/show/NCT03037931 or currently recruiting IRONMAN: https://clinicaltrials.gov/ct2/show/NCT02642562; accessed on 16 July 2022) (Table 3).
HF may be associated with high plasma copper concentrations, but also with myocardial copper depletion. Experimental models suggest that copper chelation is beneficial for HF patients. Small doses of Trientine, an alternative copper chelator, could facilitate redistribution of copper into the tissues. The ongoing TRACER-HF trial (NCT0387518—Study to Evaluate Effects of INL1 in Patients With Heart Failure and Reduced Ejection Fraction), a multicenter, randomized, double-blind, placebo-controlled, dose–response study, is evaluating the efficacy and safety of three PO INL1 (a copper-binding agent) doses in 200 patients with chronic, stable HFrEF compared to placebo. The primary outcome measure is the decrease in serum NT-proBNP level from baseline to 12 weeks. The trial will also assess the effect on echocardiographic parameters and functional status [https://clinicaltrials.gov/ct2/show/NCT03875183; accessed on 3 July 2022] (Table 3) [47].
Coenzyme Q10 is an essential component of the mitochondrial electron transport chain and has an important role in the metabolic process. Lower selenium and coenzyme Q10 concentrations have been associated with adverse HF progression. Some randomized trials and meta-analyses sustain the efficacy of coenzyme Q10 in improving functional status and echocardiographic parameters in HFrEF, though its prognostic role is still debated [48,49,50].
Apelin was discovered in 1993 as an orphan G protein-coupled receptor. Its protein structure is highly similar to that of angiotensin II receptor type 1 (AT1), although no binding to the receptor was observed with angiotensin II. Apelin attenuates ventricular hypertrophy and stimulates contractility in failing cardiac muscle by increasing the availability of intracellular calcium rather than enhancing myofilament calcium sensitivity. Therefore, it is a promising therapeutic agent in HF. Many apelin receptor agonists have been used both in vitro and in vivo. The 2021 study by Gargalovic et al. presents a new apelin receptor agonist—BMS-986224—as a potential target in HF. Its administration enhances cardiac output by different mechanisms compared to ACE-I. BMS-986224 is a selective and potent apelin agonist, with a receptor binding profile similar to that of apelin-13. In experimental models, BMS-986224 perfusion increased stroke volume and cardiac output but, unlike ACE-I, it did not prevent cardiac hypertrophy and fibrosis. The molecule is presented as a new, potent non-peptide apelin agonist, with oral bioavailability that mimics the signaling properties of apelin-13. Its oral administration induces a sustained increase in cardiac output, the unique profile supporting its further clinical evaluation in HF [34].
Omecamtiv mecarbil, a selective cardiac myosin activator that improves cardiac contractility, is another promising molecule in HFrEF. The GALACTIC-HF (Global Approach to Lowering Adverse Cardiac Outcomes Through Improving Contractility in Heart Failure), a randomized, placebo-controlled, phase 3 trial, showed a lower risk of heart-failure events and cardiovascular death with omecamtiv mecarbil compared to placebo. The trial enrolled 8256 patients with NYHA class II, III or IV symptoms, LVEF 35% or less and elevated natriuretic peptides who were then assigned to a dose of 25 mg, 37.5 mg or 50 mg twice daily or to a placebo dose. The median follow-up time was 21.8 months. The primary composite endpoint was an HF event or cardiovascular death which occurred in 37.0% of the omecamtiv mecarbil group versus 39.1% in the placebo group (hazard ratio, 0.92; 95% CI, 0.86 to 0.99; p = 0.03). Omecamtiv mecarbil produced greater therapeutic benefit and was more efficient in patients with an LVEF of 28% or less (hazard ratio, 0.84; 95% CI, 0.77–0.92) and blood pressure lower than 100 mmHg (hazard ratio, 0.81; 95% CI, 0.70–0.94), with no effect on cardiovascular death rates. In addition, omecamtiv mecarbil had no adverse effect on blood pressure, heart rate or creatinine and potassium levels. The drug is still awaiting FDA approval [51].
Cell and gene therapy is making amazing progress in experimental research, but currently in clinical trials the results remain modest. The theoretical premise is that, in humans, the cardiomyocytes lost after myocardial infarction are replaced only by fibrous scars and hypertrophy of remaining cells, while inferior species have a powerful regenerative capacity. Some phase 1, 2 and 3 studies are testing cell therapy (progenitor cells, uni- and multipotent cardiomyocytes, tissues processed by cell engineering techniques), gene therapy (noncoding RNA) or noncellular therapy such as growth factors. While current pharmacotherapy manages to prolong life and avoids unwanted major events without addressing the cause of HF—loss of myocardial contractility—cell and gene therapy mainly address this cause while aiming for the restoration of lost cardiomyocytes [52,53].
The loss of viable cardiomyocytes is a central feature of cardiac remodeling and progression to HF that occurs secondary to the process of necrosis or apoptosis. The triggers are not fully understood but several mechanisms have been suggested, including the gradual accumulation of oxidative stress-related damaged macromolecules, persistent hyperactivation of catecholamines, activation of TNF-α signaling and chronic inflammatory signaling [54]. Mitochondrial permeability transition pores (MPTPs) play a critical role in cell death, and several methods of inhibiting this process (e.g., inhibition of CaMKII31 or cyclophilin D) have been shown to prevent cell death or reduce adverse remodeling in experimental studies. Dysfunctional mitochondria resulting from the intracellular damage caused by oxidative stress are also considered to be a central player in inducing the process called sterile inflammation. Incomplete mitochondrial DNA autophagy can activate the TLR 9 receptor (Toll-like receptor), a signaling cascade that leads to the production of proinflammatory cytokines and continuous cell damage in HF. Similarly, activation of inflammasomes, multiprotein complexes that identify harmful substances resulting intracellularly through oxidative stress, are also involved in ischemic HF [55]. Therefore, targeting inflammatory receptors and cytokine signaling may provide potential resources to limit cell death in HF. Autophagy is a key process in cell renewal. Both defective and excessive autophagy have a negative impact on cardiac remodeling and progression to HF. Rapamycin complex (mTOR) is the key regulator of autophagy signaling, currently considered a potential therapeutic target. Cell viability is also determined by other cell processes, such as the ubiquitin-proteasome system (UPS) and the endoplasmic reticulum (ER) stress response, both involved in maintaining intracellular protein homeostasis. Disruption of these mechanisms also leads to the activation of cell death pathways. Modulation of UPS and ER signaling cascades, as well as their associated molecules, may be another potential target for HF therapy [56].
6. Conclusions
Regarding the development of HF strategies, what Moses Maimonides said over 800 years ago in his oath is still valid: “Grant me the strength, time, and opportunity always to correct what I have acquired, always to extend its domain; for knowledge is immense and the spirit of man can extend indefinitely to enrich itself daily with new requirements. Today he can discover his errors of yesterday and tomorrow he can obtain a new light on what he thinks himself sure of today” [57].
HF management underwent dynamic changes regarding therapeutic concepts, from the hemodynamic and neurohormonal treatment to strategies targeting maladaptive signaling pathways. Recent beneficial results with ARNI and SGLT2 inhibitors support the fact that improving HF progression is not an impossible task and significant progress is going to be made. The current paradigm is based on creating a balance between the classic hemodynamic and neurohormonal modulators and the new molecular targets in order to obtain a personalized, adapted therapy. Three different conceptual therapeutic models (hemodynamic, cardiorenal and neurohormonal models) were used to develop strategies for treating HFrEF, but none of these can explain disease progression completely. New therapeutic classes (ARNI, Vericiguat, Omecamtiv mecarbil) were also developed based on interesting observations provided by current models, whereas the pleiotropic mechanisms underlying the benefits of SGLT2 inhibitors are not fully understood. Thus, it is an urgent need to rethink or expand the existing models or develop new paradigms for additional effective and safe therapies for this population [58]. Given its complex pathophysiology, the fundamental challenge is to adapt the medication to patient phenotype. For clinicians, it is a difficult task that requires an integrative but personalized view. On the other hand, the development of valid targets for HF therapy needs appropriate and complete preclinical studies based on evolving guidelines recommendations [59].
The ultimate purpose is to facilitate the prevention and progression of HF by translating research into practice. Many innovative programs have been developed to improve clinician adherence to evidence-based clinical guidelines. The National Heart, Lung, and Blood Institute emphasizes the urgent need for the development of comprehensive guidelines reflecting real-life clinical scenarios to assist physicians in improving daily practice [60].
This review summarizes the molecules recently introduced in clinical practice guidelines for HFrEF, but also the potential of old and new targets. The purpose is to provide updated information and advocate for its implementation in clinical practice for optimal care of patients with heart failure.
Author Contributions
M.-A.M.—conceptualization, literature search, writing-original draft preparation and editing, data curation; I.-I.C.—visualization; A.N.—writing-original draft preparation; V.O.A.—conceptualization, visualization, literature search, writing-review and editing, data curation and validation, supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pellicori, P.; Khan, M.J.I.; Graham, F.J.; Cleland, J.G.F. New perspectives and future directions in the treatment of heart failure. Heart Fail. Rev. 2020, 25, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correale, M.; Tricarico, L.; Fortunato, M.; Mazzeo, P.; Nodari, S.; Di Biase, M.; Brunetti, N.D. New Targets in Heart Failure Drug Therapy. Front. Cardiovasc. Med. 2021, 8, 665797. [Google Scholar] [CrossRef] [PubMed]
- Nabeebaccus, A.; Zheng, S.; Shah, A.M. Heart failure-potential new targets for therapy. Br. Med. Bull. 2016, 1, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.S.; Tkacs, N.C. Current concepts of neurohormonal activation in heart failure: Mediators and mechanisms. AACN Adv. Crit. Care 2008, 19, 364–385. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.V.; Bacal, F. Pathophysiology and Current Terrapeutic Implication. Int. J. Cardiovasc. Sci. 2020, 33, 439–446. [Google Scholar]
- Chaggar, P.S.; Malkin, C.J.; Shaw, S.M.; Williams, S.G.; Channer, K.S. Neuroendocrine effects on the heart and targets for therapeutic manipulation in heart failure. Cardiovasc. Ther. 2009, 27, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.E.; Dunlap, M.E. Renal hemodynamics in heart failure: Implications for treatment. Curr. Opin. Nephrol. Hypertens. 2008, 17, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Lopatin, Y.M.; Rosano, G.M.; Fragasso, G.; Lopaschuk, G.D.; Seferovic, P.M.; Gowdak, L.H.; Vinereanu, D.; Hamid, M.A.; Jourdain, P.; Ponikowski, P. Rationale and benefits of trimetazidine by acting on cardiac metabolism in heart failure. Int. J. Cardiol. 2016, 203, 909–915. [Google Scholar] [CrossRef]
- Giani, D.; Chan, J.; Gwathmey, J.K.; del Monte, F.; Hajjar, J.R. SERCA2a in heart failure: Role and therapeutic prospects. J. Bioenerg. Biomembr. 2005, 37, 375–380. [Google Scholar] [CrossRef]
- McDonagh, T.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group, 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef] [PubMed]
- Maddox, T.M.; Januzzi, J.L.; Allen, L.A.; Breathett, K.; Butler, J.; Davis, L.L.; Fonarow, G.C.; Ibrahim, N.E.; Lindenfeld, J.; Masoudiet, F.A.; et al. 2021 Update to the 2017 ACC Expert Consensus Decision Pathway for Optimization of Heart Failure Treatment: Answers to 10 Pivotal Issues About Heart Failure With Reduced Ejection Fraction: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2021, 6, 772–810. [Google Scholar]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.C.; Swedberg, K.; et al. For the PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez, E.; Morrow, D.A.; DeVore, A.; Duffy, C.I.; Ambrosy, A.P.; McCague, K.; Rocha, R.; Braunwald, E.; For the PIONEER-HF Investigators. Angiotensin–Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med. 2019, 380, 539–548. [Google Scholar]
- Gaziano, T.A.; Fonarow, G.C.; Velazquez, E.J.; Morrow, D.A.; Eugene Braunwald, E.; Solomon, S.D. Cost-effectiveness of sacubitril-valsartan in hospitalized patients who have heart failure with reduced ejection fraction. JAMA Cardiol. 2020, 5, 1236–1244. [Google Scholar] [CrossRef]
- Kuchulakanti, P.K. ARNI in cardiovascular disease: Current evidence and future perspectives. Future Cardiolog. 2020, 16, 505–515. [Google Scholar] [CrossRef]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.A.; Bělohlávek, J.; et al. For the DAPA-HF Trial Committees and Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Fitchett, D.; Zinman, B.; Wanner, C.; Lachin, J.M.; Hantel, S.; Salsali, A.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Inzucchi, S.E.; et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascularrisk: Results of the EMPA-REG OUTCOME® trial. Eur. Heart J. 2016, 37, 1526–1534. [Google Scholar]
- Rosano, G.; Moura, B.; Metra, M.; Böhm, M.; Bauersachs, J.; Gal, T.B.; Adamopoulos, S.; Abdelhamid, M.; Bistola, V.; Čelutkienė, J.; et al. Patient profiling in heart failure for tailoring medical therapy. Eur. J. Heart Fail. 2021, 23, 872–881. [Google Scholar] [CrossRef]
- Deepak, L.; Bhatt, D.L.; Szarek, M.; Steg, G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar]
- Armstrong, P.W.; Roessig, L.; Patel, M.J.; Anstrom, K.J.; Butler, J.; Voors, A.A.; Lam, C.S.P.; Ponikowski, P.; Temple, T.; Pieske, B.; et al. A Multicenter, randomized, double-blind, placebo-controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator. The VICTORIATrial. JACC Heart Fail. 2018, 6, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L.; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): A randomised placebo-controlled study. Lancet 2010, 11, 875–885. [Google Scholar]
- Ahmed, A.; Rich, M.W.; Love, T.E.; Lloyd-Jones, D.M.; Aban, I.B.; Colucci, W.S.; Adams, K.F.; Gheorghiade, M. Digoxin and reduction in mortality and hospitalization in heart failure: A comprehensive post hoc analysis of the DIG trial. Eur. Heart J. 2006, 27, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Pitt, B.; Rahimtoola, S.H.; Waagstein, F.; White, M.; Love, T.E.; Braunwald, E. Effects of digoxin at low serum concentrationson mortality and hospitalization in heart failure: A propensity matched study of the DIG trial. Int. J. Cardiol. 2008, 123, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Abraham, B.; Megaly, M.; Sous, M.; Fransawyalkomos, M.; Saad, M.; Fraser, R.; Topf, J.; Goldsmith, S.; Simegn, M.; Bart, B.; et al. Meta-Analysis Comparing Torsemide Versus Furosemide in Patients with Heart Failure. Am. J. Cardiol. 2020, 1, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.J.; Velazquez, E.J.; Anstrom, K.J.; Eisenstein, E.L.; Sapp, S.; Morgan, S.; Harding, T.; Sachdev, V.; Ketema, F.; Kim, D.Y.; et al. Pragmatic Design of Randomized Clinical Trials for Heart Failure: Rationale and Design of the TRANSFORM-HF Trial. JACC Heart Fail. 2021, 5, 325–335. [Google Scholar] [CrossRef]
- Mullens, W.; Verbrugge, F.H.; Nijst, P.; Martens, P.; Tartaglia, K.; Theunissen, E.; Bruckers, L.; Droogne, W.; Troisfontaines, P.; Damman, K.; et al. Rationale and design of the ADVOR (Acetazolamide in Decompensated Heart Failure with Volume Overload) trial. Eur. Heart J. 2018, 20, 1591–1600. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.J.; Yang, J.; Yang, J.; Fan, Z.X. Arginine vasopressin antagonist tolvaptan in the treatment of heart failure: A meta-analysis of randomized controlled trials. Int. J. Clin. Exp. Med. 2015, 8, 22117–22128. [Google Scholar]
- Van Tassell, B.; Mihalick, V.; Thomas, G.; Marawan, A.; Talasaz, A.H.; Lu, J.; Le Kang, L.; Ladd, A.; Damonte, J.I.; Dixon, D.L.; et al. Rationale and design of interleukin-1 blockade in recently decompensated heart failure (REDHART2): A randomized, double blind, placebo controlled, single center, phase 2 study. J. Transl. Med. 2022, 20, 270. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin, C.J.; Filippatos, G.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; Kirwan, B.A.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [Green Version]
- Bavry, A.A.; Bhatt, D.L. Patiromer for the Management of Hyperkalemia in Subjects Receiving RAASi for HFrEF—DIAMOND. Available online: https://www.acc.org/latest-in-cardiology/clinical-trials/2022/04/02/15/56/diamond (accessed on 3 July 2022).
- Filippatos, G.; Anker, S.D.; Böhm, M.; Gheorghiade, M.; Køber, L.; Krum, H.; Maggioni, A.P.; Ponikowski, P.; Voors, A.A.; Zannad, F.; et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur. Heart J. 2016, 37, 2105–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargalovic, P.; Wong, P.; Onorato, J.; Finlay, H.; Wang, T.; Yan, M.; Crain, E.; St-Onge, S.; Héroux, M.; Bouvieret, M.; et al. In Vitro and In Vivo Evaluation of a Small-Molecule APJ (Apelin Receptor) Agonist, BMS-986224, as a Potential Treatment for Heart Failure. Circ. Heart Fail. 2021, 14, e007351. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Erdle, C.O.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 blockade in recently decompensated systolic heart failure: Results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, e004373. [Google Scholar] [CrossRef]
- Ponikowski, P.; Van Veldhuisen, D.J.; Comin, C.J.; Ertl, G.; Komajda, M.; Mareev, V.; McDonagh, T.; Parkhomenko, A.; Tavazzi, L.; Levesque, V.; et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur. Heart J. 2015, 36, 657–668. [Google Scholar] [CrossRef]
- Beadle, R.M.; Williams, L.K.; Kuehl, M.; Bowater, S.; Abozguia, K.; Leyva, F.; Yousef, Z.; Wagenmakers, A.J.M.; Thies, F.; Horowitz, J.; et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. 2015, 3, 202–211. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Suen, C.; Jiang, B.; Deng, Y.; Weldrick, J.J.; Putinski, C.; Brunette, S.; Fernando, P.; Lee, T.T.; Flynn, P.; et al. Cardiotrophin 1 stimulates beneficial myogenic and vascular remodeling of the heart. Cell Res. 2017, 27, 1195–1215. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Aliaga, M.; Pérez-Echarri, N.; Marcos-Gómez, B.; Larequi, E.; Gil-Bea, F.J.; Viollet, B.; Gimenez, I.; Martínez, J.A.; Prieto, J.; Bustos, M. Cardiotrophin-1 is a key regulator of glucose and lipid metabolism. Cell Metab. 2011, 14, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Buckley, L.F.; Abbate, A. Interleukin-1 blockade in cardiovascular diseases: A clinical update. Eur. Heart J. 2018, 39, 2063–2069. [Google Scholar] [CrossRef] [Green Version]
- Wohlford, G.; VanTassell, B.; Billingsley, H.; Kadariya, D.; Canada, J.M.; Carbone, S.; Mihalick, V.L.; Bonaventura, A.; Vecchié, A.; Chiabrando, J.G.; et al. Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. J. Cardiovasc. Pharm. 2020, 77, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; McMurray, J.J.V.; Krum, H.; Kiowski, W.; Massie, B.M.; Caspi, A.; Pratt, C.M.; Petrie, M.C.; DeMets, D.; Kobrin, I.; et al. Long-Term Effect of Endothelin Receptor Antagonism with Bosentan on the Morbidity and Mortality of Patients with Severe Chronic Heart Failure: Primary Results of the ENABLE Trials. JACC Heart Fail. 2017, 5, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Zhang, J.; Cheng, L.; Kiowski, W.; Massie, B.M.; Caspi, A.; Pratt, C.M.; Petrie, M.C.; DeMets, D.; Kobrin, I.; et al. ENABLE Investigators and Committees. A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J. Am. Coll. Cardiol. 2010, 55, 1907–1914. [Google Scholar] [CrossRef] [Green Version]
- Beusekamp, J.C.; Tromp, J.; Van der Wal, H.H.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.; van der Harst, P.; Hillege, H.L.; Lang, C.C.; et al. Potassium and the use of renin-angiotensin-aldosterone system inhibitors in heart failure with reduced ejection fraction: Data from BIOSTAT-CHF. Eur. J. Heart Fail. 2018, 20, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Kirwan, B.A.; Van Veldhuisen, D.J.; Filippatos, G.; Comin, C.J.; Ruschitzka, F.; Lüscher, T.F.; Arutyunov, G.P.; Motro, M.; Mori, C.; et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: An individual patient data meta-analysis. Eur. J. Heart Fail. 2018, 20, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Bomer, N.; Pavez-Giani, M.G.; Beverborg, N.G.; Cleland, J.G.F.; van Veldhuisen, D.J.; van der Meer, P. Micronutrient deficiencies in heart failure: Mitochondrial dysfunction as a common pathophysiological mechanism? J. Int. Med. 2022, 291, 713–731. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Iannuzzo, G.; Parlato, A.; Cuomo, G.; Crescenzo Testa, C.; Coppola, M.; D’Ambrosio, G.; Oliviero, D.A.; Sarullo, S.; Vitaleet, G.; et al. Clinical Evidence for Q10 Coenzyme Supplementation in Heart Failure: From Energetics to Functional Improvement. J. Clin. Med. 2020, 9, 1266. [Google Scholar] [CrossRef]
- Mortensen, A.L.; Rosenfeldt, F.; Filipiak, K.J. Effect of coenzyme Q10 in Europeans with chronic heart failure: A subgroup analysis of the Q-SYMBIO randomized double-blind trial. Cardiol. J. 2019, 26, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Pourmoghaddas, M.; Rabbani, M.; Shahabi, J.; Garakyaraghi, M.; Khanjani, R.; Hedayat, P. Combination of atorvastatin/coenzyme Q10 as adjunctive treatment in congestive heart failure: A double-blind randomized placebo-controlled clinical trial. ARYA Atheroscler. 2014, 10, 1–5. [Google Scholar]
- Teerlink, J.R.; Diaz, R.; Felker, M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Biering-Sørensen, T.; Böhm, M.; Bonderman, D.; Fang, J.C.; et al. Effect of Ejection Fraction on Clinical Outcomes in Patients Treated with Omecamtiv Mecarbil in GALACTIC-HF. J. Am. Coll. Cardiol. 2021, 78, 97–108. [Google Scholar] [CrossRef]
- Zsebo, K.; Yaroshinsky, A.; Rudy, J.J.; Wagner, K.; Greenberg, B.; Jessup, M.; Hajjar, R.J. Long-Term Effects of AAV1/SERCA2a Gene Transfer in Patients with Severe Heart Failure: Analysis of recurrent cardiovascular events and mortality. Circ. Res. 2014, 114, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Jessup, M.; Greenberg, B.; Mancini, D.; Cappola, T.; Pauly, D.F.; Jaski, B.; Yaroshinsky, A.; Zsebo, K.M.; Dittrich, H.; Hajjar, R.J. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID). Circulation 2011, 124, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, G.W.; Garcia, J. Role of cell death in the progression of heart failure. Heart Fail. Rev. 2016, 21, 157–167. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavandero, S.; Chiong, M.; Rothermel, B.A.; Hill, J.A. Autophagy in cardiovascular biology. J. Clin. Investig. 2015, 125, 55–64. [Google Scholar] [CrossRef]
- McGinnis, M.; Goolsby, W.A.; Olsen, L.A. Leadership Commitments to Improve Value in Health Care; National Academies Press: Washington, DC, USA, 2009; pp. 224–225. [Google Scholar]
- Mann, D.L.; Felke, M.G. Mechanisms and Models in Heart Failure. A Translational Approach. Circ. Res. 2021, 128, 1435–1450. [Google Scholar] [CrossRef]
- Lara-Pezzi, E.; Menasché, P.; Trouvin, J.H.; Ioannidis, J.P.; Wu, J.C.; Hill, J.A.; Koch, W.J.; De Felice, A.F.; de Waele, P.; Steenwinckel, V.; et al. AM Guidelines for translational research in heart failure. J. Cardiovasc. Transl. Res. 2015, 8, 3–22. [Google Scholar] [CrossRef]
- Lauer, M.S.; Skarlatos, S. Translational Research for Cardiovascular Diseases at the NHLBI: Moving from Bench to Bedside and From Bedside to Community. Circulation 2010, 121, 929–933. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).