
Prediction and Experimental Validation of a New Salinity-Responsive Cis-Regulatory Element (CRE) in a Tilapia Cell Line



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Hyperosmotic Stress Challenge and Actinomycin D Treatment
2.2. Quantitative Proteomics
2.3. Motif Discovery and Refinement
2.4. Cloning
2.5. Site-Directed Motif Mutagenesis
2.6. Quantitative Fluorescent Reporter Assay
3. Results
3.1. Transcriptional Regulation Is Required for Upregulation of Proteins in OmB Cells Exposed to Hyperosmotic Stress
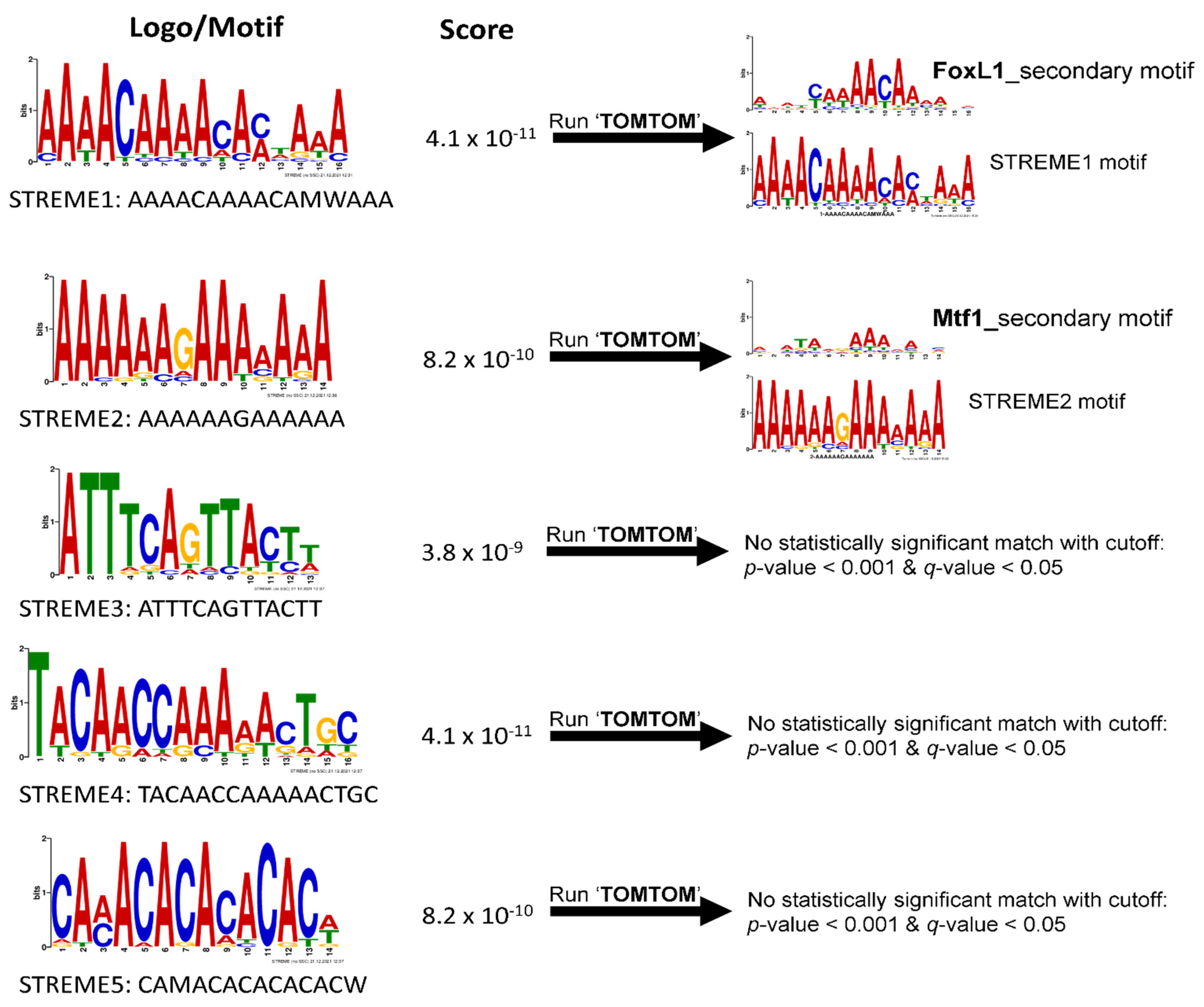
3.2. Discovery of Putative CRE Motifs That Mediate Hyperosmotic Induction of Tilapia Genes
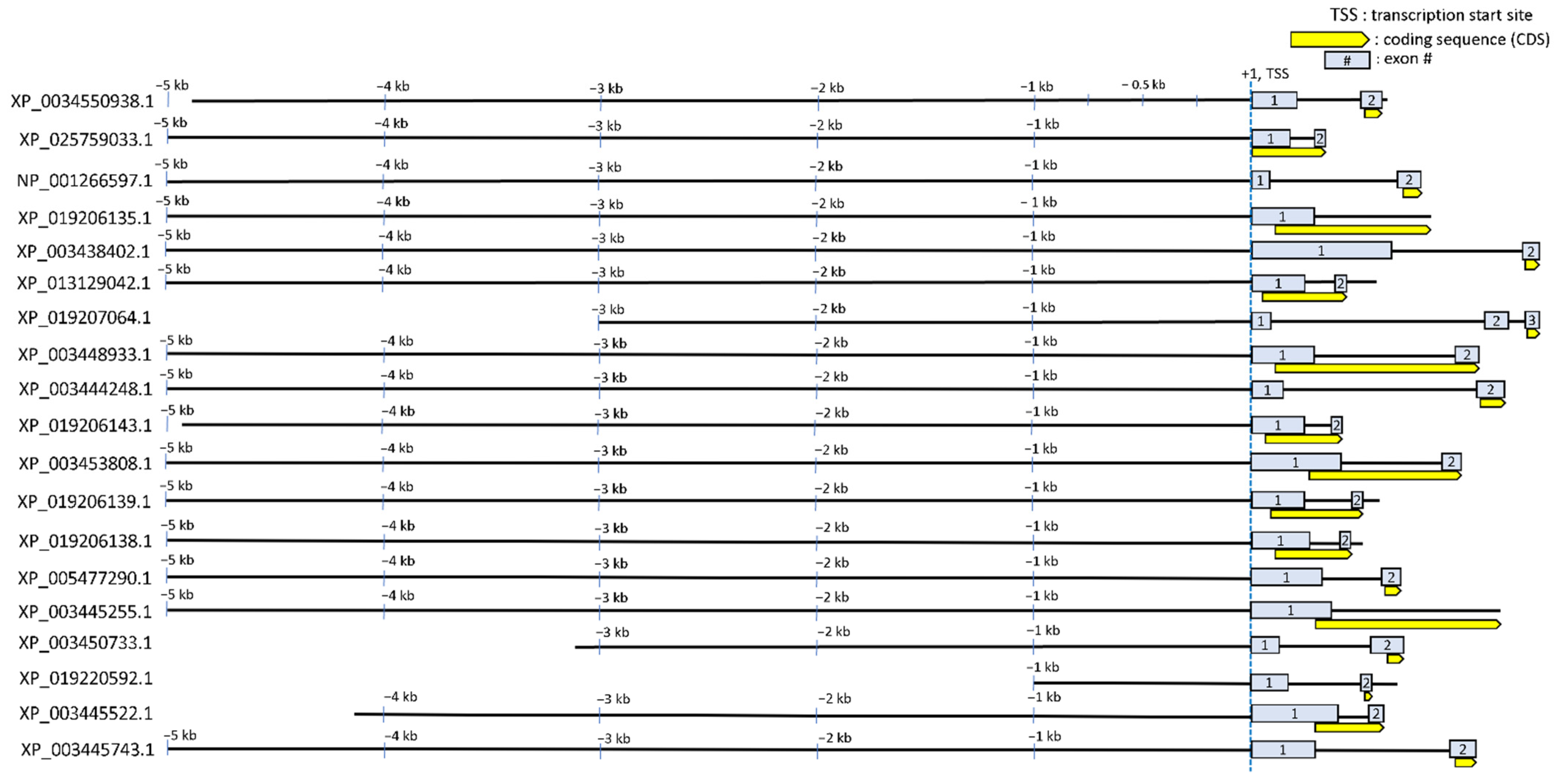
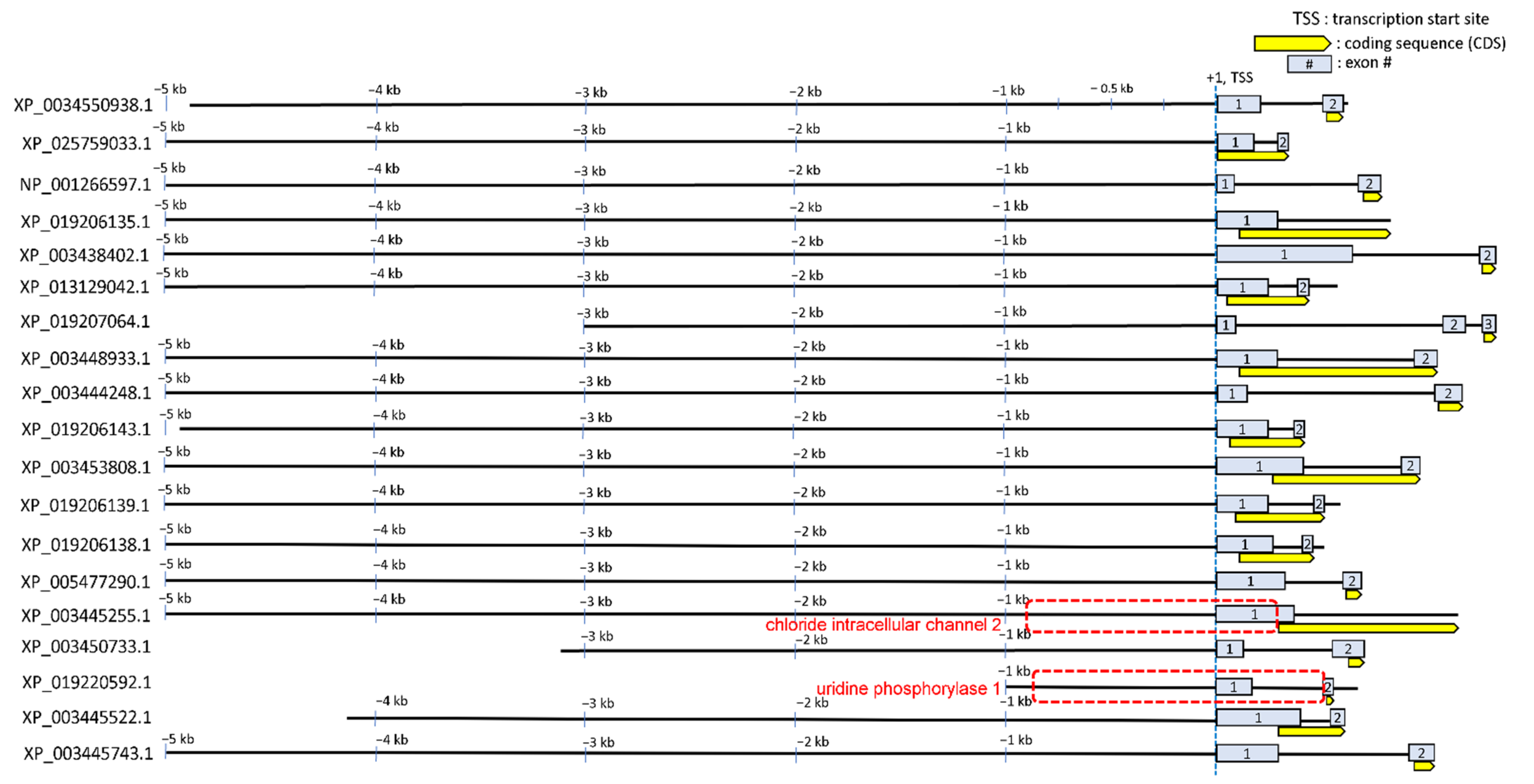
3.3. Annotating STREM1 Hit Localization on the 19 Regulatory Sequences and Selecting for Candidate Gene Regulatory Regions to Be Experimentally Tested
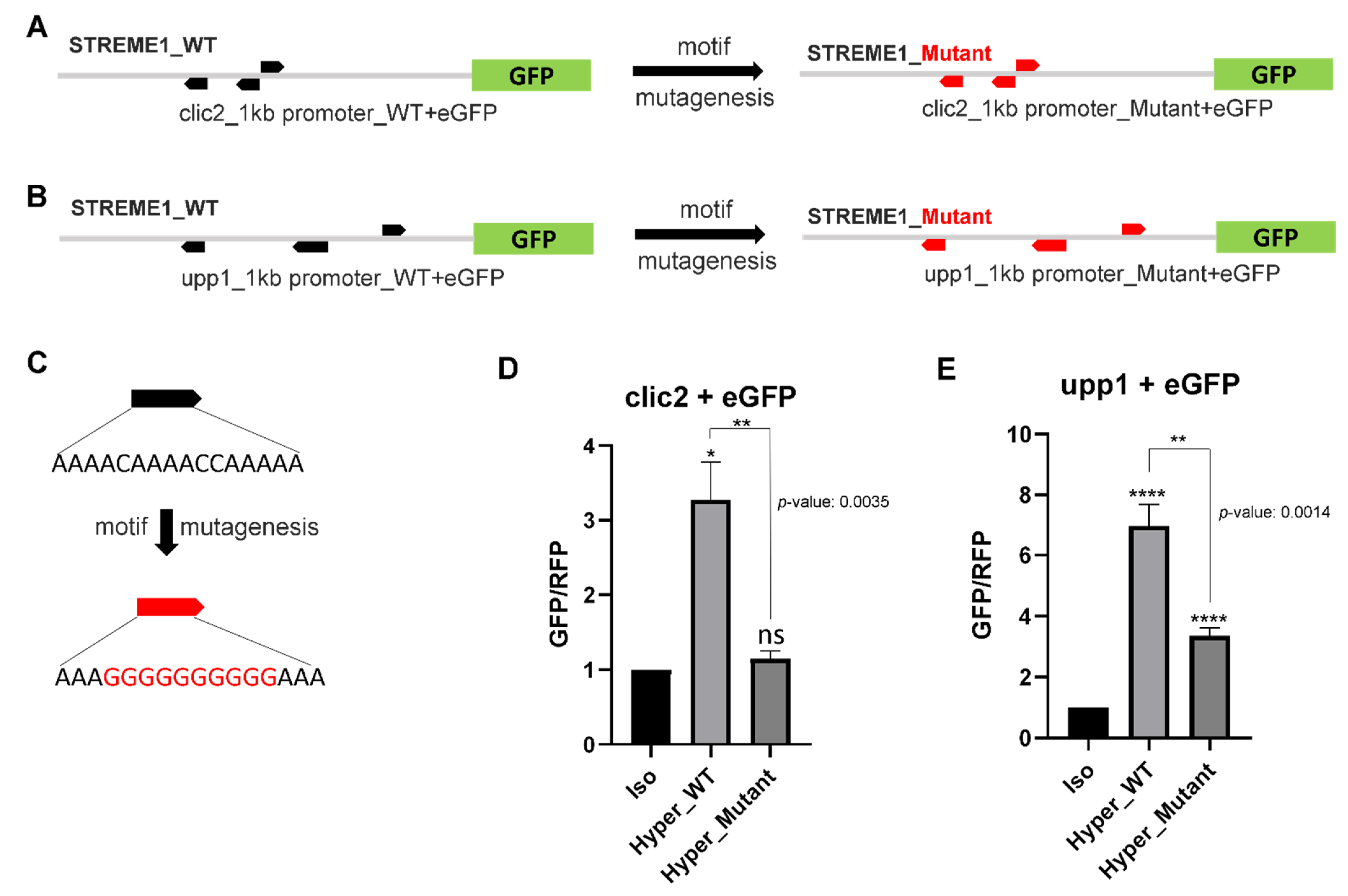
3.4. Experimental Validation of the Selected Candidate Gene STREME1 Motifs
4. Discussion
4.1. The Role of CREs in Environmental Acclimation of Fish
4.2. Transcriptional Regulation of Genes That Penetrates to Proteins and Phenotypes
4.3. STREME1 as a Novel Hyperosmotically Inducible CRE of Euryhaline Tilapia
4.4. Roles of CLIC2 and UPP1 during Hyperosmolality
4.5. Other Candidate Binding Sites for Hyperosmolality Inducible TFs
4.6. Fox L1 as a Putative Hyperosmotically Inducible TF Binding to STREME1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wray, N.R.; Yang, J.; Hayes, B.J.; Price, A.L.; Goddard, M.E.; Visscher, P.M. Pitfalls of Predicting Complex Traits from SNPs. Nat. Rev. Genet. 2013, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kültz, D. Physiological Mechanisms Used by Fish to Cope with Salinity Stress. J. Exp. Biol. 2015, 218, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Marie, A.D.; Smith, S.; Green, A.J.; Rico, C.; Lejeusne, C. Transcriptomic Response to Thermal and Salinity Stress in Introduced and Native Sympatric Palaemon Caridean Shrimps. Sci. Rep. 2017, 7, 13980. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Dalmet, S.; Litchfield, C.D.; Gillevet, P.; Baxter, B.K. Differential Gene Expression in Response to Salinity and Temperature in a Haloarcula Strain from Great Salt Lake, Utah. Genes 2018, 9, 52. [Google Scholar] [CrossRef]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Regulation of Transcription in Eukaryotes; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Wray, G.A.; Hahn, M.W.; Abouheif, E.; Balhoff, J.P.; Pizer, M.; Rockman, M.V.; Romano, L.A. The Evolution of Transcriptional Regulation in Eukaryotes. Mol. Biol. Evol. 2003, 20, 1377–1419. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, O.; Qin, J.; Liu, S.; Sun, S.; Liu, H.; Kuang, J.; Jiang, G.; Zhang, W. Cis-Acting Elements and Trans-Acting Factors in the Transcriptional Regulation of Raf Kinase Inhibitory Protein Expression. PLoS ONE 2013, 8, e83097. [Google Scholar] [CrossRef]
- Mattioli, K.; Oliveros, W.; Gerhardinger, C.; Andergassen, D.; Maass, P.G.; Rinn, J.L.; Melé, M. Cis and Trans Effects Differentially Contribute to the Evolution of Promoters and Enhancers. Genome Biol. 2020, 21, 210. [Google Scholar] [CrossRef]
- Reuveni, E.; Getselter, D.; Oron, O.; Elliott, E. Differential Contribution of Cis and Trans Gene Transcription Regulatory Mechanisms in Amygdala and Prefrontal Cortex and Modulation by Social Stress. Sci. Rep. 2018, 8, 6339. [Google Scholar] [CrossRef]
- Berman, B.P.; Pfeiffer, B.D.; Laverty, T.R.; Salzberg, S.L.; Rubin, G.M.; Eisen, M.B.; Celniker, S.E. Computational Identification of Developmental Enhancers: Conservation and Function of Transcription Factor Binding-Site Clusters in Drosophila Melanogaster and Drosophila Pseudoobscura. Genome Biol. 2004, 5, R61. [Google Scholar] [CrossRef]
- Huang, C.W.; Li, Y.H.; Hu, S.Y.; Chi, J.R.; Lin, G.H.; Lin, C.C.; Gong, H.Y.; Chen, J.Y.; Chen, R.H.; Chang, S.J.; et al. Differential Expression Patterns of Growth-Related MicroRNAs in the Skeletal Muscle of Nile Tilapia (Oreochromis niloticus). J. Anim. Sci. 2012, 90, 4266–4279. [Google Scholar] [CrossRef]
- Konstantinidis, I.; Sætrom, P.; Mjelle, R.; Nedoluzhko, A.V.; Robledo, D.; Fernandes, J.M.O. Major Gene Expression Changes and Epigenetic Remodelling in Nile Tilapia Muscle after Just One Generation of Domestication. Epigenetics 2020, 15, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Root, L.T.; Kültz, D. Gill Proteome Networks Explain Energy Homeostasis during Salinity Stress in Oreochromis Mossambicus. Authorea 2022. [Google Scholar] [CrossRef]
- Qin, H.; Yu, Z.; Zhu, Z.; Lin, Y.; Xia, J.; Jia, Y. The Integrated Analyses of Metabolomics and Transcriptomics in Gill of GIFT Tilapia in Response to Long Term Salinity Challenge. Aquac. Fish. 2021, 7, 131–139. [Google Scholar] [CrossRef]
- Kültz, D.; Li, J.; Gardell, A.; Sacchi, R. Quantitative Molecular Phenotyping of Gill Remodeling in a Cichlid Fish Responding to Salinity Stress. Mol. Cell. Proteom. 2013, 12, 3962–3975. [Google Scholar] [CrossRef] [PubMed]
- Moorman, B.P.; Yamaguchi, Y.; Lerner, D.T.; Grau, E.G.; Seale, A.P. Rearing Mozambique Tilapia in Tidally-Changing Salinities: Effects on Growth and the Growth Hormone/Insulin-like Growth Factor I Axis. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2016, 198, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kültz, D. Osmolality/Salinity-Responsive Enhancers (OSREs) Control Induction of Osmoprotective Genes in Euryhaline Fish. Proc. Natl. Acad. Sci. USA 2017, 114, E2729–E2738. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kültz, D. An Osmolality/Salinity-Responsive Enhancer 1 (OSRE1) in Intron 1 Promotes Salinity Induction of Tilapia Glutamine Synthetase. Sci. Rep. 2020, 10, 12103. [Google Scholar] [CrossRef]
- Rim, J.S.; Atta, M.G.; Dahl, S.C.; Berry, G.T.; Handler, J.S.; Kwon, H.M. Transcription of the Sodium/Myo-Inositol Cotransporter Gene Is Regulated by Multiple Tonicity-Responsive Enhancers Spread over 50 Kilobase Pairs in the 5’-Flanking Region. J. Biol. Chem. 1998, 273, 20615–20621. [Google Scholar] [CrossRef]
- Zarka, D.G.; Vogel, J.T.; Cook, D.; Thomashow, M.F. Cold Induction of Arabidopsis CBF Genes Involves Multiple ICE (Inducer of CBF Expression) Promoter Elements and a Cold-Regulatory Circuit That Is Desensitized by Low Temperature. Plant Physiol. 2003, 133, 910–918. [Google Scholar] [CrossRef]
- Hughes, J.D.; Estep, P.W.; Tavazoie, S.; Church, G.M. Computational Identification of Cis-Regulatory Elements Associated with Groups of Functionally Related Genes in Saccharomyces Cerevisiae. J. Mol. Biol. 2000, 296, 1205–1214. [Google Scholar] [CrossRef]
- Elkon, R.; Zeller, K.I.; Linhart, C.; Dang, C.V.; Shamir, R.; Shiloh, Y. In Silico Identification of Transcriptional Regulators Associated with C-Myc. Nucleic Acids Res. 2004, 32, 4955–4961. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Bohnert, H.J. Integration of Arabidopsis Thaliana Stress-Related Transcript Profiles, Promoter Structures, and Cell-Specific Expression. Genome Biol. 2007, 8, R49. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and Predictive Chromatin Signatures of Transcriptional Promoters and Enhancers in the Human Genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. ChIP-Seq Accurately Predicts Tissue-Specific Activity of Enhancers. Nature 2009, 457, 854–858. [Google Scholar] [CrossRef]
- Hu, P.; Liu, M.; Zhang, D.; Wang, J.; Niu, H.; Liu, Y.; Wu, Z.; Han, B.; Zhai, W.; Shen, Y.; et al. Global Identification of the Genetic Networks and Cis-Regulatory Elements of the Cold Response in Zebrafish. Nucleic Acids Res. 2015, 43, 9198–9213. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Liu, M.; Liu, Y.; Wang, J.; Zhang, D.; Niu, H.; Jiang, S.; Wang, J.; Zhang, D.; Han, B.; et al. Transcriptome Comparison Reveals a Genetic Network Regulating the Lower Temperature Limit in Fish. Sci. Rep. 2016, 6, 28952. [Google Scholar] [CrossRef] [PubMed]
- Cassé, C.; Giannoni, F.; Nguyen, V.T.; Dubois, M.-F.; Bensaude, O. The Transcriptional Inhibitors, Actinomycin D and α-Amanitin, Activate the HIV-1 Promoter and Favor Phosphorylation of the RNA Polymerase II C-Terminal Domain. J. Biol. Chem. 1999, 274, 16097–16106. [Google Scholar] [CrossRef]
- Koba, M.; Konopa, J. Actinomycin D and its mechanisms of action. Postepy Hig. Med. Dosw. Online 2005, 59, 290–298. [Google Scholar]
- Li, J.; Levitan, B.; Gomez-Jimenez, S.; Kültz, D. Development of a Gill Assay Library for Ecological Proteomics of Threespine Sticklebacks (Gasterosteus Aculeatus). Mol. Cell. Proteom. MCP 2018, 17, 2146–2163. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Rost, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Collins, B.C.; Gillet, L.C.; Rosenberger, G.; Rost, H.L.; Vichalkovski, A.; Gstaiger, M.; Aebersold, R. Quantifying Protein Interaction Dynamics by SWATH Mass Spectrometry: Application to the 14-3-3 System. Nat. Methods 2013, 10, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Arnhard, K.; Gottschall, A.; Pitterl, F.; Oberacher, H. Applying “Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra” (SWATH) for Systematic Toxicological Analysis with Liquid Chromatography-High-Resolution Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2015, 407, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Yang, L.; Luo, J.; Guo, L.; Wang, Z.; Yang, X.; Jin, W.; Fang, Y.; Ye, J.; Shan, B.; et al. SWATH Enables Precise Label-Free Quantification on Proteome Scale. Proteomics 2015, 15, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, F.; Ho, J.T.C.; Smit, A.B.; Li, K.W. Comparative Analyses of Data Independent Acquisition Mass Spectrometric Approaches: DIA, WiSIM-DIA, and Untargeted DIA. Proteomics 2018, 18, 1700304. [Google Scholar] [CrossRef]
- Root, L.; Campo, A.; MacNiven, L.; Con, P.; Cnaani, A.; Kültz, D. Nonlinear Effects of Environmental Salinity on the Gill Transcriptome versus Proteome of Oreochromis niloticus. Genomics 2021, 113, 3235–3249. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev. 2017, 39, 229–244. [Google Scholar] [CrossRef]
- Reiter, L.; Rinner, O.; Picotti, P.; Huttenhain, R.; Beck, M.; Brusniak, M.Y.; Hengartner, M.O.; Aebersold, R. MProphet: Automated Data Processing and Statistical Validation for Large-Scale SRM Experiments. Nat. Methods 2011, 8, 430–435. [Google Scholar] [CrossRef]
- Choi, M.; Chang, C.Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R Package for Statistical Analysis of Quantitative Mass Spectrometry-Based Proteomic Experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef]
- Sharma, V.; Eckels, J.; Taylor, G.K.; Shulman, N.J.; Stergachis, A.B.; Joyner, S.A.; Yan, P.; Whiteaker, J.R.; Halusa, G.N.; Schilling, B.; et al. Panorama: A Targeted Proteomics Knowledge Base. J Proteome Res. 2014, 13, 4205–4210. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange Consortium in 2020: Enabling ‘Big Data’ Approaches in Proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Bailey, T.L. STREME: Accurate and Versatile Sequence Motif Discovery. Bioinformatics 2021, 37, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying Similarity between Motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for Occurrences of a given Motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, T.; Dror, I.; Mathelier, A.; Wasserman, W.W.; Gordân, R.; Rohs, R. TFBSshape: A Motif Database for DNA Shape Features of Transcription Factor Binding Sites. Nucleic Acids Res. 2014, 42, D148–D155. [Google Scholar] [CrossRef]
- Salekin, S.; Zhang, J.M.; Huang, Y. Base-Pair Resolution Detection of Transcription Factor Binding Site by Deep Deconvolutional Network. Bioinformatics 2018, 34, 3446–3453. [Google Scholar] [CrossRef]
- Hena, A.; Kamal, M.; Mair, G.C. Salinity Tolerance in Superior Genotypes of Tilapia, Oreochromis niloticus, Oreochromis Mossambicus and Their Hybrids. Aquaculture 2005, 247, 189–201. [Google Scholar] [CrossRef]
- Firmat, C.; Alibert, P.; Losseau, M.; Baroiller, J.-F.; Schliewen, U.K. Successive Invasion-Mediated Interspecific Hybridizations and Population Structure in the Endangered Cichlid Oreochromis Mossambicus. PLoS ONE 2013, 8, e63880. [Google Scholar] [CrossRef]
- Sheshadri, S.A.; Nishanth, M.J.; Simon, B. Stress-Mediated Cis-Element Transcription Factor Interactions Interconnecting Primary and Specialized Metabolism in Planta. Front. Plant Sci. 2016, 7, 1725. [Google Scholar] [CrossRef]
- Lin, L.-H.; Lee, H.-C.; Li, W.-H.; Chen, B.-S. A Systematic Approach to Detecting Transcription Factors in Response to Environmental Stresses. BMC Bioinform. 2007, 8, 473. [Google Scholar] [CrossRef]
- Estruch, F. Stress-Controlled Transcription Factors, Stress-Induced Genes and Stress Tolerance in Budding Yeast. FEMS Microbiol. Rev. 2000, 24, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.C.; Capellini, T.D.; Guenther, C.A.; Chan, Y.F.; Infante, C.R.; Menke, D.B.; Kingsley, D.M. A Novel Enhancer near the Pitx1 Gene Influences Development and Evolution of Pelvic Appendages in Vertebrates. eLife 2018, 7, e38555. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Fuentes, L.; Santos-Pereira, J.M.; Tena, J.J. Functional Conservation of Divergent P63-Bound Cis-Regulatory Elements. Front. Genet. 2020, 11, 339. [Google Scholar] [CrossRef] [PubMed]
- Fiol, D.F.; Chan, S.Y.; Kültz, D. Regulation of Osmotic Stress Transcription Factor 1 (Ostf1) in Tilapia (Oreochromis Mossambicus) Gill Epithelium during Salinity Stress. J. Exp. Biol. 2006, 209, 3257–3265. [Google Scholar] [CrossRef][Green Version]
- Fiol, D.F.; Kültz, D. Rapid Hyperosmotic Coinduction of Two Tilapia (Oreochromis Mossambicus) Transcription Factors in Gill Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 927–932. [Google Scholar] [CrossRef]
- Fiol, D.F.; Mak, S.K.; Kültz, D. Specific TSC22 Domain Transcripts Are Hypertonically Induced and Alternatively Spliced to Protect Mouse Kidney Cells during Osmotic Stress. FEBS J. 2007, 274, 109–124. [Google Scholar] [CrossRef]
- Tse, W.K.F. The Role of Osmotic Stress Transcription Factor 1 in Fishes. Front. Zool. 2014, 11, 86. [Google Scholar] [CrossRef][Green Version]
- Wong, M.K.-S.; Ozaki, H.; Suzuki, Y.; Iwasaki, W.; Takei, Y. Discovery of Osmotic Sensitive Transcription Factors in Fish Intestine via a Transcriptomic Approach. BMC Genom. 2014, 15, 1134. [Google Scholar] [CrossRef]
- Verta, J.-P.; Jones, F.C. Predominance of Cis-Regulatory Changes in Parallel Expression Divergence of Sticklebacks. eLife 2019, 8, e43785. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Suhre, K.; McCarthy, M.I.; Schwenk, J.M. Genetics Meets Proteomics: Perspectives for Large Population-Based Studies. Nat. Rev. Genet. 2021, 22, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Buccitelli, C.; Selbach, M. MRNAs, Proteins and the Emerging Principles of Gene Expression Control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Pascal, L.E.; True, L.D.; Campbell, D.S.; Deutsch, E.W.; Risk, M.; Coleman, I.M.; Eichner, L.J.; Nelson, P.S.; Liu, A.Y. Correlation of MRNA and Protein Levels: Cell Type-Specific Gene Expression of Cluster Designation Antigens in the Prostate. BMC Genom. 2008, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.G.; Somero, G.N. A Microarray-Based Transcriptomic Time-Course of Hyper- and Hypo-Osmotic Stress Signaling Events in the Euryhaline Fish Gillichthys Mirabilis: Osmosensors to Effectors. J. Exp. Biol. 2008, 211, 3636–3649. [Google Scholar] [CrossRef]
- Zhang, X.; Wen, H.; Wang, H.; Ren, Y.; Zhao, J.; Li, Y. RNA-Seq Analysis of Salinity Stress–Responsive Transcriptome in the Liver of Spotted Sea Bass (Lateolabrax maculatus). PLoS ONE 2017, 12, e0173238. [Google Scholar] [CrossRef]
- Su, H.; Ma, D.; Zhu, H.; Liu, Z.; Gao, F. Transcriptomic Response to Three Osmotic Stresses in Gills of Hybrid Tilapia (Oreochromis Mossambicus Female × O. Urolepis Hornorum Male). BMC Genom. 2020, 21, 110. [Google Scholar] [CrossRef]
- Tse, W.K.F.; Chow, S.C.; Wong, C.K.C. The Cloning of Eel Osmotic Stress Transcription Factor and the Regulation of Its Expression in Primary Gill Cell Culture. J. Exp. Biol. 2008, 211, 1964–1968. [Google Scholar] [CrossRef]
- Gardell, A.M.; Qin, Q.; Rice, R.H.; Li, J.; Kültz, D. Derivation and Osmotolerance Characterization of Three Immortalized Tilapia (Oreochromis mossambicus) Cell Lines. PLoS ONE 2014, 9, e95919. [Google Scholar] [CrossRef]
- Reiter, F.; Wienerroither, S.; Stark, A. Combinatorial Function of Transcription Factors and Cofactors. Curr. Opin. Genet. Dev. 2017, 43, 73–81. [Google Scholar] [CrossRef]
- Kato, M.; Hata, N.; Banerjee, N.; Futcher, B.; Zhang, M.Q. Identifying Combinatorial Regulation of Transcription Factors and Binding Motifs. Genome Biol. 2004, 5, R56. [Google Scholar] [CrossRef]
- Sacilotto, N.; Monteiro, R.; Fritzsche, M.; Becker, P.W.; Sanchez-del-Campo, L.; Liu, K.; Pinheiro, P.; Ratnayaka, I.; Davies, B.; Goding, C.R.; et al. Analysis of Dll4 Regulation Reveals a Combinatorial Role for Sox and Notch in Arterial Development. Proc. Natl. Acad. Sci. USA 2013, 110, 11893–11898. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.N.; Lawson, N.D.; Mugford, J.W.; Dye, L.; Castranova, D.; Lo, B.; Weinstein, B.M. Combinatorial Function of ETS Transcription Factors in the Developing Vasculature. Dev. Biol. 2007, 303, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, J.D.; Williams, C.K.; Jung, K.-Y.; Bedford, J.J.; Burg, M.B.; García-Pérez, A. ORE, a Eukaryotic Minimal Essential Osmotic Response Element THE ALDOSE REDUCTASE GENE IN HYPEROSMOTIC STRESS. J. Biol. Chem. 1996, 271, 18318–18321. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.C.B.; Ruepp, B.; Bohren, K.M.; Gabbay, K.H.; Chung, S.S.M. Identification and Characterization of Multiple Osmotic Response Sequences in the Human Aldose Reductase Gene. J. Biol. Chem. 1997, 272, 16431–16437. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Sandelin, A. Determinants of Enhancer and Promoter Activities of Regulatory Elements. Nat. Rev. Genet. 2019, 21, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Khamici, H.A.; Brown, L.J.; Hossain, K.R.; Hudson, A.L.; Sinclair-Burton, A.A.; Ng, J.P.M.; Daniel, E.L.; Hare, J.E.; Cornell, B.A.; Curmi, P.M.G.; et al. Members of the Chloride Intracellular Ion Channel Protein Family Demonstrate Glutaredoxin-Like Enzymatic Activity. PLoS ONE 2015, 10, e115699. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Ozaki, S.; Umakoshi, A.; Yano, H.; Choudhury, M.E.; Abe, N.; Sumida, Y.; Kuwabara, J.; Uchida, R.; Islam, A.; et al. Chloride Intracellular Channel Protein 2 in Cancer and Non-Cancer Human Tissues: Relationship with Tight Junctions. Tissue Barriers 2019, 7, 1593775. [Google Scholar] [CrossRef] [PubMed]
- Tipsmark, C.K.; Baltzegar, D.A.; Ozden, O.; Grubb, B.J.; Borski, R.J. Salinity Regulates Claudin MRNA and Protein Expression in the Teleost Gill. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2008, 294, R1004–R1014. [Google Scholar] [CrossRef]
- Tang, V.W.; Goodenough, D.A. Paracellular Ion Channel at the Tight Junction. Biophys. J. 2003, 84, 1660–1673. [Google Scholar] [CrossRef]
- Pizzorno, G.; Cao, D.; Leffert, J.J.; Russell, R.L.; Zhang, D.; Handschumacher, R.E. Homeostatic Control of Uridine and the Role of Uridine Phosphorylase: A Biological and Clinical Update. Biochim. Biophys. Acta 2002, 1587, 133–144. [Google Scholar] [CrossRef]
- Watanabe, S.; Uchida, T. Cloning and Expression of Human Uridine Phosphorylase. Biochem. Biophys. Res. Commun. 1995, 216, 265–272. [Google Scholar] [CrossRef]
- Kültz, D. Molecular and Evolutionary Basis of the Cellular Stress Response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.J.; Simmons, L.A. Hydroxyurea Induces a Stress Response That Alters DNA Replication and Nucleotide Metabolism in Bacillus Subtilis. J. Bacteriol. 2021, 203, e0017121. [Google Scholar] [CrossRef] [PubMed]
- Kilstrup, M.; Hammer, K.; Ruhdal Jensen, P.; Martinussen, J. Nucleotide Metabolism and Its Control in Lactic Acid Bacteria. FEMS Microbiol. Rev. 2005, 29, 555–590. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.; Wen, H.; Li, Y.; Li, J.; Zhao, J.; Zhang, S.; Song, M.; Wang, X. Deep Transcriptomic Analysis of Black Rockfish (Sebastes schlegelii) Provides New Insights on Responses to Acute Temperature Stress. Sci. Rep. 2018, 8, 9113. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.; Denis, V.; Yum, S. Transcriptional Changes Caused by Bisphenol A in Oryzias Javanicus, a Fish Species Highly Adaptable to Environmental Salinity. Mar. Drugs 2014, 12, 983–998. [Google Scholar] [CrossRef]
- O’Shields, B.; McArthur, A.G.; Holowiecki, A.; Kamper, M.; Tapley, J.; Jenny, M.J. Inhibition of Endogenous MTF-1 Signaling in Zebrafish Embryos Identifies Novel Roles for MTF-1 in Development. Biochim. Biophys. Acta 2014, 1843, 1818–1833. [Google Scholar] [CrossRef]
- Van Loo, K.M.J.; Schaub, C.; Pitsch, J.; Kulbida, R.; Opitz, T.; Ekstein, D.; Dalal, A.; Urbach, H.; Beck, H.; Yaari, Y.; et al. Zinc Regulates a Key Transcriptional Pathway for Epileptogenesis via Metal-Regulatory Transcription Factor 1. Nat. Commun. 2015, 6, 8688. [Google Scholar] [CrossRef]
- Stevanovic, M.; Drakulic, D.; Lazic, A.; Ninkovic, D.S.; Schwirtlich, M.; Mojsin, M. SOX Transcription Factors as Important Regulators of Neuronal and Glial Differentiation During Nervous System Development and Adult Neurogenesis. Front. Mol. Neurosci. 2021, 14, 51. [Google Scholar] [CrossRef]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription Factor-DNA Binding: Beyond Binding Site Motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef]
- Geertz, M.; Maerkl, S.J. Experimental Strategies for Studying Transcription Factor-DNA Binding Specificities. Brief. Funct. Genom. 2010, 9, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Philippakis, A.A.; Qureshi, A.M.; He, F.S.; Estep, P.W.; Bulyk, M.L. Compact, Universal DNA Microarrays to Comprehensively Determine Transcription-Factor Binding Site Specificities. Nat. Biotechnol. 2006, 24, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Andrilenas, K.K.; Penvose, A.; Siggers, T. Using Protein-Binding Microarrays to Study Transcription Factor Specificity: Homologs, Isoforms and Complexes. Brief. Funct. Genom. 2015, 14, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Gordân, R.; Murphy, K.F.; McCord, R.P.; Zhu, C.; Vedenko, A.; Bulyk, M.L. Curated Collection of Yeast Transcription Factor DNA Binding Specificity Data Reveals Novel Structural and Gene Regulatory Insights. Genome Biol. 2011, 12, R125. [Google Scholar] [CrossRef]
- Nakagawa, S.; Gisselbrecht, S.S.; Rogers, J.M.; Hartl, D.L.; Bulyk, M.L. DNA-Binding Specificity Changes in the Evolution of Forkhead Transcription Factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12349–12354. [Google Scholar] [CrossRef]
- Badis, G.; Berger, M.F.; Philippakis, A.A.; Talukder, S.; Gehrke, A.R.; Jaeger, S.A.; Chan, E.T.; Metzler, G.; Vedenko, A.; Chen, X.; et al. Diversity and Complexity in DNA Recognition by Transcription Factors. Science 2009, 324, 1720–1723. [Google Scholar] [CrossRef]
- Chen, A.; Zhong, L.; Lv, J. FOXL1 Overexpression Is Associated with Poor Outcome in Patients with Glioma. Oncol. Lett. 2019, 18, 751–757. [Google Scholar] [CrossRef]
- Aoki, R.; Shoshkes-Carmel, M.; Gao, N.; Shin, S.; May, C.L.; Golson, M.L.; Zahm, A.M.; Ray, M.; Wiser, C.L.; Wright, C.V.E.; et al. Foxl1-Expressing Mesenchymal Cells Constitute the Intestinal Stem Cell Niche. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 175–188. [Google Scholar] [CrossRef]
- Perreault, N.; Katz, J.P.; Sackett, S.D.; Kaestner, K.H. Foxl1 Controls the Wnt/β-Catenin Pathway by Modulating the Expression of Proteoglycans in the Gut. J. Biol. Chem. 2001, 276, 43328–43333. [Google Scholar] [CrossRef]
- Zhang, G.; He, P.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.G.; Lee, D.H.; Hanna, N.; Alexander, H.R.; Hussain, S.P. FOXL1, a Novel Candidate Tumor Suppressor, Inhibits Tumor Aggressiveness and Predicts Outcome in Human Pancreatic Cancer. Cancer Res. 2013, 73, 5416–5425. [Google Scholar] [CrossRef]
- Nakada, C.; Satoh, S.; Tabata, Y.; Arai, K.; Watanabe, S. Transcriptional Repressor Foxl1 Regulates Central Nervous System Development by Suppressing Shh Expression in Zebra Fish. Mol. Cell. Biol. 2006, 26, 7246–7257. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, C.; Wang, X.; Kültz, D. Prediction and Experimental Validation of a New Salinity-Responsive Cis-Regulatory Element (CRE) in a Tilapia Cell Line. Life 2022, 12, 787. https://doi.org/10.3390/life12060787
Kim C, Wang X, Kültz D. Prediction and Experimental Validation of a New Salinity-Responsive Cis-Regulatory Element (CRE) in a Tilapia Cell Line. Life. 2022; 12(6):787. https://doi.org/10.3390/life12060787
Chicago/Turabian StyleKim, Chanhee, Xiaodan Wang, and Dietmar Kültz. 2022. "Prediction and Experimental Validation of a New Salinity-Responsive Cis-Regulatory Element (CRE) in a Tilapia Cell Line" Life 12, no. 6: 787. https://doi.org/10.3390/life12060787
APA StyleKim, C., Wang, X., & Kültz, D. (2022). Prediction and Experimental Validation of a New Salinity-Responsive Cis-Regulatory Element (CRE) in a Tilapia Cell Line. Life, 12(6), 787. https://doi.org/10.3390/life12060787