
Identification of Known and Novel Arundo donax L. MicroRNAs and Their Targets Using High-Throughput Sequencing and Degradome Analysis


 , , ,
, , ,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. sRNAs and Degradome Sequencing
2.3. Data Analysis
2.4. 5′ RACE for Cleavage Site Identification of miRNA Targets
3. Results
3.1. sRNA Profiles of A. donax
3.2. Identification of Known miRNAs in A. donax
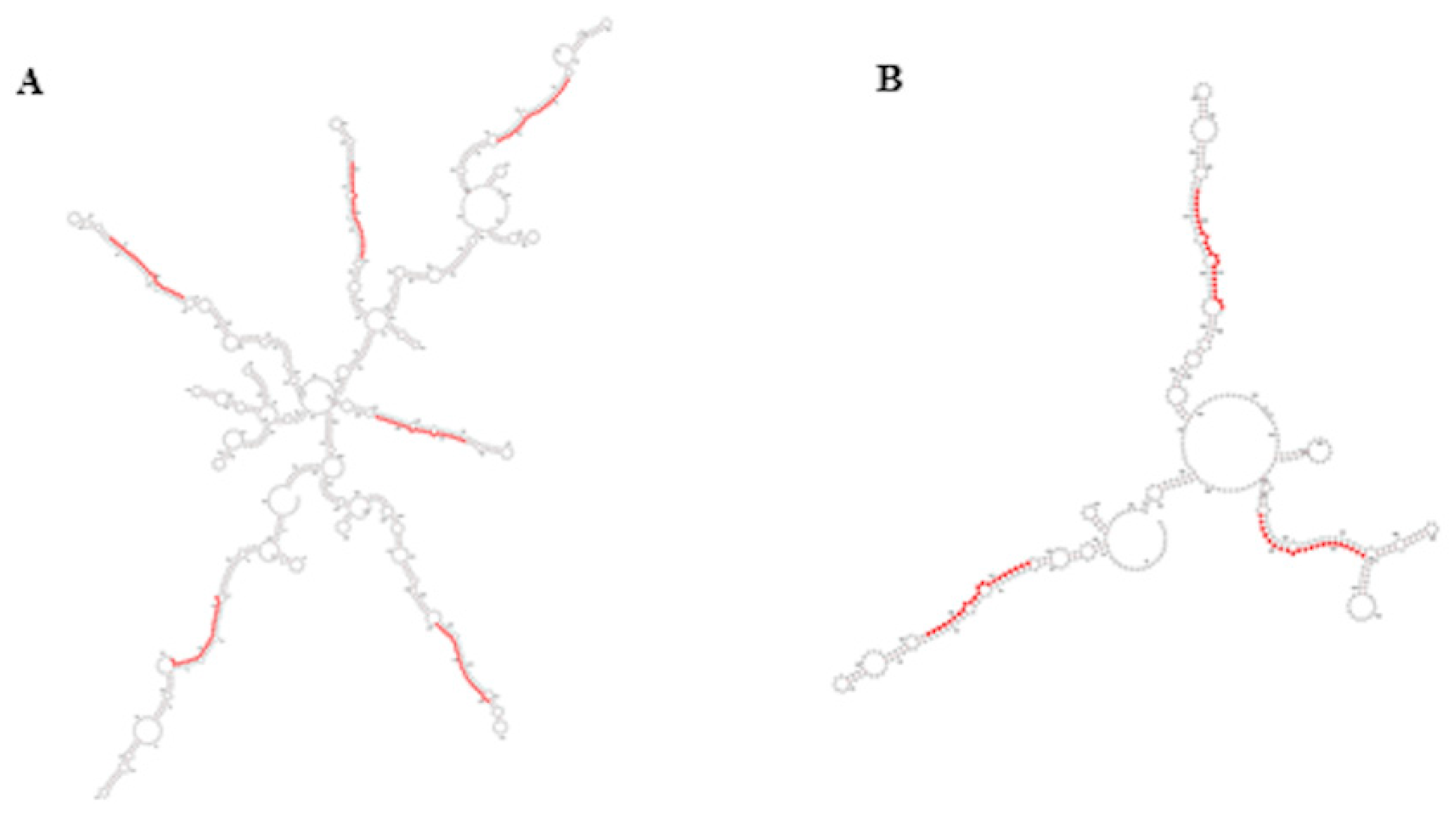
3.3. Identification of Novel Candidate miRNAs in A. donax
3.4. miRNAs and miRCs Targets Identification and Functional Characterization
3.5. Validation of miRNAs and miRCs Cleavage Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corno, L.; Pilu, R.; Adani, F. Arundo donax L.: A non-food crop for bioenergy and bio-compound production. Biotechnol. Adv. 2014, 32, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- D’Imporzano, G.; Pilu, R.; Corno, L.; Adani, F. Arundo donax L. can substitute traditional energy crops for more efficient, environmentally-friendly production of biogas: A Life Cycle Assessment approach. Bioresour. Technol. 2018, 267, 249–256. [Google Scholar] [CrossRef]
- Ren, L.; Eller, F.; Lambertini, C.; Guo, W.Y.; Brix, H.; Sorrell, B.K. Assessing nutrient responses and biomass quality for selection of appropriate paludiculture crops. Sci. Total Environ. 2019, 664, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Corno, L.; Lonati, S.; Riva, C.; Pilu, R.; Adani, F. Giant cane (Arundo donax L.) can substitute traditional energy crops in producing energy by anaerobic digestion, reducing surface area and costs: A full-scale approach. Bioresour. Technol. 2016, 218, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Cristaldi, A.; Oliveri Conti, G.; Cosentino, S.L.; Mauromicale, G.; Copat, C.; Grasso, A.; Zuccarello, P.; Fiore, M.; Restuccia, C.; Ferrante, M. Phytoremediation potential of Arundo donax (Giant Reed) in contaminated soil by heavy metals. Environ. Res. 2020, 185, 109427. [Google Scholar] [CrossRef] [PubMed]
- Cano-Ruiz, J.; Ruiz Galea, M.; Amorós, M.C.; Alonso, J.; Mauri, P.V.; Lobo, M.C. Assessing Arundo donax L. in vitro-tolerance for phytoremediation purposes. Chemosphere 2020, 252, 126576. [Google Scholar] [CrossRef]
- Shaheen, S.; Ahmad, R.; Mahmood, Q.; Mubarak, H.; Mirza, N.; Hayat, M.T. Physiology and selected genes expression under cadmium stress in Arundo donax L. Int. J. Phytoremed. 2018, 20, 1162–1167. [Google Scholar] [CrossRef]
- Cocozza, C.; Brilli, F.; Pignattelli, S.; Pollastri, S.; Brunetti, C.; Gonnelli, C.; Tognetti, R.; Centritto, M.; Loreto, F. The excess of phosphorus in soil reduces physiological performances over time but enhances prompt recovery of salt-stressed Arundo donax plants. Plant Physiol. Biochem. 2020, 151, 556–565. [Google Scholar] [CrossRef]
- Cocozza, C.; Brilli, F.; Miozzi, L.; Pignattelli, S.; Rotunno, S.; Brunetti, C.; Giordano, C.; Pollastri, S.; Centritto, M.; Accotto, G.P.; et al. Impact of high or low levels of phosphorus and high sodium in soils on productivity and stress tolerance of Arundo donax plants. Plant Sci. 2019, 289, 110260. [Google Scholar] [CrossRef]
- Docimo, T.; De Stefano, R.; De Palma, M.; Cappetta, E.; Villano, C.; Aversano, R.; Tucci, M. Transcriptional, metabolic and DNA methylation changes underpinning the response of Arundo donax ecotypes to NaCl excess. Planta 2019, 251, 34. [Google Scholar] [CrossRef] [PubMed]
- Haworth, M.; Marino, G.; Riggi, E.; Avola, G.; Brunetti, C.; Scordia, D.; Testa, G.; Thiago Gaudio Gomes, M.; Loreto, F.; Luciano Cosentino, S.; et al. The effect of summer drought on the yield of Arundo donax is reduced by the retention of photosynthetic capacity and leaf growth later in the growing season. Ann. Bot. 2019, 124, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Manavella, P.A.; Yang, S.W.; Palatnik, J. Keep calm and carry on: miRNA biogenesis under stress. Plant J. 2019, 99, 832–843. [Google Scholar] [CrossRef]
- Pegler, J.L.; Grof, C.P.L.; Eamens, A.L. The Plant microRNA Pathway: The Production and Action Stages. Methods Mol. Biol. 2019, 1932, 15–39. [Google Scholar] [CrossRef]
- Li, S.; Castillo-González, C.; Yu, B.; Zhang, X. The functions of plant small RNAs in development and in stress responses. Plant J. 2017, 90, 654–670. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Lin, L.; Sui, N. Regulation mechanism of microRNA in plant response to abiotic stress and breeding. Mol. Biol. Rep. 2019, 46, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Danelli, T.; Laura, M.; Savona, M.; Landoni, M.; Adani, F.; Pilu, R. Genetic Improvement of Arundo donax L.: Opportunities and Challenges. Plants 2020, 9, 1584. [Google Scholar] [CrossRef]
- Sablok, G.; Fu, Y.; Bobbio, V.; Laura, M.; Rotino, G.; Bagnaresi, P.; Allavena, A.; Velikova, V.; Viola, R.; Loreto, F.; et al. Fuelling genetic and metabolic exploration of C-3 bioenergy crops through the first reference transcriptome of Arundo donax L. Plant Biotechnol. J. 2014, 12, 554–567. [Google Scholar] [CrossRef]
- Fu, Y.; Poli, M.; Sablok, G.; Wang, B.; Liang, Y.C.; La Porta, N.; Velikova, V.; Loreto, F.; Li, M.A.; Varotto, C. Dissection of early transcriptional responses to water stress in Arundo donax L. by unigene-based RNA-seq. Biotechnol. Biofuels 2016, 9, 54. [Google Scholar] [CrossRef]
- Sicilia, A.; Santoro, D.F.; Testa, G.; Cosentino, S.L.; Lo Piero, A.R. Transcriptional response of giant reed (Arundo donax L.) low ecotype to long-term salt stress by unigene-based RNAseq. Phytochemistry 2020, 177, 112436. [Google Scholar] [CrossRef] [PubMed]
- Barrero, R.A.; Guerrero, F.D.; Moolhuijzen, P.; Goolsby, J.A.; Tidwell, J.; Bellgard, S.E.; Bellgard, M.I. Shoot transcriptome of the giant reed, Arundo donax. Data Brief 2015, 3, 1–6. [Google Scholar] [CrossRef][Green Version]
- Evangelistella, C.; Valentini, A.; Ludovisi, R.; Firrincieli, A.; Fabbrini, F.; Scalabrin, S.; Cattonaro, F.; Morgante, M.; Mugnozza, G.S.; Keurentjes, J.J.B.; et al. De novo assembly, functional annotation, and analysis of the giant reed (Arundo donax L.) leaf transcriptome provide tools for the development of a biofuel feedstock. Biotechnol. Biofuels 2017, 10, 138. [Google Scholar] [CrossRef]
- Jike, W.; Sablok, G.; Bertorelle, G.; Li, M.G.; Varotto, C. In silico identification and characterization of a diverse subset of conserved microRNAs in bioenergy crop Arundo donax L. Sci. Rep. 2018, 8, 16667. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.J.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Campagna, D.; Albiero, A.; Bilardi, A.; Caniato, E.; Forcato, C.; Manavski, S.; Vitulo, N.; Valle, G. PASS: A program to align short sequences. Bioinformatics 2009, 25, 967–968. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.J.; Budak, H. Sorting the wheat from the chaff: Identifying miRNAs in genomic survey sequences of Triticum aestivum chromosome 1AL. PLoS ONE 2012, 7, e40859. [Google Scholar] [CrossRef]
- Alptekin, B.; Akpinar, B.A.; Budak, H. A Comprehensive Prescription for Plant miRNA Identification. Front. Plant Sci. 2016, 7, 2058. [Google Scholar] [CrossRef] [PubMed]
- Mathelier, A.; Carbone, A. MIReNA: Finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data. Bioinformatics 2010, 26, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; Wuyts, J.; Rouzé, P.; Van de Peer, Y. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 2004, 20, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef]
- Sunkar, R.; Jagadeeswaran, G. In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biol. 2008, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.B.; Balyan, S.; Jha, S.; Mathur, S. Novel insights into expansion and functional diversification of MIR169 family in tomato. Planta 2020, 251, 17. [Google Scholar] [CrossRef] [PubMed]
- Guddeti, S.; Zhang, D.C.; Li, A.L.; Leseberg, C.H.; Kang, H.; Li, X.G.; Zhai, W.X.; Johns, M.A.; Mao, L. Molecular evolution of the rice miR395 gene family. Cell Res. 2005, 15, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Singh, N.; Kumar, S.; Kumari, J.; Singh, R.; Gaba, S.; Yadav, M.C.; Grover, M.; Chaurasia, S.; Kumar, R. Identification and evolutionary analysis of polycistronic miRNA clusters in domesticated and wild wheat. Genomics 2020, 112, 2334–2348. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Meyers, B.C. Revisiting Criteria for Plant MicroRNA Annotation in the Era of Big Data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Song, C.; Wang, C.; Zhang, C.; Korir, N.K.; Yu, H.; Ma, Z.; Fang, J. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genom. 2010, 11, 431. [Google Scholar] [CrossRef]
- Mao, W.; Li, Z.; Xia, X.; Li, Y.; Yu, J. A combined approach of high-throughput sequencing and degradome analysis reveals tissue specific expression of microRNAs and their targets in cucumber. PLoS ONE 2012, 7, e33040. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Zheng, Y.; Qin, L.; Wang, Y.; Chen, L.F.; He, Y.J.; Fei, Z.J.; Lu, G. Identification of miRNAs and their targets through high-throughput sequencing and degradome analysis in male and female Asparagus officinalis. BMC Plant Biol. 2016, 16, 19. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of MIRNA genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Green, P.J. Methods for validation of miRNA sequence variants and the cleavage of their targets. Methods 2012, 58, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Fernando, A.L.; Barbosa, B.; Costa, J.; Papazoglou, E.G. Chapter 4—Giant Reed (Arundo donax L.): A Multipurpose Crop Bridging Phytoremediation with Sustainable Bioeconomy. In Bioremediation and Bioeconomy; Prasad, M.N.V., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 77–95. [Google Scholar]
- Jámbor, A.; Török, Á. The Economics of Arundo donax—A Systematic Literature Review. Sustainability 2019, 11, 4225. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, K.; Liu, G.R.; Li, S.S.; Zhao, S.H.; Liu, X.W.; Yang, X.J.; Xiao, K. Global identification and characterization of miRNA family members responsive to potassium deprivation in wheat (Triticum aestivum L.). Sci. Rep. 2020, 10, 13. [Google Scholar] [CrossRef]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Magrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18, 571–584. [Google Scholar] [CrossRef]
- Fu, R.; Zhang, M.; Zhao, Y.C.; He, X.C.; Ding, C.Y.; Wang, S.K.; Feng, Y.; Song, X.L.; Li, P.; Wang, B.H. Identification of Salt Tolerance-related microRNAs and Their Targets in Maize (Zea mays L.) Using High-throughput Sequencing and Degradome Analysis. Front. Plant Sci. 2017, 8, 13. [Google Scholar] [CrossRef]
- Huang, B.S.; Gan, L.; Chen, D.J.; Zhang, Y.C.; Zhang, Y.J.; Liu, X.L.; Chen, S.; Wei, Z.S.; Tong, L.Q.; Song, Z.J.; et al. Integration of small RNA, degradome and proteome sequencing in Oryza sativa reveals a delayed senescence network in tetraploid rice seed. PLoS ONE 2020, 15, e0242260. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Li, S.; Zhou, H.; Long, C.; et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5’ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef]
- Millar, A.A.; Waterhouse, P.M. Plant and animal microRNAs: Similarities and differences. Funct. Integr. Genom. 2005, 5, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Blevins, T.; Ailhas, J.; Boller, T.; Meins, F. Evolution of Arabidopsis MIR genes generates novel microRNA classes. Nucleic Acids Res. 2008, 36, 6429–6438. [Google Scholar] [CrossRef]
- Pantaleo, V.; Szittya, G.; Moxon, S.; Miozzi, L.; Moulton, V.; Dalmay, T.; Burgyan, J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Achkar, N.P.; Cambiagno, D.A.; Manavella, P.A. miRNA Biogenesis: A Dynamic Pathway. Trends Plant Sci. 2016, 21, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, P.; Li, X.; Liu, C.; Cao, S.; Chu, C.; Cao, X. Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiol. 2005, 139, 296–305. [Google Scholar] [CrossRef]
- Calviño, M.; Bruggmann, R.; Messing, J. Characterization of the small RNA component of the transcriptome from grain and sweet sorghum stems. BMC Genom. 2011, 12, 356. [Google Scholar] [CrossRef]
- Unver, T.; Budak, H. Conserved microRNAs and their targets in model grass species Brachypodium distachyon. Planta 2009, 230, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.G. mRNA 3’ end formation in plants: Novel connections to growth, development and environmental responses. Wiley Interdiscip. Rev. RNA 2020, 11, e1575. [Google Scholar] [CrossRef]
- Ganie, S.A.; Ahammed, G.J.; Wani, S.H. Vascular plant one zinc-finger (VOZ) transcription factors: Novel regulators of abiotic stress tolerance in rice (Oryza sativa L.). Genet. Resour. Crop Evol. 2020, 67, 799–807. [Google Scholar] [CrossRef]
- Rehman, S.U.; Qanmber, G.; Tahir, M.H.N.; Irshad, A.; Fiaz, S.; Ahmad, F.; Ali, Z.; Sajjad, M.; Shees, M.; Usman, M.; et al. Characterization of Vascular plant One-Zinc finger (VOZ) in soybean (Glycine max and Glycine soja) and their expression analyses under drought condition. PLoS ONE 2021, 16, e0253836. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.V.S.K.; Xing, D.; Reddy, A.S.N. Vascular Plant One-Zinc-Finger (VOZ) Transcription Factors Are Positive Regulators of Salt Tolerance in Arabidopsis. Int. J. Mol. Sci. 2018, 19, 3731. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zheng, J.C.; Wang, T.T.; Min, D.H.; Wei, W.L.; Chen, J.; Zhou, Y.B.; Chen, M.; Xu, Z.S.; Ma, Y.Z. Expression Analyses of Soybean VOZ Transcription Factors and the Role of. Int. J. Mol. Sci. 2020, 21, 2177. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Guo, W.; Li, J.; Pan, X.; Pan, L.; Zhao, J.; Zhang, Y.; Cai, S.; Huang, X.; Wang, A.; et al. The miR528-AO Module Confers Enhanced Salt Tolerance in Rice by Modulating the Ascorbic Acid and Abscisic Acid Metabolism and ROS Scavenging. J. Agric. Food Chem. 2021, 69, 8634–8648. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; He, S.; Liu, D.; Patil, G.B.; Zhai, H.; Wang, F.; Stephenson, T.J.; Wang, Y.; Wang, B.; Valliyodan, B.; et al. A Sweetpotato Geranylgeranyl Pyrophosphate Synthase Gene, IbGGPS, Increases Carotenoid Content and Enhances Osmotic Stress Tolerance in Arabidopsis thaliana. PLoS ONE 2015, 10, e0137623. [Google Scholar] [CrossRef]
- Jirage, D.; Tootle, T.L.; Reuber, T.L.; Frost, L.N.; Feys, B.J.; Parker, J.E.; Ausubel, F.M.; Glazebrook, J. Arabidopsis thaliana PAD4 encodes a lipase-like gene that is important for salicylic acid signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 13583–13588. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Fatma, M.; Per, T.S.; Anjum, N.A.; Khan, N.A. Salicylic acid-induced abiotic stress tolerance and underlying mechanisms in plants. Front. Plant Sci. 2015, 6, 462. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA Family | A. donax | O. sativa | A. donax [23] |
|---|---|---|---|
| miR156 | 3 | 12 | 8 |
| miR159 | 2 | 6 | - |
| miR160 | 1 | 6 | 4 |
| miR162 | 1 | 2 | - |
| miR164 | 2 | 6 | - |
| miR167 | 3 | 10 | 5 |
| miR169 | 10 | 18 | 16 |
| miR172 | 1 | 4 | 3 |
| miR319 | 2 | 2 | 2 |
| miR393 | 3 | 2 | 4 |
| miR395 | 8 | 24 | - |
| miR396 | 1 | 8 | 2 |
| miR398 | 1 | 4 | - |
| miR444 | 5 | 6 | 12 |
| miR528 | 2 | 1 | - |
| miR6222 | 1 | - | - |
| miR9774 | 1 | - | - |
| Name | Target Id | Target Annotation | Category | p-Value | |
|---|---|---|---|---|---|
| Known miRNAs | miR1128 | TR24300 | Senescence-associated protein (partial) | 2 | 0.082 |
| miR156d-3p | TR4471 | 32 kDa dirigent-like protein | 2 | 0.037 | |
| miR156j-5p.2 | TR14474 | Hypothetical protein | 0 | 0.007 | |
| miR159a-3p | TR5918 | n/a | 3 | 0.055 | |
| miR159b | TR24324 | n/a | 3 | 0.058 | |
| miR159b | TR30640 | n/a | 0 | 0.059 | |
| miR159e | TR11309 | BEL1-like homeodomain 4 containing protein | 2 | 0.025 | |
| miR159e | TR4218 | Uncharacterized protein | 3 | 0.046 | |
| miR159e | TR6528 | Ankyrin repeat domain-containing 13B | 0 | 0.066 | |
| miR167c-5p | TR29148 | ATP synthase delta chloroplastic protein | 2 | 0.005 | |
| miR169c-3p | TR24300 | Senescence-associated protein (partial) | 2 | 0.051 | |
| miR172b | TR25857 | Hypothetical protein | 3 | 0.086 | |
| miR172d-5p | TR29998 | Pentatricopeptide repeat-containing protein | 3 | 0.036 | |
| miR319a | TR1733 | Calcineurin B 1 | 3 | 0.034 | |
| miR319b-3p | TR24557 | n/a | 0 | 0.026 | |
| miR396b | TR20898 | Chlorophyll a-b binding chloroplastic-like protein | 2 | 0.062 | |
| miR528-3p | TR1686 | n/a | 3 | 0.266 | |
| miR528-3p | TR8922 | Peptidyl-prolyl cis-trans isomerase-like 3 protein | 0 | 0.313 | |
| miR529a | TR20898 | n/a | 3 | 0.163 | |
| miR6253 | TR13145 | Serine carboxypeptidase II-3 | 3 | 0.066 | |
| miR9774 | TR9615 | F-box SKIP22 | 3 | 0.022 | |
| Candidate miRNAs | miRC174433-3 | TR10651 | Oxygen-evolving enhancer chloroplastic protein | 3 | 0.553 |
| miRC174433-3 | TR27080 | Gamma-glutamyl peptidase 5-like | 2 | 0.063 | |
| miRC174433-3 | TR8896 | Hypothetical protein | 0 | 0.141 | |
| miRC366946-2 | TR28663 | Cytochrome P450 | 2 | 0.005 | |
| miRC71512-6 | TR24307 | B-box zinc finger 24 | 2 | 0.064 | |
| miRC808846-2 | TR19738 | n/a | 3 | 0.083 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotunno, S.; Cocozza, C.; Pantaleo, V.; Leonetti, P.; Bertoldi, L.; Valle, G.; Accotto, G.P.; Loreto, F.; Scippa, G.S.; Miozzi, L. Identification of Known and Novel Arundo donax L. MicroRNAs and Their Targets Using High-Throughput Sequencing and Degradome Analysis. Life 2022, 12, 651. https://doi.org/10.3390/life12050651
Rotunno S, Cocozza C, Pantaleo V, Leonetti P, Bertoldi L, Valle G, Accotto GP, Loreto F, Scippa GS, Miozzi L. Identification of Known and Novel Arundo donax L. MicroRNAs and Their Targets Using High-Throughput Sequencing and Degradome Analysis. Life. 2022; 12(5):651. https://doi.org/10.3390/life12050651
Chicago/Turabian StyleRotunno, Silvia, Claudia Cocozza, Vitantonio Pantaleo, Paola Leonetti, Loris Bertoldi, Giorgio Valle, Gian Paolo Accotto, Francesco Loreto, Gabriella Stefania Scippa, and Laura Miozzi. 2022. "Identification of Known and Novel Arundo donax L. MicroRNAs and Their Targets Using High-Throughput Sequencing and Degradome Analysis" Life 12, no. 5: 651. https://doi.org/10.3390/life12050651
APA StyleRotunno, S., Cocozza, C., Pantaleo, V., Leonetti, P., Bertoldi, L., Valle, G., Accotto, G. P., Loreto, F., Scippa, G. S., & Miozzi, L. (2022). Identification of Known and Novel Arundo donax L. MicroRNAs and Their Targets Using High-Throughput Sequencing and Degradome Analysis. Life, 12(5), 651. https://doi.org/10.3390/life12050651