
Dystrophin Dp71 Subisoforms Localize to the Mitochondria of Human Cells

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Primary Cultured Myocytes
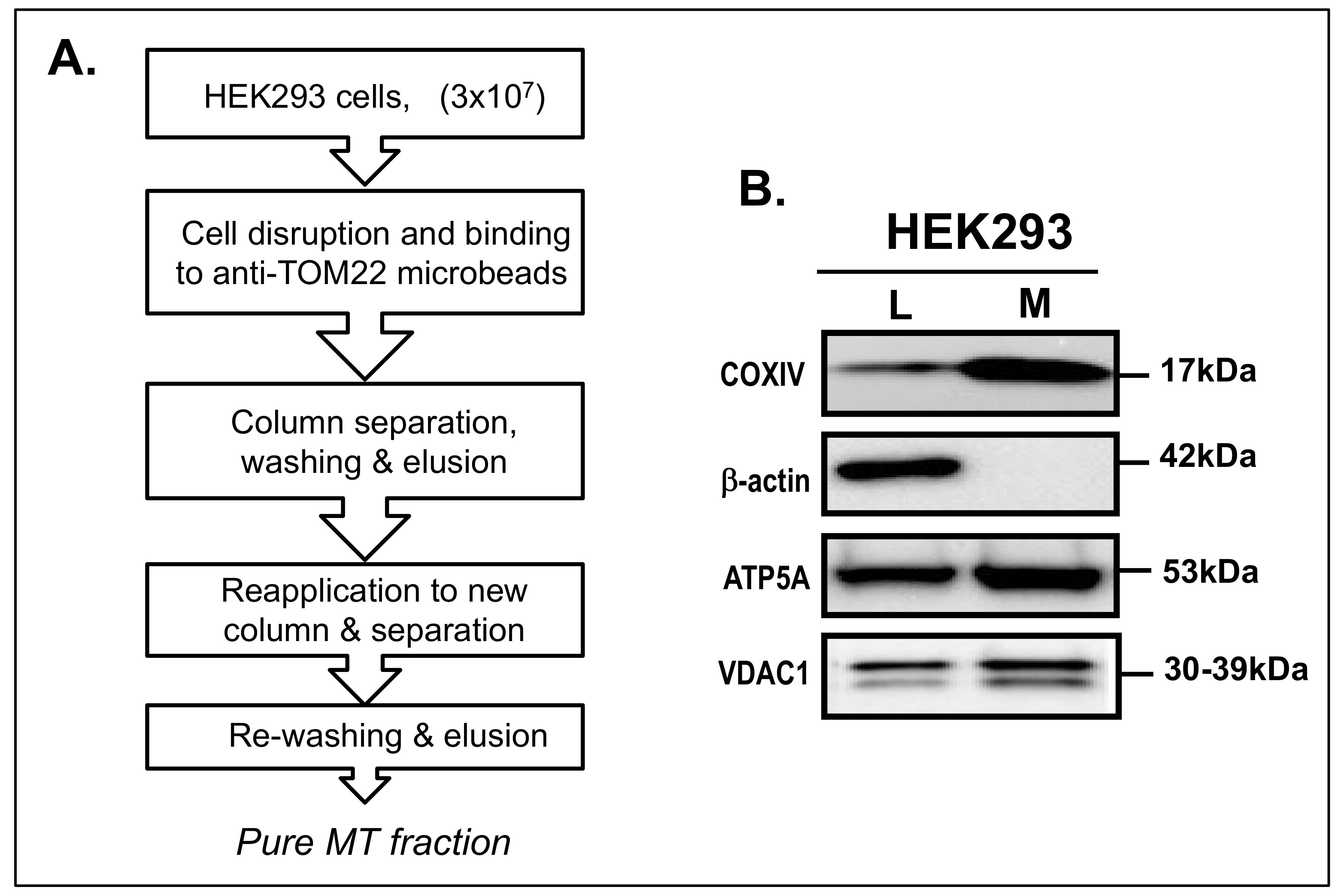
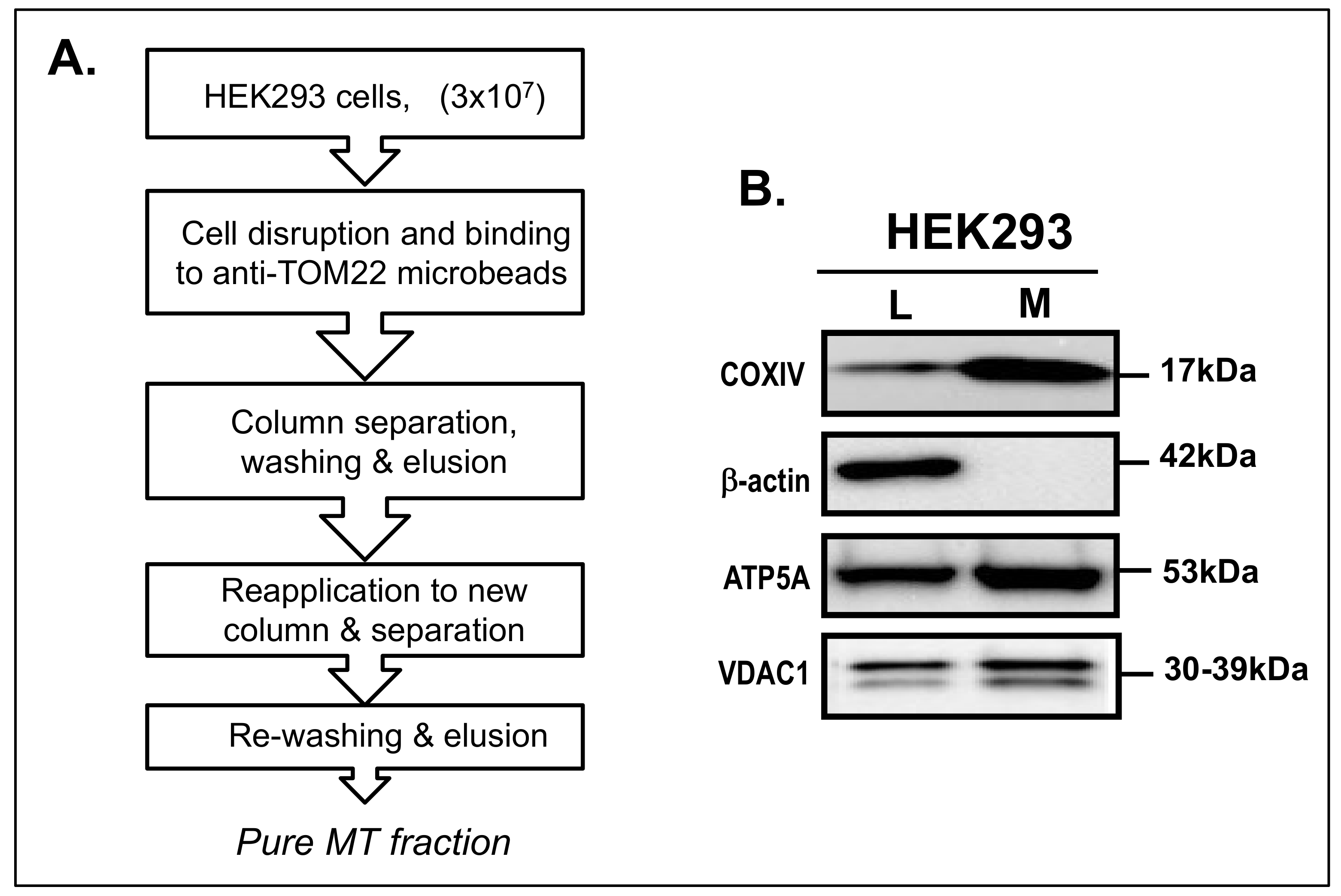
2.2. Isolation of MT Fraction
2.3. Isolation of Nuclear and Cytoplasmic Fractions
2.4. Protein Analysis by Western Blotting
2.5. RNA Isolation and RT-PCR
2.6. DNA Sequencing
2.7. Immunoprecipitation
2.8. Statistical Analysis
3. Results
3.1. Isolation of MT Fraction from HEK293 Cells
3.2. Dp71 Is Detected in MT Fraction of HEK293 Cells
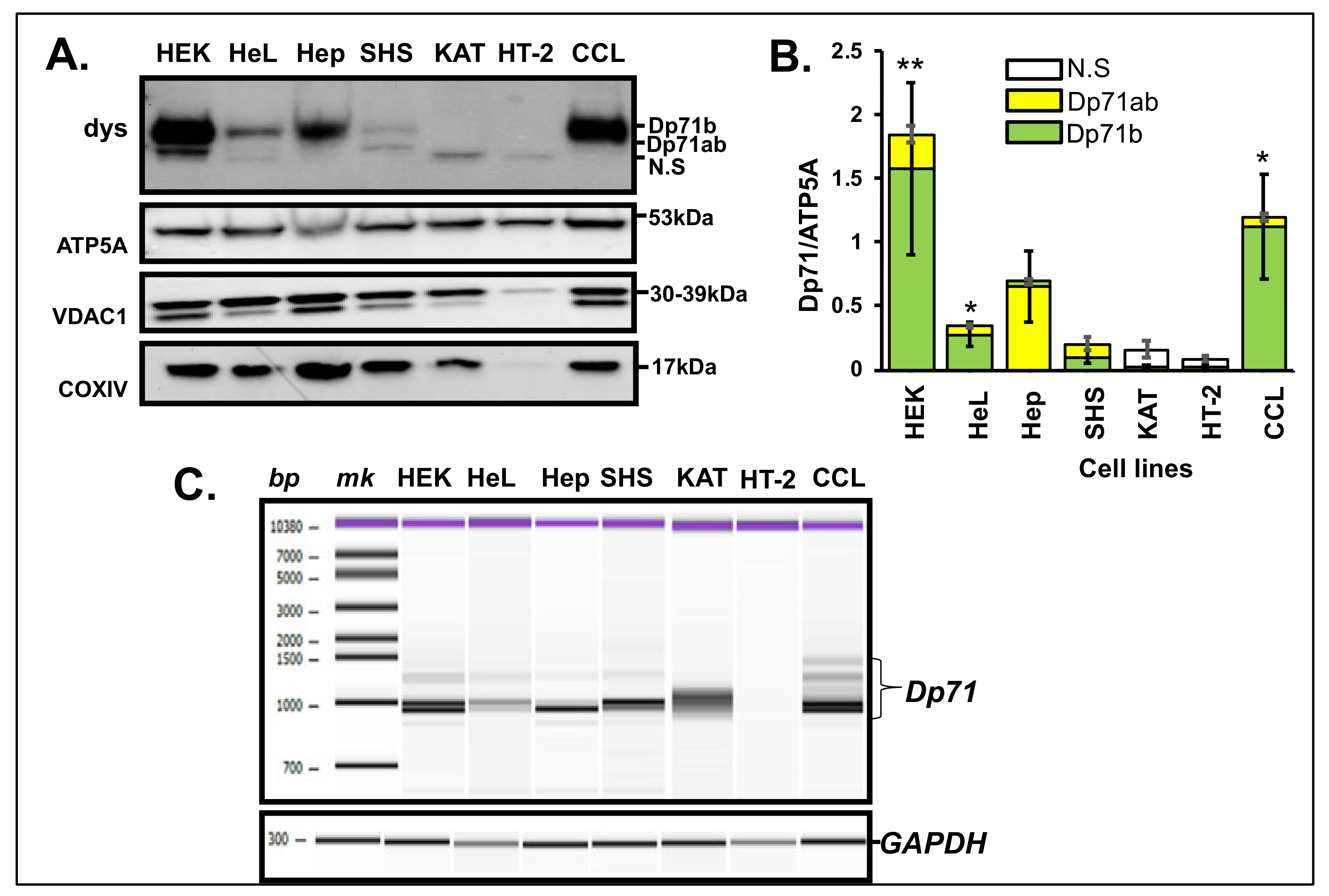
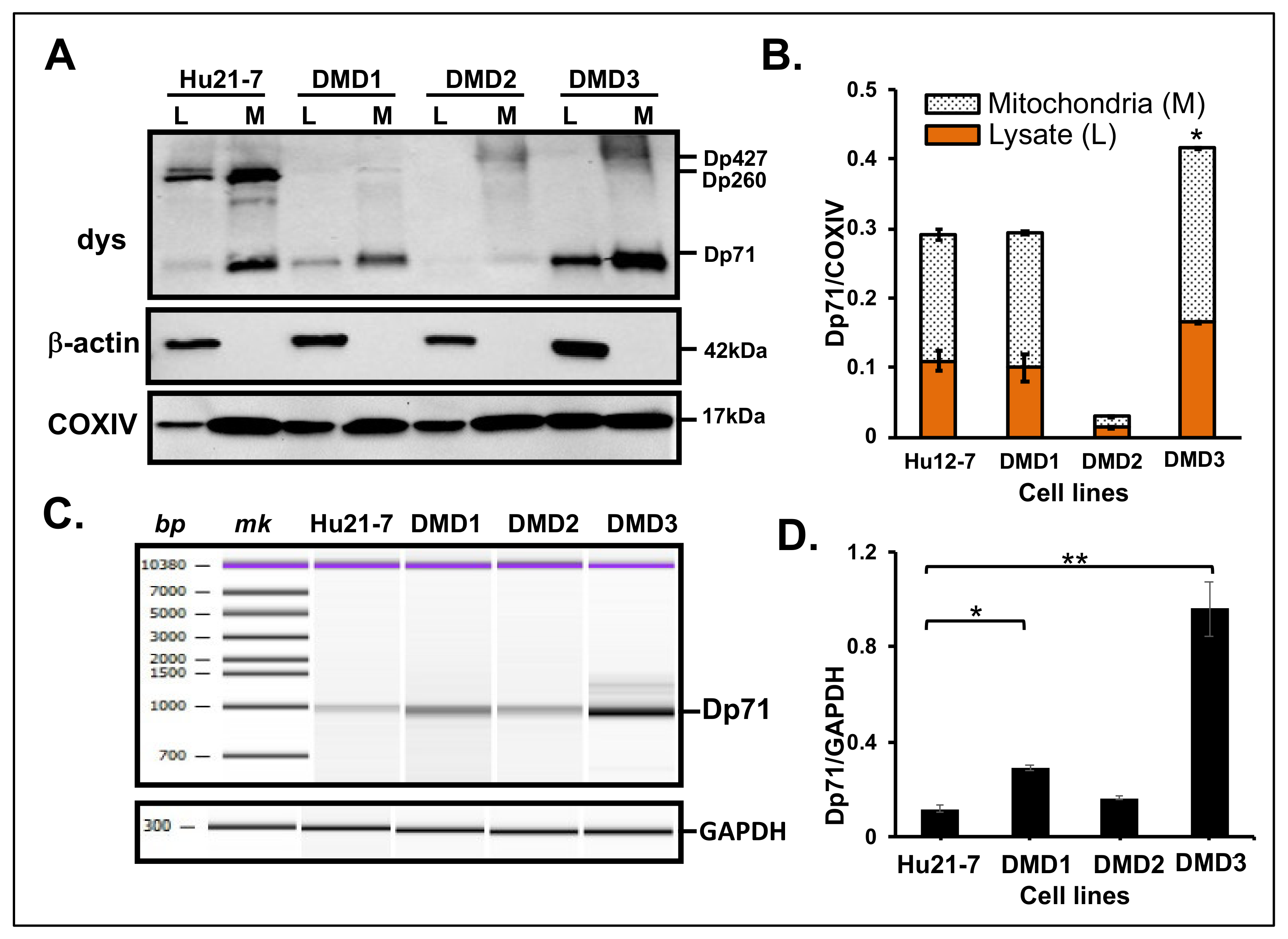
3.3. Dp71 Expression in MT Fraction of Other Human Cancer Cells
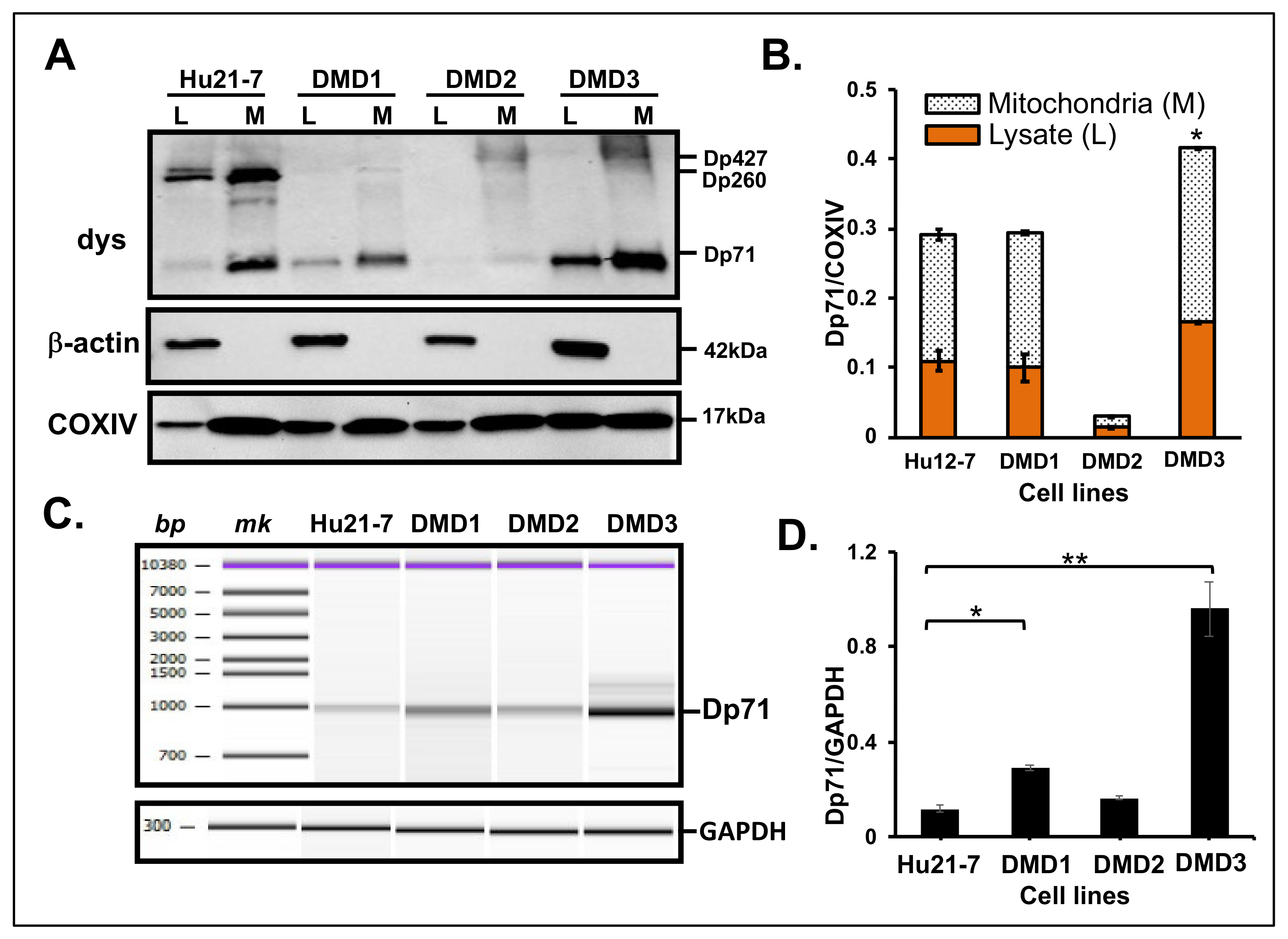
3.4. Dp71 Expression in MT Fraction from Primary Cultured Myocytes
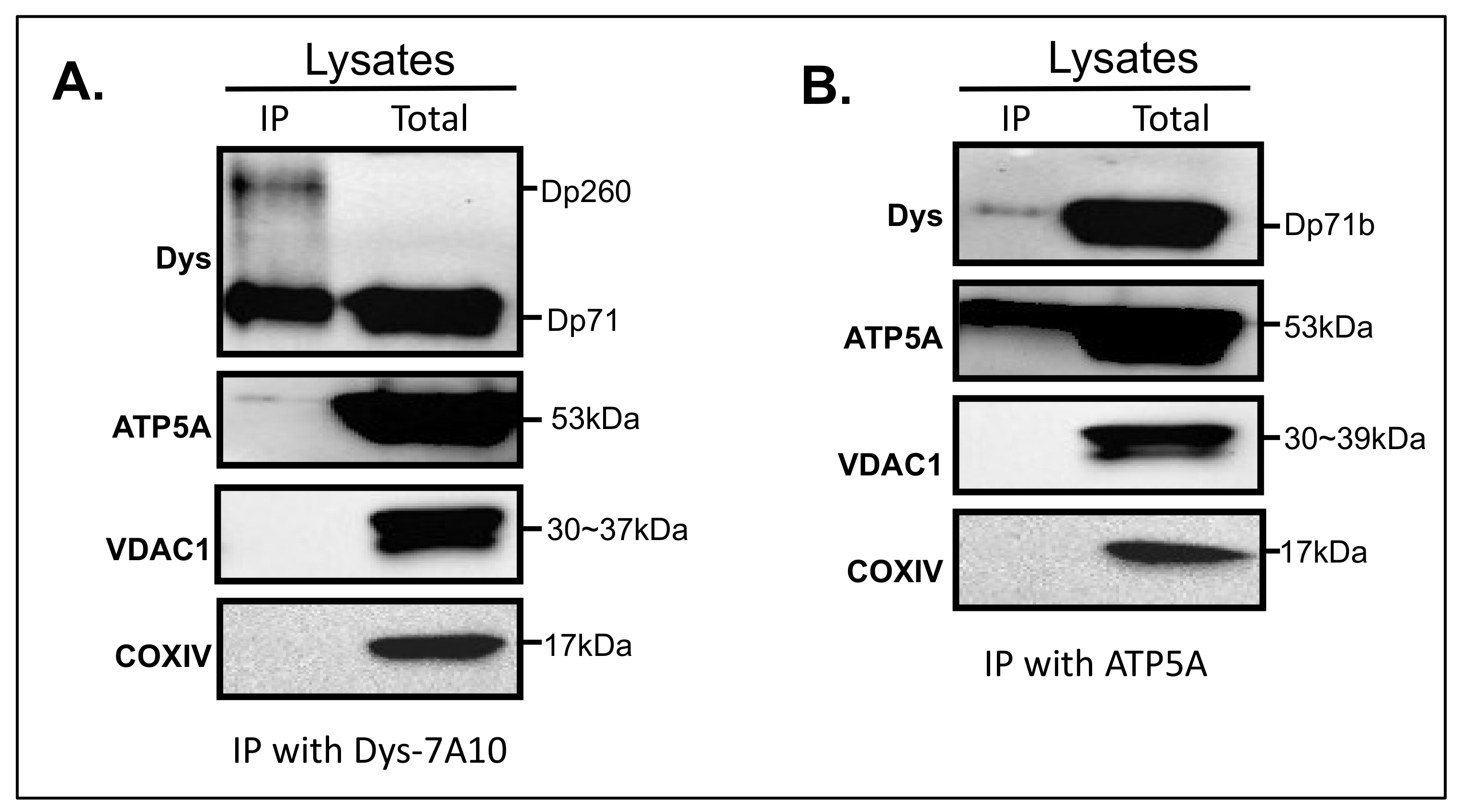
3.5. Interaction of Dystrophin with ATP5A
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and Genetics of Dystrophin and Dystrophin-Related Proteins in Muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [Green Version]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete Cloning of the Duchenne Muscular Dystrophy (DMD) CDNA and Preliminary Genomic Organization of the DMD Gene in Normal and Affected Individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and Mutations: One Gene, Several Proteins, Multiple Phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Ahn, A.H.; Kunkel, L.M. The Structural and Functional Diversity of Dystrophin. Nat. Genet. 1993, 3, 283–291. [Google Scholar] [CrossRef]
- Massouridès, E.; Polentes, J.; Mangeot, P.-E.; Mournetas, V.; Nectoux, J.; Deburgrave, N.; Nusbaum, P.; Leturcq, F.; Popplewell, L.; Dickson, G.; et al. Dp412e: A Novel Human Embryonic Dystrophin Isoform Induced by BMP4 in Early Differentiated Cells. Skelet. Muscle 2015, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Aragón, J.; Martínez-Herrera, A.; Bermúdez-Cruz, R.M.; Bazán, M.L.; Soid-Raggi, G.; Ceja, V.; Coy-Arechavaleta, A.S.; Alemán, V.; Depardón, F.; Montañez, C. EF-Hand Domains Are Involved in the Differential Cellular Distribution of Dystrophin Dp40. Neurosci. Lett. 2015, 600, 115–120. [Google Scholar] [CrossRef]
- D’Souza, V.N.; Man, N.T.; Morris, G.E.; Karges, W.; Pillers, D.-A.M.; Ray, P.N. A Novel Dystrophin Isoform Is Required for Normal Retinal Electrophysiology. Hum. Mol. Genet. 1995, 4, 837–842. [Google Scholar] [CrossRef]
- Ervasti, J.; Campbell, K. A Role for the Dystrophin-Glycoprotein Complex as a Transmembrane Linker between Laminin and Actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Ohlendieck, K. Mass Spectrometric Identification of Dystrophin, the Protein Product of the Duchenne Muscular Dystrophy Gene, in Distinct Muscle Surface Membranes. Int. J. Mol. Med. 2017, 40, 1078–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, L.M.; Hejtmancik, J.F.; Caskey, C.T.; Speer, A.; Monaco, A.P.; Middlesworth, W.; Colletti, C.A.; Bertelson, C.; Müller, U.; Bresnan, M.; et al. Analysis of Deletions in DNA from Patients with Becker and Duchenne Muscular Dystrophy. Nature 1986, 322, 73–77. [Google Scholar] [CrossRef]
- Tadayoni, R.; Rendon, A.; Soria-Jasso, L.E.; Cisneros, B. Dystrophin Dp71: The Smallest but Multifunctional Product of the Duchenne Muscular Dystrophy Gene. Mol. Neurobiol. 2012, 45, 43–60. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2020, 57, 1748–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Mathari, B.; Sene, A.; Charles-Messance, H.; Vacca, O.; Guillonneau, X.; Grepin, C.; Sennlaub, F.; Sahel, J.-A.; Rendon, A.; Tadayoni, R. Dystrophin Dp71 Gene Deletion Induces Retinal Vascular Inflammation and Capillary Degeneration. Hum. Mol. Genet. 2015, 24, 3939–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerna, J.; Cerecedo, D.; Ortega, A.; García-Sierra, F.; Centeno, F.; Garrido, E.; Mornet, D.; Cisneros, B. Dystrophin Dp71f Associates with the Β1-Integrin Adhesion Complex to Modulate PC12 Cell Adhesion. J. Mol. Biol. 2006, 362, 954–965. [Google Scholar] [CrossRef] [Green Version]
- Villarreal-Silva, M.; Centeno-Cruz, F.; Suárez-Sánchez, R.; Garrido, E.; Cisneros, B. Knockdown of Dystrophin Dp71 Impairs PC12 Cells Cycle: Localization in the Spindle and Cytokinesis Structures Implies a Role for Dp71 in Cell Division. PLoS ONE 2011, 6, e23504. [Google Scholar] [CrossRef]
- Benabdesselam, R.; Rendon, A.; Dorbani-Mamine, L.; Hardin-Pouzet, H. Effect of Dp71 Deficiency on the Oxytocin Hypothalamic Axis in Osmoregulation Function in Mice. Acta Histochem. 2019, 121, 268–276. [Google Scholar] [CrossRef]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.-J.B.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genom. Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in Heart Mitochondria in the Early Stages of Duchenne Muscular Dystrophy. Biochim. Biophys. Acta (BBA) Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Bonsett, C.A.; Rudman, A. The Dystrophin Connection—ATP? Med. Hypotheses 1992, 38, 139–154. [Google Scholar] [CrossRef]
- Carberry, S.; Brinkmeier, H.; Zhang, Y.; Winkler, C.K.; Ohlendieck, K. Comparative Proteomic Profiling of Soleus, Extensor Digitorum Longus, Flexor Digitorum Brevis and Interosseus Muscles from the Mdx Mouse Model of Duchenne Muscular Dystrophy. Int. J. Mol. Med. 2013, 32, 544–556. [Google Scholar] [CrossRef] [Green Version]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in Mitochondrial ATP Synthesis in Dystrophin-Deficient Mdx Skeletal Muscles May Be Caused by Complex I Insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [Green Version]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the Dystrophin-ATP Connection: How Half a Century of Research Still Implicates Mitochondrial Dysfunction in Duchenne Muscular Dystrophy Aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in Mitochondrial Localization and ATP Synthesis in the Mdx Mouse Model of Duchenne Muscular Dystrophy Are Not Alleviated by PDE5 Inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Chávez, O.; Harricane, M.C.; Alemán, V.; Dorbani, L.; Larroque, C.; Mornet, D.; Rendon, A.; Martínez-Rojas, D. Mitochondrial Expression of a Short Dystrophin-like Product with Molecular Weight of 71 KDa. Biochem. Biophys. Res. Commun. 2000, 274, 275–280. [Google Scholar] [CrossRef]
- Nishida, A.; Yasuno, S.; Takeuchi, A.; Awano, H.; Lee, T.; Niba, E.T.E.; Fujimoto, T.; Itoh, K.; Takeshima, Y.; Nishio, H.; et al. HEK293 Cells Express Dystrophin Dp71 with Nucleus-Specific Localization of Dp71ab. Histochem. Cell Biol. 2016, 146, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Farea, M.; Rani, A.Q.M.; Maeta, K.; Nishio, H.; Matsuo, M. Dystrophin Dp71ab Is Monoclonally Expressed in Human Satellite Cells and Enhances Proliferation of Myoblast Cells. Sci. Rep. 2020, 10, 17123. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.A.; Howard, P.L.; D’Souza, V.N.; Klamut, H.J.; Ray, P.N. Cloning and Characterization of Alternatively Spliced Isoforms of Dp71. Hum. Mol. Genet. 1995, 4, 1475–1483. [Google Scholar] [CrossRef]
- Aragón, J.; González-Reyes, M.; Romo-Yáñez, J.; Vacca, O.; Aguilar-González, G.; Rendón, A.; Vaillend, C.; Montañez, C. Dystrophin Dp71 Isoforms Are Differentially Expressed in the Mouse Brain and Retina: Report of New Alternative Splicing and a Novel Nomenclature for Dp71 Isoforms. Mol. Neurobiol. 2018, 55, 1376–1386. [Google Scholar] [CrossRef]
- Herrera-Salazar, A.; García-Villegas, R.; Aragón, J.; Sánchez-Trujillo, A.; Ceja, V.; Martínez-Herrera, A.; Merino-Jiménez, C.; Montañez, C. Overexpression of Mutant Dystrophin Dp71 [INCREMENT] 78–79 Stimulates Cell Proliferation. NeuroReport 2016, 27, 6–11. [Google Scholar] [CrossRef]
- Hornig-Do, H.-T.; Günther, G.; Bust, M.; Lehnartz, P.; Bosio, A.; Wiesner, R.J. Isolation of Functional Pure Mitochondria by Superparamagnetic Microbeads. Anal. Biochem. 2009, 389, 1–5. [Google Scholar] [CrossRef]
- Nishida, A.; Minegishi, M.; Takeuchi, A.; Awano, H.; Niba, E.T.E.; Matsuo, M. Neuronal SH-SY5Y Cells Use the C-Dystrophin Promoter Coupled with Exon 78 Skipping and Display Multiple Patterns of Alternative Splicing Including Two Intronic Insertion Events. Hum. Genet. 2015, 134, 993–1001. [Google Scholar] [CrossRef]
- Nishida, A.; Minegishi, M.; Takeuchi, A.; Niba, E.T.E.; Awano, H.; Lee, T.; Iijima, K.; Takeshima, Y.; Matsuo, M. Tissue- and Case-Specific Retention of Intron 40 in Mature Dystrophin MRNA. J. Hum. Genet. 2015, 60, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Niba, E.T.E.; Yamanaka, R.; Rani, A.Q.M.; Awano, H.; Matsumoto, M.; Nishio, H.; Matsuo, M. DMD Transcripts in CRL-2061 Rhabdomyosarcoma Cells Show High Levels of Intron Retention by Intron-Specific PCR Amplification. Cancer Cell Int. 2017, 17, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, M.P.; Bosworth, C.M.; McMahon, S.; Grandhi, S.; Grimerg, B.T.; LaFramboise, T. PCR-Free Enrichment of Mitochondrial DNA from Human Blood and Cell Lines for High Quality Next-Generation DNA Sequencing. PLoS ONE 2015, 10, e0139253. [Google Scholar] [CrossRef]
- Satori, C.P.; Kostal, V.; Arriaga, E.A. Review on Recent Advances in the Analysis of Isolated Organelles. Anal. Chim. Acta 2012, 753, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zeng, F.; Wu, H.; Hu, C.; Yu, C.; Wu, S. Preparation of a Mitochondria-Targeted and NO-Releasing Nanoplatform and Its Enhanced Pro-Apoptotic Effect on Cancer Cells. Small 2014, 10, 3750–3760. [Google Scholar] [CrossRef]
- Kappler, L.; Li, J.; Häring, H.-U.; Weigert, C.; Lehmann, R.; Xu, G.; Hoene, M. Purity Matters: A Workflow for the Valid High-Resolution Lipid Profiling of Mitochondria from Cell Culture Samples. Sci. Rep. 2016, 6, 21107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria Mediate Cell Membrane Repair and Contribute to Duchenne Muscular Dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early Myopathy in Duchenne Muscular Dystrophy Is Associated with Elevated Mitochondrial H2O2 Emission during Impaired Oxidative Phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in Mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Wang, Y.; Marino-Enriquez, A.; Bennett, R.R.; Zhu, M.; Shen, Y.; Eilers, G.; Lee, J.-C.; Henze, J.; Fletcher, B.S.; Gu, Z.; et al. Dystrophin Is a Tumor Suppressor in Human Cancers with Myogenic Programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Tan, S.; Chen, Z.; Cheng, K.; Chen, Z.; Wang, W.; Wen, Q.; Zhang, W. Knocking down Dp71 Expression in A549 Cells Reduces Its Malignancy in Vivo and in Vitro. Cancer Investig. 2016, 34, 16–25. [Google Scholar] [CrossRef]
- Mauduit, O.; Delcroix, V.; Lesluyes, T.; Pérot, G.; Lagarde, P.; Lartigue, L.; Blay, J.-Y.; Chibon, F. Recurrent DMD Deletions Highlight Specific Role of Dp71 Isoform in Soft-Tissue Sarcomas. Cancers 2019, 11, 922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doddihal, H.; Jalali, R. Medulloblastoma in a Child with Duchenne Muscular Dystrophy. Childs Nerv. Syst. 2007, 23, 595–597. [Google Scholar] [CrossRef] [PubMed]
- van den Akker, M.; Northcott, P.; Taylor, M.D.; Halliday, W.; Bartels, U.; Bouffet, E. Anaplastic Medulloblastoma in a Child with Duchenne Muscular Dystrophy: Case Report. PED 2012, 10, 21–24. [Google Scholar] [CrossRef]
- Niba, E.T.E.; Awano, H.; Nagai, M.; Taniguchi, M.; Itoh, K.; Nishimura, N.; Iijima, K.; Nishio, H.; Matsuo, M. Pilocytic Astrocytoma Tissue from a 25-Year-Old Patient with Duchenne Muscular Dystrophy Expresses the DMD Transcript but Not Protein. Clin. Res. Pediatr. 2020, 3, 1–9. [Google Scholar]
- Nishida, A.; Kataoka, N.; Takeshima, Y.; Yagi, M.; Awano, H.; Ota, M.; Itoh, K.; Hagiwara, M.; Matsuo, M. Chemical Treatment Enhances Skipping of a Mutated Exon in the Dystrophin Gene. Nat. Commun. 2011, 2, 308. [Google Scholar] [CrossRef] [Green Version]
- Radley-Crabb, H.G.; Marini, J.C.; Sosa, H.A.; Castillo, L.I.; Grounds, M.D.; Fiorotto, M.L. Dystropathology Increases Energy Expenditure and Protein Turnover in the Mdx Mouse Model of Duchenne Muscular Dystrophy. PLoS ONE 2014, 9, e89277. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Niba, E.; Rani, A.; Onishi, Y.; Koizumi, M.; Awano, H.; Matsumoto, M.; Nagai, M.; Yoshida, S.; Sakakibara, S.; et al. Detection of Dystrophin Dp71 in Human Skeletal Muscle Using an Automated Capillary Western Assay System. Int. J. Mol. Sci. 2018, 19, 1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daoud, F.; Angeard, N.; Demerre, B.; Martie, I.; Benyaou, R.; Leturcq, F.; Cossee, M.; Deburgrave, N.; Saillour, Y.; Tuffery, S.; et al. Analysis of Dp71 Contribution in the Severity of Mental Retardation through Comparison of Duchenne and Becker Patients Differing by Mutation Consequences on Dp71 Expression. Hum. Mol. Genet. 2009, 18, 3779–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial Dysfunction as a Central Actor in Intellectual Disability-Related Diseases: An Overview of Down Syndrome, Autism, Fragile X and Rett Syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Ricotti, V.; Mandy, W.P.L.; Scoto, M.; Pane, M.; Deconinck, N.; Messina, S.; Mercuri, E.; Skuse, D.H.; Muntoni, F. Neurodevelopmental, Emotional, and Behavioural Problems in Duchenne Muscular Dystrophy in Relation to Underlying Dystrophin Gene Mutations. Dev. Med. Child Neurol. 2016, 58, 77–84. [Google Scholar] [CrossRef]
- Legido, A.; Jethva, R.; Goldenthal, M.J. Mitochondrial Dysfunction in Autism. Semin. Pediatr. Neurol. 2013, 20, 163–175. [Google Scholar] [CrossRef]
- Wood, C.L.; Straub, V.; Guglieri, M.; Bushby, K.; Cheetham, T. Short Stature and Pubertal Delay in Duchenne Muscular Dystrophy. Arch. Dis. Child. 2016, 101, 101–106. [Google Scholar] [CrossRef]
- Nagel, B.H.; Mortier, W.; Elmlinger, M.; Wollmann, H.A.; Schmitt, K.; Ranke, M.B. Short Stature in Duchenne Muscular Dystrophy: A Study of 34 Patients. Acta Paediatr. 1999, 88, 62–65. [Google Scholar] [CrossRef]
- Matsumoto, M.; Awano, H.; Lee, T.; Takeshima, Y.; Matsuo, M.; Iijima, K. Patients with Duchenne Muscular Dystrophy Are Significantly Shorter than Those with Becker Muscular Dystrophy, with the Higher Incidence of Short Stature in Dp71 Mutated Subgroup. Neuromuscul. Disord. 2017, 27, 1023–1028. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An Updated Mitochondrial Proteome Now with Sub-Organelle Localization and Pathway Annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Carregari, V.C.; Greco, V.; Ronci, M.; Iavarone, F.; Persichilli, S.; Castagnola, M.; Urbani, A.; Pieroni, L. Exploring the HeLa Dark Mitochondrial Proteome. Front. Cell Dev. Biol. 2020, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.G.; Wu, Y.; Wolski, W.; Kim, J.Y.; Lan, J.; Hasan, M.; Halter, C.; Jha, P.; Ryu, D.; Auwerx, J.; et al. Quantifying and Localizing the Mitochondrial Proteome Across Five Tissues in A Mouse Population. Mol. Cell. Proteom. 2018, 17, 1766–1777. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.C.; Robinson, A.J. MitoMiner v3.1, an Update on the Mitochondrial Proteomics Database. Nucleic Acids Res. 2016, 44, D1258–D1261. [Google Scholar] [CrossRef] [Green Version]
- Lefort, N.; Yi, Z.; Bowen, B.; Glancy, B.; De Filippis, E.A.; Mapes, R.; Hwang, H.; Flynn, C.R.; Willis, W.T.; Civitarese, A.; et al. Proteome Profile of Functional Mitochondria from Human Skeletal Muscle Using One-Dimensional Gel Electrophoresis and HPLC-ESI-MS/MS. J. Proteom. 2009, 72, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An Updated Inventory of Mammalian Mitochondrial Proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Satrialdi; Hibino, M.; Sasaki, D.; Abe, J.; Harashima, H. Power of Mitochondrial Drug Delivery Systems to Produce Innovative Nanomedicines. Adv. Drug Deliv. Rev. 2020, 154–155, 187–209. [Google Scholar] [CrossRef]
- Lu, P.; Bruno, B.J.; Rabenau, M.; Lim, C.S. Delivery of Drugs and Macromolecules to the Mitochondria for Cancer Therapy. J. Control. Release 2016, 240, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Lamhonwah, A.-M.; Tein, I. Expression of the Organic Cation/Carnitine Transporter Family (Octn1,-2 and-3) in Mdx Muscle and Heart: Implications for Early Carnitine Therapy in Duchenne Muscular Dystrophy to Improve Cellular Carnitine Homeostasis. Clin. Chim. Acta 2020, 505, 92–97. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Oltmann, M.; Rinaldi, C.; Myung, K.H.; Tidball, J.G. Loss of Positive Allosteric Interactions between Neuronal Nitric Oxide Synthase and Phosphofructokinase Contributes to Defects in Glycolysis and Increased Fatigability in Muscular Dystrophy. Hum. Mol. Genet. 2009, 18, 3439–3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, P.; Gargan, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Proteomic Profiling of Fatty Acid Binding Proteins in Muscular Dystrophy. Expert Rev. Proteom. 2020, 17, 137–148. [Google Scholar] [CrossRef]
- Gaglianone, R.B.; Santos, A.T.; Bloise, F.F.; Ortiga-Carvalho, T.M.; Costa, M.L.; Quirico-Santos, T.; da Silva, W.S.; Mermelstein, C. Reduced Mitochondrial Respiration and Increased Calcium Deposits in the EDL Muscle, but Not in Soleus, from 12-Week-Old Dystrophic Mdx Mice. Sci. Rep. 2019, 9, 1986. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DMD1 | DMD2 | DMD3 | |
|---|---|---|---|
| Mutation | c.2308A > T (Exon 19) p.Lys770 * | c.2677C >T (Exon 21) p.Gln893 * | c.4303G >T (Exon 31) p.Glu1436 * |
| Phenotype | DMD | DMD | BMD |
| Symptoms | Calf pseudohypertrophy (8yo) Waddling gait (<8yo) Climb stairs with support (<8yo) Gowers’s sign (8yo) | Calf pseudohypertrophy (3yo) Waddling gait (6yo) Climb stairs with support (7yo) | None related to muscle weakness (2yo) |
| Muscle biopsy | No staining with DYS1-3 ab (8yo) | No staining with DYS1-3 ab (8 yo) | Patchy and faint staining with DYS1-3 ab (4yo) |
| Present situation | 11yo; Ambulant | 15yo; Non ambulant | 14yo; Runs normally, riding daily to school |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niba, E.T.E.; Awano, H.; Lee, T.; Takeshima, Y.; Shinohara, M.; Nishio, H.; Matsuo, M. Dystrophin Dp71 Subisoforms Localize to the Mitochondria of Human Cells. Life 2021, 11, 978. https://doi.org/10.3390/life11090978
Niba ETE, Awano H, Lee T, Takeshima Y, Shinohara M, Nishio H, Matsuo M. Dystrophin Dp71 Subisoforms Localize to the Mitochondria of Human Cells. Life. 2021; 11(9):978. https://doi.org/10.3390/life11090978
Chicago/Turabian StyleNiba, Emma Tabe Eko, Hiroyuki Awano, Tomoko Lee, Yasuhiro Takeshima, Masakazu Shinohara, Hisahide Nishio, and Masafumi Matsuo. 2021. "Dystrophin Dp71 Subisoforms Localize to the Mitochondria of Human Cells" Life 11, no. 9: 978. https://doi.org/10.3390/life11090978
APA StyleNiba, E. T. E., Awano, H., Lee, T., Takeshima, Y., Shinohara, M., Nishio, H., & Matsuo, M. (2021). Dystrophin Dp71 Subisoforms Localize to the Mitochondria of Human Cells. Life, 11(9), 978. https://doi.org/10.3390/life11090978