
Non-Invasive Respiratory Assessment in Duchenne Muscular Dystrophy: From Clinical Research to Outcome Measures



{kind=link}
Abstract
1. Introduction
2. The Act of Breathing
3. Non-Invasive Assessment of Respiratory Function
3.1. Pulmonary Function Tests
3.1.1. Lung Volume
3.1.2. Pressure
3.1.3. Flow
3.1.4. Ventilation Inhomogeneity
3.1.5. Transcutaneous Gas Monitoring
3.2. Ventilatory Pattern
3.3. Thoraco-Abdominal Breathing Motion
3.4. Imaging
3.4.1. Ultrasound (US)
3.4.2. Magnetic Resonance Imaging (MRI)
4. Respiratory Assessment in Duchenne Muscular Dystrophy
4.1. Respiratory System
4.1.1. Lung Volume
4.1.2. Flow
4.2. Ventilation
4.2.1. Ventilatory Pattern
4.2.2. Thoraco-Abdominal Breathing Motion
4.2.3. Ventilation Inhomogeneity
4.3. Gas Exchange
Transcutaneous Gas Monitoring
4.4. Respiratory Muscles
4.4.1. Pressure
4.4.2. Diaphragm Excursion
4.4.3. Diaphragm Thickness and Thickening Ratio
4.4.4. Fatty Infiltration
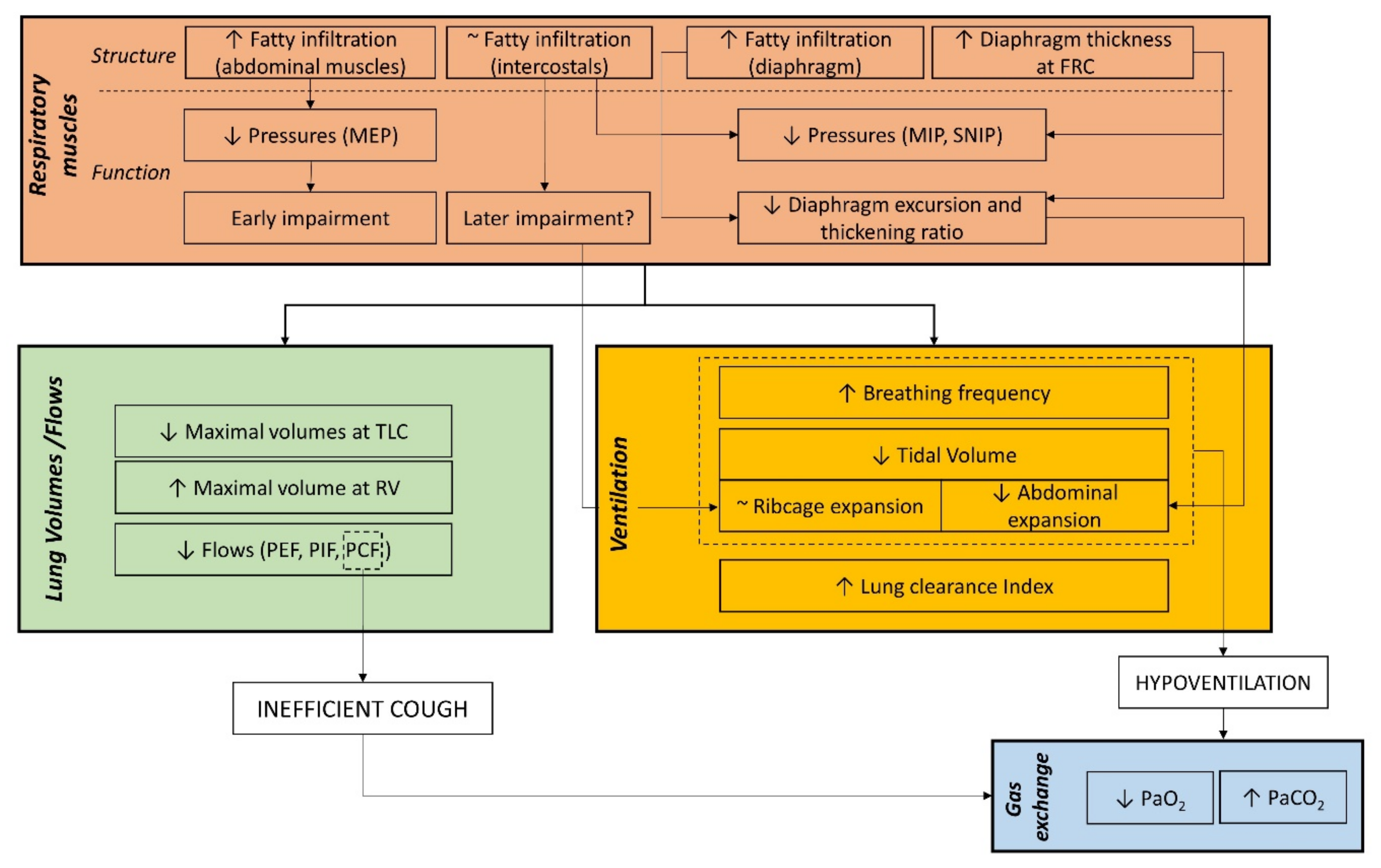
5. Respiratory Outcomes to Link Structural and Functional Respiratory Impairment
6. Respiratory Outcomes for the Clinical Management of Patients with DMD
7. Respiratory Outcomes for Clinical Trials
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bushby, K.; Finkel, R.; Birnkrant, D.; Case, L.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Barbé, F.; Quera-Salva, M.; McCann, C.; Gajdos, P.; Raphael, J.; De Lattre, J.; Agustí, A. Sleep-related respiratory disturbances in patients with Duchenne muscular dystrophy. Eur. Respir. J. 1994, 7, 1403–1408. [Google Scholar] [CrossRef]
- Dohna-Schwake, C.; Ragette, R.; Teschler, H.; Voit, T.; Mellies, U. Predictors of severe chest infections in pediatric neuromuscular disorders. Neuromuscul. Disord. 2006, 16, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.M.; Amin, R.S.; Bach, J.R.; Benditt, J.O.; Eagle, M.; Finder, J.D.; Kalra, M.S.; Kissel, J.T.; Koumbourlis, A.C.; et al. The respiratory management of patients with duchenne muscular dystrophy: A DMD care considerations working group specialty article. Pediatr. Pulmonol. 2010, 45, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef]
- Ledford, H. US government approves controversial drug for muscular dystrophy. Nature 2016. [Google Scholar] [CrossRef]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef]
- Finder, J.D.; Birnkrant, D.; Carl, J.; Farber, H.J.; Gozal, D.; Iannaccone, S.T.; Kovesi, T.; Kravitz, R.M.; Panitch, H.; Schramm, C.; et al. Respiratory Care of the Patient with Duchenne Muscular Dystrophy. Am. J. Respir. Crit. Care Med. 2004, 170, 456–465. [Google Scholar] [CrossRef]
- Phillips, M.F.; Quinlivan, R.C.M.; Edwards, R.H.T.; Calverley, P.M.A. Changes in Spirometry over Time as a Prognostic Marker in Patients with Duchenne Muscular Dystrophy. Am. J. Respir. Crit. Care Med. 2001, 164, 2191–2194. [Google Scholar] [CrossRef]
- Ward, M.; Macklem, P.T. The Act of Breathing and How It Fails. Chest 1990, 97, 36S–39S. [Google Scholar] [CrossRef] [PubMed]
- Macklem, P.T. Normal and abnormal function of the diaphragm. Thorax 1981, 36, 161–163. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allen, J. Pulmonary complications of neuromuscular disease: A Respiratory mechanics perspective. Paediatr. Respir. Rev. 2010, 11, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Mauro, M.A.L.; Aliverti, A. Physiology of respiratory disturbances in muscular dystrophies. Breathe 2016, 12, 318–327. [Google Scholar] [CrossRef]
- Ueki, J.; De Bruin, P.F.; Pride, N.B. In vivo assessment of diaphragm contraction by ultrasound in normal subjects. Thorax 1995, 50, 1157–1161. [Google Scholar] [CrossRef]
- Testa, A.; Soldati, G.; Giannuzzi, R.; Berardi, S.; Portale, G.; Silveri, N.G. Ultrasound M-Mode Assessment of Diaphragmatic Kinetics by Anterior Transverse Scanning in Healthy Subjects. Ultrasound Med. Biol. 2011, 37, 44–52. [Google Scholar] [CrossRef]
- Gauthier, A.P.; Verbanck, S.; Estenne, M.; Segebarth, C.; Macklem, P.T.; Paiva, M. Three-dimensional reconstruction of the in vivo human diaphragm shape at different lung volumes. J. Appl. Physiol. 1994, 76, 495–506. [Google Scholar] [CrossRef]
- Boussuges, A.; Rives, S.; Julie, F.; Brégeon, F. Assessment of diaphragmatic function by ultrasonography: Current approach and perspectives. World J. Clin. Cases 2020, 8, 2408–2424. [Google Scholar] [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; Van Der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Quanjer, P.; Stanojevic, S.; Cole, T.; Baur, X.; Hall, G.; Culver, B.; Enright, P.; Hankinson, J.L.; Ip, M.S.; Zheng, J.; et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: The global lung function 2012 equations. Eur. Respir. J. 2012, 40, 1324–1343. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, P.; Lebecque, P.; Corey, M.; Coates, A.L. Comparison of spirometric reference values. Pediatr. Pulmonol. 2004, 37, 515–522. [Google Scholar] [CrossRef]
- Loth, D.W.; Ittermann, T.; Lahousse, L.; Hofman, A.; Leufkens, H.G.M.; Brusselle, G.; Stricker, B.H. Normal spirometry values in healthy elderly: The Rotterdam Study. Eur. J. Epidemiol. 2013, 28, 329–334. [Google Scholar] [CrossRef]
- Hull, J. British Thoracic Society guideline for respiratory management of children with neuromuscular weakness: Commentary. Thorax 2012, 67, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Wanger, J.; Clausen, J.L.; Coates, A.; Pedersen, O.F.; Brusasco, V.; Burgos, F.; Casaburi, R.; Crapo, R.; Enright, P.; Van Der Grinten, C.P.M.; et al. Standardisation of the measurement of lung volumes. Eur. Respir. J. 2005, 26, 511–522. [Google Scholar] [CrossRef]
- Laveneziana, P.; Albuquerque, A.; Aliverti, A.; Babb, T.; Barreiro, E.; Dres, M.; Dubé, B.-P.; Fauroux, B.; Gea, J.; Guenette, J.A.; et al. ERS statement on respiratory muscle testing at rest and during exercise. Eur. Respir. J. 2019, 53, 1801214. [Google Scholar] [CrossRef]
- Fauroux, B.; Quijano-Roy, S.; Desguerre, I.; Khirani, S. The Value of Respiratory Muscle Testing in Children with Neuromuscular Disease. Chest 2015, 147, 552–559. [Google Scholar] [CrossRef]
- Esau, S.A.; Bellemare, F.; Grassino, A.; Permutt, S.; Roussos, C.; Pardy, R.L. Changes in relaxation rate with diaphragmatic fatigue in humans. J. Appl. Physiol. 1983, 54, 1353–1360. [Google Scholar] [CrossRef]
- Nava, S.; Ambrosino, N.; Crotti, P.; Fracchia, C.; Rampulla, C. Recruitment of some respiratory muscles during three maximal inspiratory manoeuvres. Thorax 1993, 48, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, M.; Abe, T.; Yokoba, M.; Dobashi, Y.; Tomita, T.; Easton, P. Neck and abdominal muscle activity during a sniff. Respir. Med. 2003, 97, 1027–1035. [Google Scholar] [CrossRef][Green Version]
- Laroche, C.M.; Mier, A.K.; Moxham, J.; Green, M. The Value of Sniff Esophageal Pressures in the Assessment of Global Inspiratory Muscle Strength. Am. Rev. Respir. Dis. 1988, 138, 598–603. [Google Scholar] [CrossRef] [PubMed]
- European, Respiratory Society, and American Thoracic Society. ATS/ERS Statement on Respiratory Muscle Testing. Am. J. Respir. Crit. Care Med. 2002, 166, 518–624. [Google Scholar] [CrossRef]
- LoMauro, A.; Privitera, E.; Aliverti, A.; Nosotti, M.; Palleschi, A. Sniff test: Does what we measure at the nose reflect what happens in the chest wall? Clin. Respir. J. 2020, 14, 589–591. [Google Scholar] [CrossRef]
- Singer, F.; Houltz, B.; Latzin, P.; Robinson, P.; Gustafsson, P. A Realistic Validation Study of a New Nitrogen Multiple-Breath Washout System. PLoS ONE 2012, 7, e36083. [Google Scholar] [CrossRef]
- Bedi, P.K.; Castro-Codesal, M.L.; Featherstone, R.; AlBalawi, M.M.; Alkhaledi, B.; Kozyrskyj, A.L.; Flores-Mir, C.; MacLean, J.E. Long-term Non-Invasive Ventilation in Infants: A Systematic Review and Meta-Analysis. Front. Pediatr. 2018, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Amaddeo, A.; Fauroux, B.; Information, P.E.K.F.C. Oxygen and carbon dioxide monitoring during sleep. Paediatr. Respir. Rev. 2016, 20, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Cala, S.J.; Kenyon, C.M.; Ferrigno, G.; Carnevali, P.; Aliverti, A.; Pedotti, A.; Macklem, P.T.; Rochester, D.F. Chest wall and lung volume estimation by optical reflectance motion analysis. J. Appl. Physiol. 1996, 81, 2680–2689. [Google Scholar] [CrossRef]
- Aliverti, A.; Dellaca’, R.; Pelosi, P.; Chiumello, D.A.; Gattinoni, L.; Pedotti, A. Compartmental Analysis of Breathing in the Supine and Prone Positions by Optoelectronic Plethysmography. Ann. Biomed. Eng. 2001, 29, 60–70. [Google Scholar] [CrossRef]
- Neumann, P.; Zinserling, J.; Haase, C.; Sydow, M.; Burchardi, H. Evaluation of Respiratory Inductive Plethysmography in Controlled Ventilation. Chest 1998, 113, 443–451. [Google Scholar] [CrossRef]
- Chadha, S.; Watson, H.; Birch, S.; Jenouri, G.A.; Schneider, A.W.; Cohn, M.A.; Sackner, M.A. Validation of respiratory inductive plethysmography using different calibration procedures. Am. Rev. Respir. Dis. 1982, 125, 644–649. [Google Scholar] [CrossRef]
- Fleck, D.; Curry, C.; Donnan, K.; Logue, O.; Graham, K.; Jackson, K.; Keown, K.; Winder, J.; Shields, M.D.; Hughes, C.M. Investigating the clinical use of structured light plethysmography to assess lung function in children with neuromuscular disorders. PLoS ONE 2019, 14, e0221207. [Google Scholar] [CrossRef] [PubMed]
- Motamedi-Fakhr, S.; Iles, R.; Barney, A.; De Boer, W.; Conlon, J.; Khalid, A.; Wilson, R.C. Evaluation of the agreement of tidal breathing parameters measured simultaneously using pneumotachography and structured light plethysmography. Physiol. Rep. 2017, 5, e13124. [Google Scholar] [CrossRef] [PubMed]
- Cretikos, M.A.; Bellomo, R.; Hillman, K.; Chen, J.; Finfer, S.; Flabouris, A. Respiratory rate: The neglected vital sign. Med. J. Aust. 2008, 188, 657–659. [Google Scholar] [CrossRef]
- Sarmento, A.; Vignati, C.; Paolillo, S.; Lombardi, C.; Scoccia, A.; Nicoli, F.; Mapelli, M.; Leonardi, A.; Ossola, D.; Rigoni, R.; et al. Qualitative and quantitative evaluation of a new wearable device for ECG and respiratory Holter monitoring. Int. J. Cardiol. 2018, 272, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Guilizzoni, D.; Angelucci, A.; Melloni, G.; Mazza, F.; Stanzi, A.; Venturino, M.; Kuller, D.; Aliverti, A. Comparison between the Airgo™ Device and a Metabolic Cart during Rest and Exercise. Sensors 2020, 20, 3943. [Google Scholar] [CrossRef]
- Chu, M.; Nguyen, T.; Pandey, V.; Zhou, Y.; Pham, H.N.; Bar-Yoseph, R.; Radom-Aizik, S.; Jain, R.; Cooper, D.M.; Khine, M. Respiration rate and volume measurements using wearable strain sensors. NPJ Digit. Med. 2019, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Naranjo-Hernández, D.; Talaminos-Barroso, A.; Reina-Tosina, J.; Roa, L.M.; Barbarov-Rostan, G.; Cejudo-Ramos, P.; Márquez-Martín, E.; Ortega-Ruiz, F. Smart Vest for Respiratory Rate Monitoring of COPD Patients Based on Non-Contact Capacitive Sensing. Sensors 2018, 18, 2144. [Google Scholar] [CrossRef]
- Liu, G.-Z.; Guo, Y.-W.; Zhu, Q.-S.; Huang, B.-Y.; Wang, L. Estimation of Respiration Rate from Three-Dimensional Acceleration Data Based on Body Sensor Network. Telemed. e-Health 2011, 17, 705–711. [Google Scholar] [CrossRef]
- Fekr, A.R.; Janidarmian, M.; Radecka, K.; Zilic, Z. A Medical Cloud-Based Platform for Respiration Rate Measurement and Hierarchical Classification of Breath Disorders. Sensors 2014, 14, 11204–11224. [Google Scholar] [CrossRef]
- Hung, P.; Bonnet, S.; Guillemaud, R.; Castelli, E.; Yen, P.T.N. Estimation of Respiratory Waveform Using an Accelerometer. In Proceedings of the 2008 5th IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Paris, France, 14–17 May 2008; pp. 1493–1496. [Google Scholar]
- Cesareo, A.; Previtali, Y.; Biffi, E.; Aliverti, A. Assessment of Breathing Parameters Using an Inertial Measurement Unit (IMU)-Based System. Sensors 2018, 19, 88. [Google Scholar] [CrossRef]
- Cesareo, A.; Biffi, E.; Cuesta-Frau, D.; D’Angelo, M.G.; Aliverti, A. A novel acquisition platform for long-term breathing frequency monitoring based on inertial measurement units. Med Biol. Eng. Comput. 2020, 58, 785–804. [Google Scholar] [CrossRef]
- Niérat, M.-C.; Dubé, B.-P.; Llontop, C.; Bellocq, A.; Ben Mohamed, L.L.; Rivals, I.; Straus, C.; Similowski, T.; Laveneziana, P. Measuring Ventilatory Activity with Structured Light Plethysmography (SLP) Reduces Instrumental Observer Effect and Preserves Tidal Breathing Variability in Healthy and COPD. Front. Physiol. 2017, 8, 316. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, P.F.C.; Ueki, J.; Bush, A.; Khan, Y.; Watson, A.; Pride, N.B. Diaphragm thickness and inspiratory strength in patients with Duchenne muscular dystrophy. Thorax 1997, 52, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Laviola, M.; Priori, R.; D’Angelo, M.G.; Aliverti, A. Assessment of diaphragmatic thickness by ultrasonography in Duchenne muscular dystrophy (DMD) patients. PLoS ONE 2018, 13, e0200582. [Google Scholar] [CrossRef]
- Van Doorn, J.L.; Pennati, F.; Hansen, H.H.; van Engelen, B.G.; Aliverti, A.; Doorduin, J. Respiratory muscle imaging by ultrasound and MRI in neuromuscular disorders. Eur. Respir. J. 2021, 58, 2100137. [Google Scholar] [CrossRef] [PubMed]
- Pennati, F.; Arrigoni, F.; LoMauro, A.; Gandossini, S.; Russo, A.; D’Angelo, M.G.; Aliverti, A. Diaphragm Involvement in Duchenne Muscular Dystrophy (DMD): An MRI Study. J. Magn. Reson. Imaging 2020, 51, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Wens, S.C.A.; Ciet, P.; Perez-Rovira, A.; Logie, K.; Salamon, E.; Wielopolski, P.; de Bruijne, M.; Kruijshaar, M.E.; Tiddens, H.A.W.M.; van Doorn, P.A.; et al. Lung MRI and impairment of diaphragmatic function in Pompe disease. BMC Pulm. Med. 2015, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, M.; Musumeci, O.; Mondello, S.; Ruggeri, P.; Montagnese, F.; Cucinotta, M.; Vinci, S.; Milardi, D.; Toscano, A. Clinical and pathophysiological clues of respiratory dysfunction in late-onset Pompe disease: New insights from a comparative study by MRI and respiratory function assessment. Neuromuscul. Disord. 2015, 25, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Mogalle, K.; Perez-Rovira, A.; Ciet, P.; Wens, S.C.A.; Van Doorn, P.A.; Tiddens, H.A.W.M.; Van Der Ploeg, A.T.; De Bruijne, M. Quantification of Diaphragm Mechanics in Pompe Disease Using Dynamic 3D MRI. PLoS ONE 2016, 11, e0158912. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.M.; Lott, D.J.; Batra, A.; Triplett, W.T.; Forbes, S.C.; Riehl, S.L.; Willcocks, R.J.; Smith, B.K.; Vandenborne, K.; Walter, G.A. Imaging respiratory muscle quality and function in Duchenne muscular dystrophy. J. Neurol. 2019, 266, 2752–2763. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Kovacs, W.; Norato, G.; Hsieh, N.; Bandettini, W.P.; Bishop, C.A.; Shimellis, H.; Newbould, R.D.; Kim, E.; Fischbeck, K.H.; et al. Respiratory magnetic resonance imaging biomarkers in Duchenne muscular dystrophy. Ann. Clin. Transl. Neurol. 2017, 4, 655–662. [Google Scholar] [CrossRef]
- Bishop, C.A.; Ricotti, V.; Sinclair, C.D.J.; Evans, M.R.B.; Butler, J.W.; Morrow, J.M.; Hanna, M.G.; Matthews, P.M.; Yousry, T.A.; Muntoni, F.; et al. Semi-Automated Analysis of Diaphragmatic Motion with Dynamic Magnetic Resonance Imaging in Healthy Controls and Non-Ambulant Subjects with Duchenne Muscular Dystrophy. Front. Neurol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Mercuri, E.; Talim, B.; Moghadaszadeh, B.; Petit, N.; Brockington, M.; Counsell, S.; Guicheney, P.; Muntoni, F.; Merlini, L. Clinical and imaging findings in six cases of congenital muscular dystrophy with rigid spine syndrome linked to chromosome 1p (RSMD1). Neuromuscul. Disord. 2002, 12, 631–638. [Google Scholar] [CrossRef]
- Gaeta, M.; Barca, E.; Ruggeri, P.; Minutoli, F.; Rodolico, C.; Mazziotti, S.; Milardi, D.; Musumeci, O.; Toscano, A. Late-onset Pompe disease (LOPD): Correlations between respiratory muscles CT and MRI features and pulmonary function. Mol. Genet. Metab. 2013, 110, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Dixon, W.T. Simple proton spectroscopic imaging. Radiology 1984, 153, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Guichoux, F.; Vuillemin, V.; Morvan, G.; Zins, M.; Thevenin, F.; Guerini, H.; Omoumi, P.; Drape, J.L. Fat Suppression with Dixon Techniques in Musculoskeletal Magnetic Resonance Imaging: A Pictorial Review. Semin. Musculoskelet. Radiol. 2015, 19, 335–347. [Google Scholar] [CrossRef]
- Eggers, H.; Börnert, P. Chemical shift encoding-based water-fat separation methods. J. Magn. Reson. Imaging 2014, 40, 251–268. [Google Scholar] [CrossRef]
- Güttsches, A.-K.; Rehmann, R.; Schreiner, A.; Rohm, M.; Forsting, J.; Froeling, M.; Tegenthoff, M.; Vorgerd, M.; Schlaffke, L. Quantitative Muscle-MRI Correlates with Histopathology in Skeletal Muscle Biopsies. J. Neuromuscul. Dis. 2021, 8, 669–678. [Google Scholar] [CrossRef]
- Mayer, O.; Finkel, R.; Rummey, C.; Benton, M.; Glanzman, A.; Flickinger, J.; Lindström, B.; Meier, T. Characterization of pulmonary function in Duchenne Muscular Dystrophy. Pediatr. Pulmonol. 2015, 50, 487–494. [Google Scholar] [CrossRef]
- Miller, F.; Moseley, C.F.; Koreska, J.; Levison, H. Pulmonary function and scoliosis in Duchenne dystrophy. J. Pediatr. Orthop. 1988, 8, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.M.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.-M.; Finkel, R.S.; Goemans, N.; McDonald, C.M.; et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef]
- Hahn, A.; Bach, J.R.; Delaubier, A.; Renardel-Irani, A.; Guillou, C.; Rideau, Y. Clinical implications of maximal respiratory pressure determinations for individuals with duchenne muscular dystrophy. Arch. Phys. Med. Rehabil. 1997, 78, 1–6. [Google Scholar] [CrossRef]
- McDonald, C.M.; Gordish-Dressman, H.; Henricson, E.K.; Duong, T.; Joyce, N.C.; Jhawar, S.; Leinonen, M.; Hsu, F.; Connolly, A.M.; Cnaan, A.; et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: Long-term natural history with and without glucocorticoids. Neuromuscul. Disord. 2018, 28, 897–909. [Google Scholar] [CrossRef]
- LoMauro, A.; Romei, M.; Gandossini, S.; Pascuzzo, R.; Vantini, S.; D’Angelo, M.G.; Aliverti, A. Evolution of respiratory function in Duchenne muscular dystrophy from childhood to adulthood. Eur. Respir. J. 2018, 51, 1701418. [Google Scholar] [CrossRef]
- Meier, T.; Rummey, C.; Leinonen, M.; Spagnolo, P.; Mayer, O.H.; Buyse, G.M.; Bernert, G.; Knipp, F.; Goemans, N.; Hauwe, M.V.D.; et al. Characterization of pulmonary function in 10–18 year old patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2017, 27, 307–314. [Google Scholar] [CrossRef]
- De Bruin, P.F.; Ueki, J.; Bush, A.; Manzur, A.Y.; Watson, A.; Pride, N.B. Inspiratory flow reserve in boys with Duchenne muscular dystrophy. Pediatr. Pulmonol. 2001, 31, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Goemans, N.; Hauwe, M.V.D.; Thijs, D.; de Groot, I.J.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.L.; D’Angelo, M.G.; Romei, M.; Motta, F.; Colombo, D.; Comi, G.; Pedotti, A.; Marchi, E.; Turconi, A.C.; Bresolin, N.; et al. Abdominal volume contribution to tidal volume as an early indicator of respiratory impairment in Duchenne muscular dystrophy. Eur. Respir. J. 2009, 35, 1118–1125. [Google Scholar] [CrossRef]
- Cesareo, A.; Nido, S.A.; Biffi, E.; Gandossini, S.; D’Angelo, M.G.; Aliverti, A. A Wearable Device for Breathing Frequency Monitoring: A Pilot Study on Patients with Muscular Dystrophy. Sensors 2020, 20, 5346. [Google Scholar] [CrossRef]
- Romei, M.; D’Angelo, M.G.; LoMauro, A.; Gandossini, S.; Bonato, S.; Brighina, E.; Marchi, E.; Comi, G.; Turconi, A.C.; Pedotti, A.; et al. Low abdominal contribution to breathing as daytime predictor of nocturnal desaturation in adolescents and young adults with Duchenne Muscular Dystrophy. Respir. Med. 2012, 106, 276–283. [Google Scholar] [CrossRef]
- LoMauro, A.; Romei, M.; D’Angelo, M.G.; Aliverti, A. Determinants of cough efficiency in Duchenne muscular dystrophy. Pediatr. Pulmonol. 2014, 49, 357–365. [Google Scholar] [CrossRef]
- Stehling, F.; Dohna-Schwake, C.; Mellies, U.; Grosse-Onnebrink, J.; Große-Onnebrink, J. Decline in Lung Volume with Duchenne Muscular Dystrophy Is Associated with Ventilation Inhomogeneity. Respir. Care 2015, 60, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Wales, P.; Dakin, C.; Harris, M.-A.; Cooper, D. (Gus) M. Sleep-related breathing disorder in Duchenne muscular dystrophy: Disease spectrum in the paediatric population. J. Paediatr. Child Health 2005, 41, 500–503. [Google Scholar] [CrossRef]
- Sawnani, H.; Thampratankul, L.; Szczesniak, R.D.; Fenchel, M.C.; Simakajornboon, N. Sleep Disordered Breathing in Young Boys with Duchenne Muscular Dystrophy. J. Pediatr. 2015, 166, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Nozoe, K.; Moreira, G.A.; Tolino, J.R.C.; Pradella-Hallinan, M.; Tufik, S.; Andersen, M.L. The sleep characteristics in symptomatic patients with Duchenne muscular dystrophy. Sleep Breath. 2015, 19, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Hukins, C.A.; Hillman, D. Daytime Predictors of Sleep Hypoventilation in Duchenne Muscular Dystrophy. Am. J. Respir. Crit. Care Med. 2000, 161, 166–170. [Google Scholar] [CrossRef]
- Toussaint, M.; Steens, M.; Soudon, P. Lung Function Accurately Predicts Hypercapnia in Patients with Duchenne Muscular Dystrophy. Chest 2007, 131, 368–375. [Google Scholar] [CrossRef]
- Gayraud, J.; Ramonatxo, M.; Rivier, F.; Humberclaude, V.; Petrof, B.; Matecki, S. Ventilatory parameters and maximal respiratory pressure changes with age in Duchenne muscular dystrophy patients. Pediatr. Pulmonol. 2010, 45, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Khirani, S.; Ramirez, A.; Aubertin, G.; Boulé, M.; Chemouny, C.; Forin, V.; Fauroux, B. Respiratory muscle decline in duchenne muscular dystrophy. Pediatr. Pulmonol. 2014, 49, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Nève, V.; Cuisset, J.-M.; Edmé, J.-L.; Carpentier, A.; Howsam, M.; Leclerc, O.; Matran, R. Sniff nasal inspiratory pressure in the longitudinal assessment of young Duchenne muscular dystrophy children. Eur. Respir. J. 2012, 42, 671–680. [Google Scholar] [CrossRef]
- Nève, V.; Edmé, J.-L.; Matran, R. Earlier decline in sniff nasal inspiratory pressure than peak expiratory flow in children with Duchenne muscular dystrophy. Eur. Respir. J. 2014, 44, 1361–1363. [Google Scholar] [CrossRef]
- Fayssoil, A.; Nguyen, L.S.; Ogna, A.; Stojkovic, T.; Meng, P.; Mompoint, D.; Carlier, R.; Prigent, H.; Clair, B.; Behin, A.; et al. Diaphragm sniff ultrasound: Normal values, relationship with sniff nasal pressure and accuracy for predicting respiratory involvement in patients with neuromuscular disorders. PLoS ONE 2019, 14, e0214288. [Google Scholar] [CrossRef] [PubMed]
- Fischmann, A.; Hafner, P.; Gloor, M.; Schmid, M.; Klein, A.; Pohlman, U.; Waltz, T.; Gonzalez, R.; Haas, T.; Bieri, O.; et al. Quantitative MRI and loss of free ambulation in Duchenne muscular dystrophy. J. Neurol. 2013, 260, 969–974. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Tzelepis, G.E.; Athanasopoulos, G.; Maounis, T.; Douskou, M.; Papavasiliou, A.; Cokkinos, D.V. Cardiac and Sternocleidomastoid Muscle Involvement in Duchenne Muscular Dystrophy. Chest 2005, 127, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Melissinos, C.G.; Bruce, E.N.; Goldman, M.D.; Elliott, E.; Mead, J. Pattern of diaphragmatic activity during forced expiratory vital capacity. J. Appl. Physiol. 1981, 51, 1515–1525. [Google Scholar] [CrossRef]
- Gandevia, S.C. Spinal and Supraspinal Factors in Human Muscle Fatigue. Physiol. Rev. 2001, 81, 1725–1789. [Google Scholar] [CrossRef]
- Pennati, F.; Quirk, J.D.; Yablonskiy, D.A.; Castro, M.; Aliverti, A.; Woods, J.C. Assessment of regional lung function with multivolume 1H MR imaging in health and obstructive lung disease: Comparison with 3He MR imaging. Radiology 2014, 273, 580–590. [Google Scholar] [CrossRef][Green Version]
- Bauman, G.; Bieri, O. Matrix pencil decomposition of time-resolved proton MRI for robust and improved assessment of pulmonary ventilation and perfusion. Magn. Reson. Med. 2017, 77, 336–342. [Google Scholar] [CrossRef]
- Bach, J.R.; Martínez-González, D. Duchenne Muscular Dystrophy: Continuous Noninvasive Ventilatory Support Prolongs Survival. Respir. Care 2011, 56, 744–750. [Google Scholar] [CrossRef]
- Gomez-Merino, E.; Bach, J.R. Duchenne Muscular Dystrophy. Am. J. Phys. Med. Rehabil. 2002, 81, 411–415. [Google Scholar] [CrossRef]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Molgat-Seon, Y.; Hannan, L.M.; Dominelli, P.B.; Peters, C.M.; Fougere, R.J.; McKim, D.A.; Sheel, A.W.; Road, J.D. Lung volume recruitment acutely increases respiratory system compliance in individuals with severe respiratory muscle weakness. ERJ Open Res. 2017, 3, 135–2016. [Google Scholar] [CrossRef] [PubMed]
- McKim, D.A.; Katz, S.; Barrowman, N.; Ni, A.; LeBlanc, C. Lung Volume Recruitment Slows Pulmonary Function Decline in Duchenne Muscular Dystrophy. Arch. Phys. Med. Rehabil. 2012, 93, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.L.; Monsour, A.; Su, S.; Hoey, L.; McKim, D.; Barrowman, N. Long-Term Effects of Lung Volume Recruitment on Maximal Inspiratory Capacity and Vital Capacity in Duchenne Muscular Dystrophy. Ann. Am. Thorac. Soc. 2015, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Wanke, T.; Toifl, K.; Merkle, M.; Formanek, D.; Lahrmann, H.; Zwick, H. Inspiratory Muscle Training in Patients with Duchenne Muscular Dystrophy. Chest 1994, 105, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Gozal, D.; Thiriet, P. Respiratory muscle training in neuromuscular disease: Long-term effects on strength and load perception. Med. Sci. Sports Exerc. 1999, 31, 1522. [Google Scholar] [CrossRef] [PubMed]
- Winkler, G.; Zifko, U.; Nader, A.; Frank, W.; Zwick, H.; Toifl, K.; Wanke, T. Dose-dependent effects of inspiratory muscle training in neuromuscular disorders. Muscle Nerve 2000, 23, 1257–1260. [Google Scholar] [CrossRef]
- Senesac, C.R.; Barnard, A.M.; Lott, D.J.; Nair, K.S.; Harrington, A.T.; Willcocks, R.J.; Zilke, K.L.; Rooney, W.D.; Walter, G.A.; Vandenborne, K. Magnetic Resonance Imaging Studies in Duchenne Muscular Dystrophy: Linking Findings to the Physical Therapy Clinic. Phys. Ther. 2020, 100, 2035–2048. [Google Scholar] [CrossRef]
- Szeinberg, A.; Tabachnik, E.; Rashed, N.; McLaughlin, F.J.; England, S.; Bryan, C.A.; Levison, H. Cough Capacity in Patients with Muscular Dystrophy. Chest 1988, 94, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.R.; Ishikawa, Y.; Kim, H. Prevention of Pulmonary Morbidity for Patients with Duchenne Muscular Dystrophy. Chest 1997, 112, 1024–1028. [Google Scholar] [CrossRef]
- Kravitz, R.M. Airway Clearance in Duchenne Muscular Dystrophy. Pediatrics 2009, 123, S231–S235. [Google Scholar] [CrossRef]
- Chatwin, M.; Toussaint, M.; Gonçalves, M.; Sheers, N.; Mellies, U.; Gonzales-Bermejo, J.; Sancho, J.; Fauroux, B.; Andersen, T.; Hov, B.; et al. Airway clearance techniques in neuromuscular disorders: A state of the art review. Respir. Med. 2018, 136, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.R.; Bianchi, C.; Vidigal-Lopes, M.; Turi, S.; Felisari, G. Lung Inflation by Glossopharyngeal Breathing and “Air Stacking” in Duchenne Muscular Dystrophy. Am. J. Phys. Med. Rehabil. 2007, 86, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Bach, J.R.; Komaroff, E.; Miura, T.; Jackson-Parekh, R. Cough Augmentation in Duchenne Muscular Dystrophy. Am. J. Phys. Med. Rehabil. 2008, 87, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-W.; Kang, Y.-S.; Sohn, H.-S.; Park, J.H.; Moon, J.-H. Respiratory Muscle Strength and Cough Capacity in Patients with Duchenne Muscular Dystrophy. Yonsei Med. J. 2006, 47, 184–190. [Google Scholar] [CrossRef]
- Ward, S.; Chatwin, M.; Heather, S.; Simonds, A.K. Randomised controlled trial of non-invasive ventilation (NIV) for nocturnal hypoventilation in neuromuscular and chest wall disease patients with daytime normocapnia. Thorax 2005, 60, 1019–1024. [Google Scholar] [CrossRef]
- Toussaint, M.; Chatwin, M.; Soudon, P. Review Article: Mechanical ventilation in Duchenne patients with chronic respiratory insufficiency: Clinical implications of 20 years published experience. Chronic Respir. Dis. 2007, 4, 167–177. [Google Scholar] [CrossRef]
- Bach, J.R.; Mahajan, K.; Lipa, B.; Saporito, L.; Goncalves, M.; Komaroff, E. Lung Insufflation Capacity in Neuromuscular Disease. Am. J. Phys. Med. Rehabil. 2008, 87, 720–725. [Google Scholar] [CrossRef]
- Koga, T.; Watanabe, K.; Sano, M.; Ishikawa, Y.; Bach, J.R. Breathing Intolerance Index. Am. J. Phys. Med. Rehabil. 2006, 85, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Lofaso, F.; Orlikowski, D.; Raphael, J.-C. Ventilatory assistance in patients with Duchenne muscular dystrophy. Eur. Respir. J. 2006, 28, 468–469. [Google Scholar] [CrossRef]
- Rodger, S.; Woods, K.; Bladen, C.L.; Stringer, A.; Vry, J.; Gramsch, K.; Kirschner, J.; Thompson, R.; Bushby, K.; Lochmüller, H. Adult care for Duchenne muscular dystrophy in the UK. J. Neurol. 2015, 262, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, M.; Steens, M.; Wasteels, G.; Soudon, P. Diurnal ventilation via mouthpiece: Survival in end-stage Duchenne patients. Eur. Respir. J. 2006, 28, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.R.; Gonçalves, M.; Hon, A.; Ishikawa, Y.; De Vito, E.L.; Prado, F.; Dominguez, M.E. Changing Trends in the Management of End-Stage Neuromuscular Respiratory Muscle Failure. Am. J. Phys. Med. Rehabil. 2013, 92, 267–277. [Google Scholar] [CrossRef] [PubMed]
- McKim, A.D.; Griller, N.; Leblanc, C.; Woolnough, A.; King, J. Twenty-Four Hour Noninvasive Ventilation in Duchenne Muscular Dystrophy: A Safe Alternative to Tracheostomy. Can. Respir. J. 2013, 20, e5–e9. [Google Scholar] [CrossRef]
- Fayssoil, A.; Chaffaut, C.; Prigent, H.; Laforet, P.; Clair, B.; Orlikowski, D.; Ogna, A.; Chevret, S.; Meng, P.; Annane, D.; et al. Nutritional status, swallowing disorders, and respiratory prognosis in adult Duchenne muscular dystrophy patients. Pediatr. Pulmonol. 2021, 56, 2146–2154. [Google Scholar] [CrossRef]
- Lee, J.W.; Oh, H.J.; Choi, W.A.; Kim, D.J.; Kang, S.-W. Relationship between Eating and Digestive Symptoms and Respiratory Function in Advanced Duchenne Muscular Dystrophy Patients. J. Neuromuscul. Dis. 2020, 7, 101–107. [Google Scholar] [CrossRef]
- Arora, N.S.; Rochester, D.F. Effect of body weight and muscularity on human diaphragm muscle mass, thickness, and area. J. Appl. Physiol. 1982, 52, 64–70. [Google Scholar] [CrossRef]
- Terzi, N.; Orlikowski, D.; Aegerter, P.; Lejaille, M.; Ruquet, M.; Zalcman, G.; Fermanian, C.; Raphael, J.-C.; Lofaso, F. Breathing–Swallowing Interaction in Neuromuscular Patients. Am. J. Respir. Crit. Care Med. 2007, 175, 269–276. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, 4, CD003725. [Google Scholar] [CrossRef]
- Henricson, E.K.; Ms, R.T.A.; Cnaan, A.; Hu, F.; Duong, T.; Ms, A.A.; Han, J.; Escolar, D.M.; Dpt, J.M.F.; Clemens, P.R.; et al. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and othe. Muscle Nerve 2013, 48, 55–67. [Google Scholar] [CrossRef]
- Buyse, G.M.; Goemans, N.; Hauwe, M.V.D.; Meier, T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr. Pulmonol. 2013, 48, 912–920. [Google Scholar] [CrossRef]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.-M.; Finkel, R.S.; Goemans, N.; Rummey, C.; et al. Treatment effect of idebenone on inspiratory function in patients with Duchenne muscular dystrophy. Pediatr. Pulmonol. 2016, 52, 508–515. [Google Scholar] [CrossRef]
- Servais, L.; Straathof, C.S.; Schara, U.; Klein, A.; Leinonen, M.; Hasham, S.; Meier, T.; De Waele, L.; Gordish-Dressman, H.; McDonald, C.M.; et al. Long-term data with idebenone on respiratory function outcomes in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2020, 30, 5–16. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 10 July 2021).
- Ebrahimi-Fakhari, D.; Dillmann, U.; Flotats-Bastardas, M.; Poryo, M.; Abdul-Khaliq, H.; Shamdeen, M.G.; Mischo, B.; Zemlin, M.; Meyer, S. Off-Label Use of Ataluren in Four Non-ambulatory Patients with Duchenne Muscular Dystrophy: Effects on Cardiac and Pulmonary Function and Muscle Strength. Front. Pediatr. 2018, 6, 316. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F.; Osorio, A.N.; Tulinius, M.; Buccella, F.; Morgenroth, L.P.; Gordish-Dressman, H.; Jiang, J.; Trifillis, P.; Zhu, J.; et al. Safety and effectiveness of ataluren: Comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J. Comp. Eff. Res. 2020, 9, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Kinane, T.B.; Mayer, O.H.; Duda, P.W.; Lowes, L.P.; Moody, S.L.; Mendell, J.R. Long-Term Pulmonary Function in Duchenne Muscular Dystrophy: Comparison of Eteplirsen-Treated Patients to Natural History. J. Neuromuscul. Dis. 2018, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Eliopoulos, H.; Han, L.; Kinane, T.B.; Lowes, L.P.; Mendell, J.R.; Gordish-Dressman, H.; Henricson, E.K.; McDonald, C.M. Eteplirsen Treatment Attenuates Respiratory Decline in Ambulatory and Non-Ambulatory Patients with Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2019, 6, 213–225. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Shieh, P.B.; Abdel-Hamid, H.Z.; Connolly, A.M.; Ciafaloni, E.; Wagner, K.R.; Goemans, N.; Mercuri, E.; Khan, N.; Koenig, E.; et al. Open-Label Evaluation of Eteplirsen in Patients with Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping: PROMOVI Trial. J. Neuromuscul. Dis. 2021, 1–13, in press. [Google Scholar] [CrossRef]
- Orde, S.R.; Boon, A.J.; Firth, D.G.; Villarraga, H.R.; Sekiguchi, H. Diaphragm assessment by two dimensional speckle tracking imaging in normal subjects. BMC Anesthesiol. 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Lacourpaille, L.; Nordez, A.; Hug, F.; Doguet, V.; Andrade, R.J.; Guilhem, G. Early detection of exercise-induced muscle damage using elastography. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 117, 2047–2056. [Google Scholar] [CrossRef]
- Pichiecchio, A.; Alessandrino, F.; Bortolotto, C.; Cerica, A.; Rosti, C.; Raciti, M.V.; Rossi, M.; Berardinelli, A.; Baranello, G.; Bastianello, S.; et al. Muscle ultrasound elastography and MRI in preschool children with Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 476–483. [Google Scholar] [CrossRef]
- Dres, M.; Dubé, B.-P.; Goligher, E.; Vorona, S.; Demiri, S.; Morawiec, E.; Mayaux, J.; Brochard, L.; Similowski, T.; Demoule, A. Usefulness of Parasternal Intercostal Muscle Ultrasound during Weaning from Mechanical Ventilation. Anesthesiology 2020, 132, 1114–1125. [Google Scholar] [CrossRef]
- Cala, S.J.; Kenyon, C.M.; Lee, A.; Watkin, K.; Macklem, P.T.; Rochester, D.F. Respiratory Ultrasonography of Human Parasternal Intercostal Muscle In Vivo. Ultrasound Med. Biol. 1998, 24, 313–326. [Google Scholar] [CrossRef]
- Tahan, N.; Khademi-Kalantari, K.; Mohseni-Bandpei, M.A.; Mikaili, S.; Baghban, A.A.; Jaberzadeh, S. Measurement of superficial and deep abdominal muscle thickness: An ultrasonography study. J. Physiol. Anthr. 2016, 35, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.C.; Walter, G.A.; Rooney, W.D.; Wang, D.-J.; Devos, S.; Pollaro, J.; Triplett, W.; Lott, D.J.; Willcocks, R.J.; Senesac, C.; et al. Skeletal Muscles of Ambulant Children with Duchenne Muscular Dystrophy: Validation of Multicenter Study of Evaluation with MR Imaging and MR Spectroscopy. Radiology 2013, 269, 198–207. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennati, F.; LoMauro, A.; D’Angelo, M.G.; Aliverti, A. Non-Invasive Respiratory Assessment in Duchenne Muscular Dystrophy: From Clinical Research to Outcome Measures. Life 2021, 11, 947. https://doi.org/10.3390/life11090947
Pennati F, LoMauro A, D’Angelo MG, Aliverti A. Non-Invasive Respiratory Assessment in Duchenne Muscular Dystrophy: From Clinical Research to Outcome Measures. Life. 2021; 11(9):947. https://doi.org/10.3390/life11090947
Chicago/Turabian StylePennati, Francesca, Antonella LoMauro, Maria Grazia D’Angelo, and Andrea Aliverti. 2021. "Non-Invasive Respiratory Assessment in Duchenne Muscular Dystrophy: From Clinical Research to Outcome Measures" Life 11, no. 9: 947. https://doi.org/10.3390/life11090947
APA StylePennati, F., LoMauro, A., D’Angelo, M. G., & Aliverti, A. (2021). Non-Invasive Respiratory Assessment in Duchenne Muscular Dystrophy: From Clinical Research to Outcome Measures. Life, 11(9), 947. https://doi.org/10.3390/life11090947