
A Conceptual Framework for Integrating Cellular Protein Folding, Misfolding and Aggregation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Relationship between Protein Folding and Aggregation
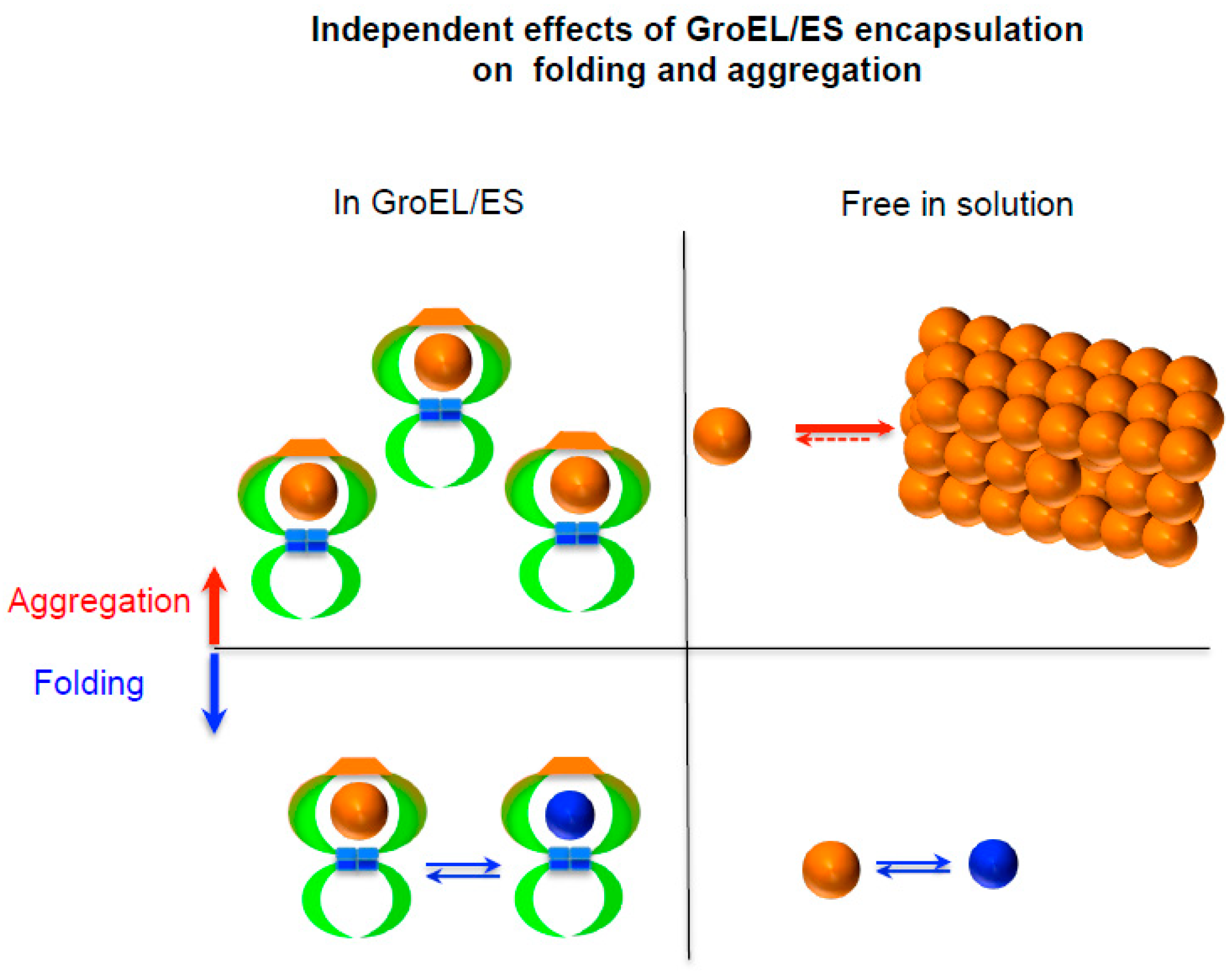
2.1. Independency between Protein Folding and Aggregation
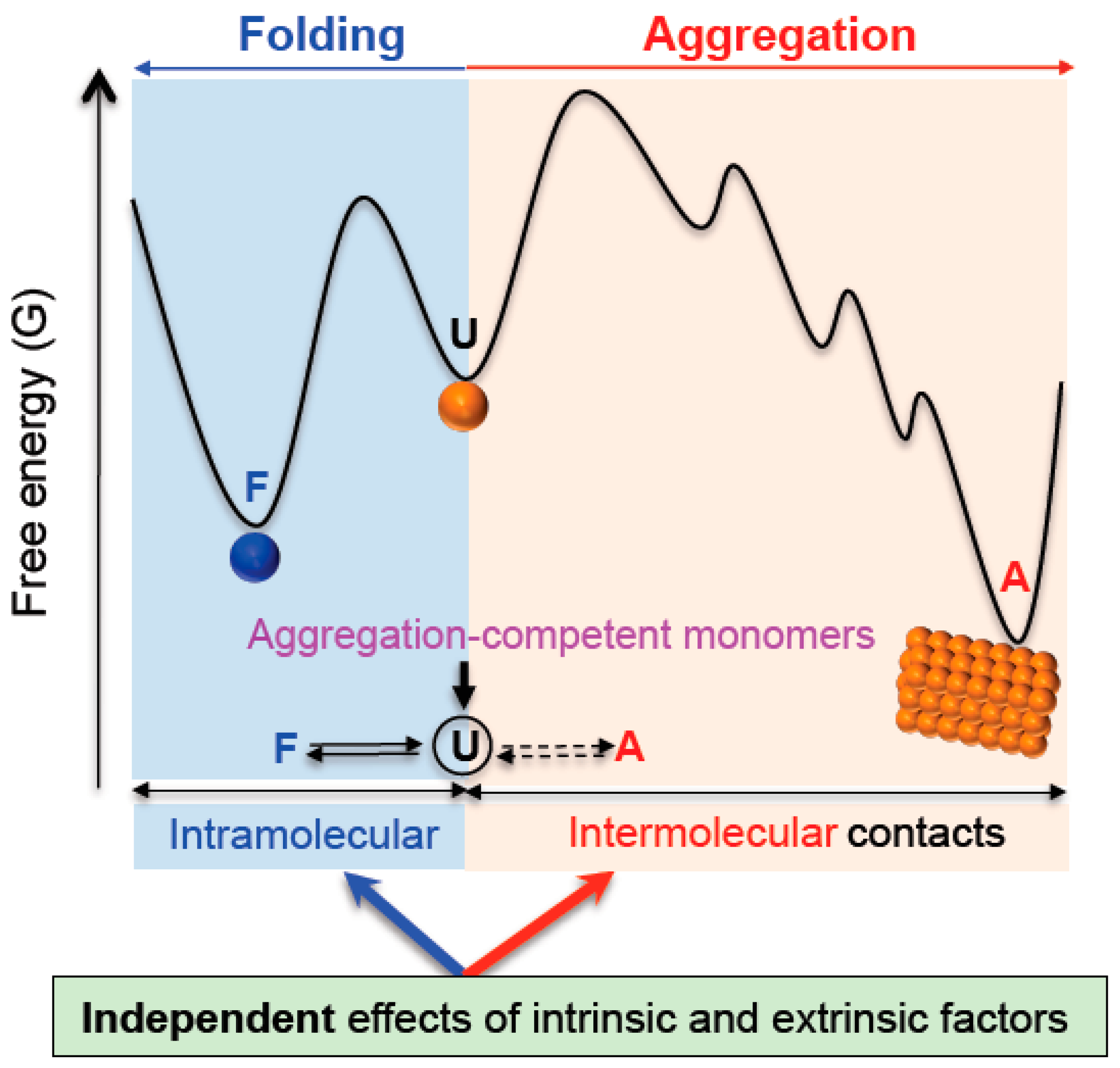
2.2. A Conceptual Framework for Integrating Protein Folding, Misfolding and Aggregation
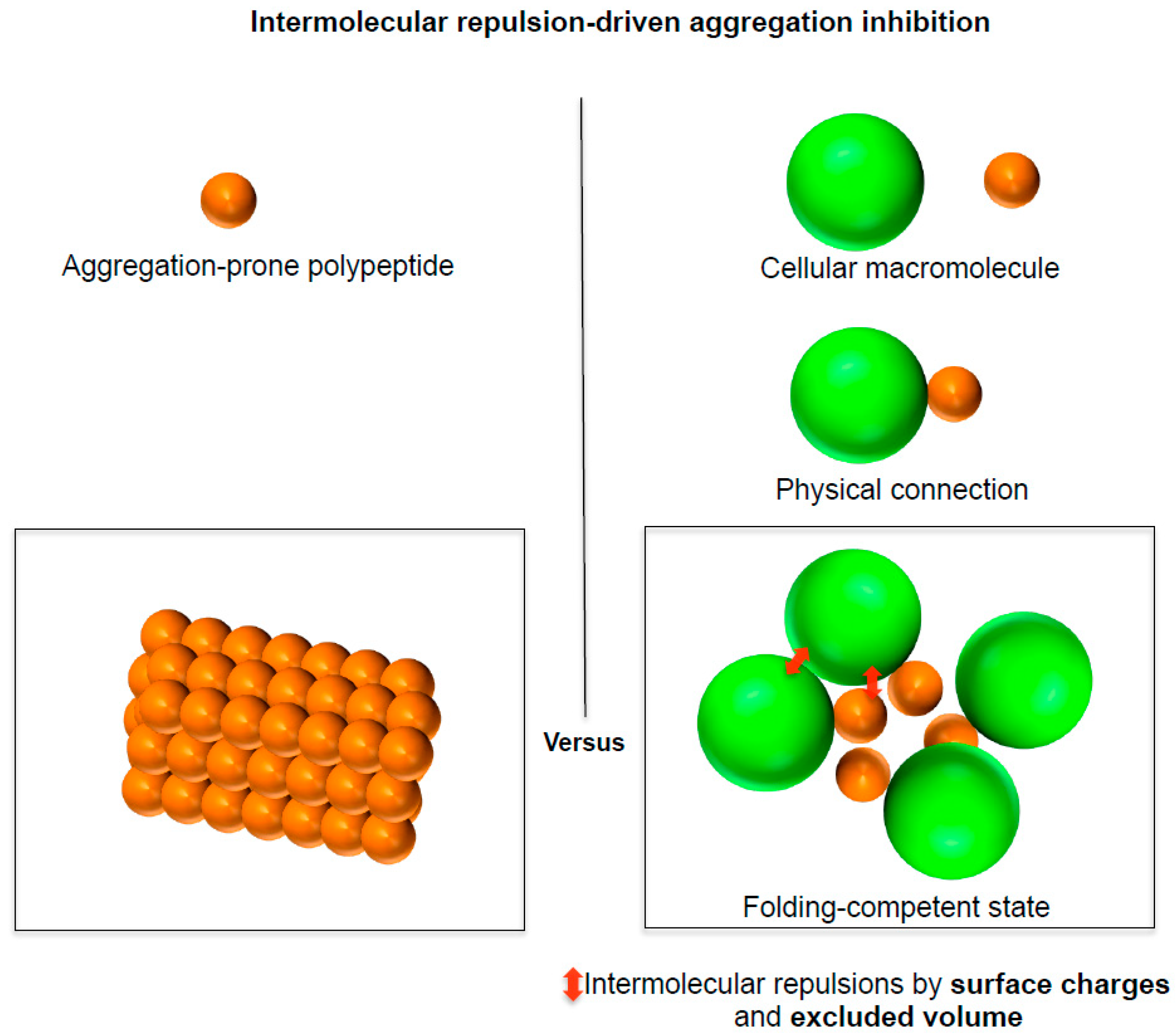
3. A Social Distancing Measure Underlying the Generic Intrinsic Chaperone Activity of Cellular Macromolecules
3.1. A Social Distancing Measure by Chaperones
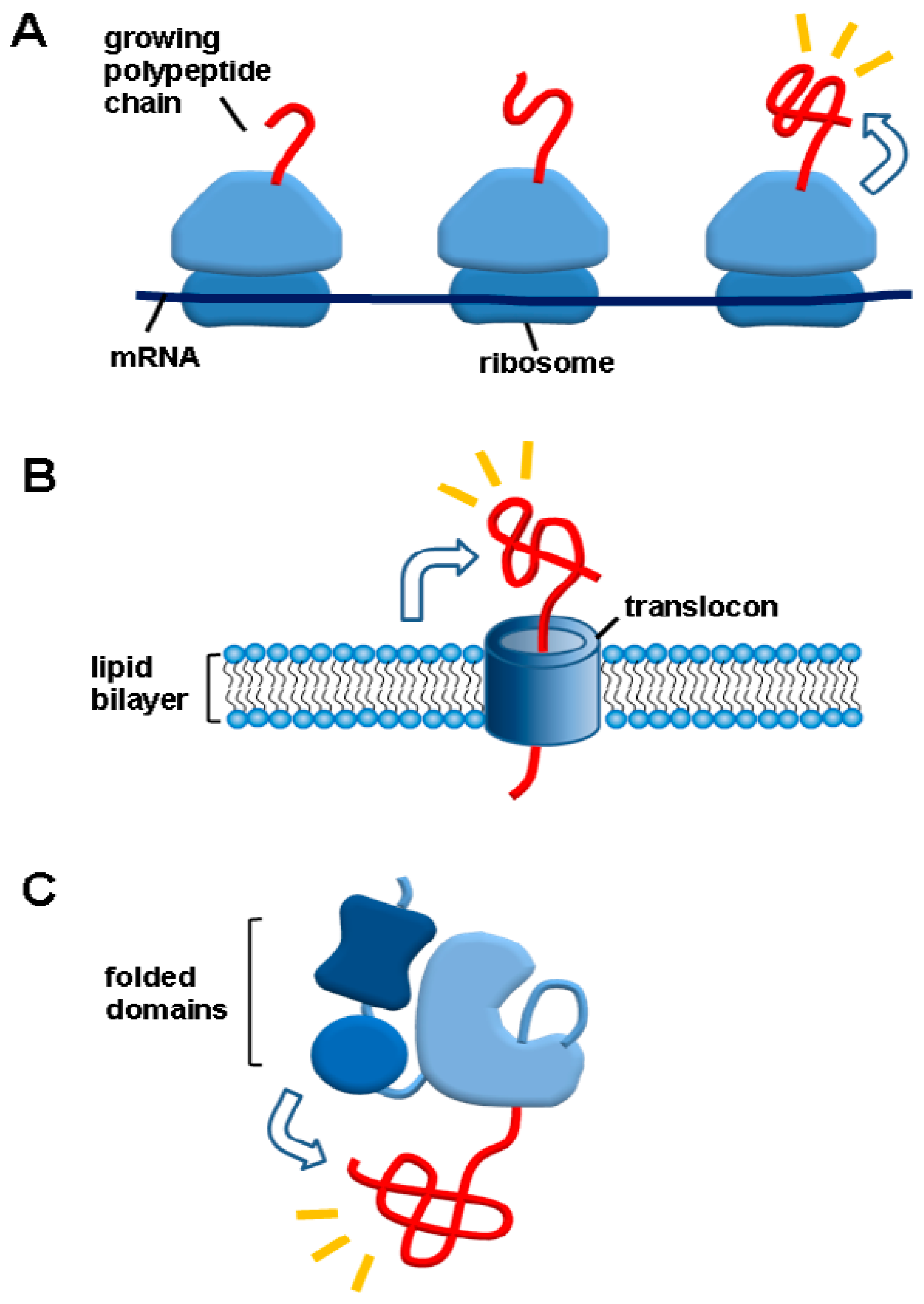
3.2. A Social Distancing Measure by Macromolecular Tethering: A Hallmark of De Novo Protein Folding Environments
3.3. Conversion of a Protein into a Potent Chaperone: Cis/Trans Conversion
3.4. Implications of a Social Distancing Measure for the Development of Therapeutic Strategies for Aggregation-Associated Diseases
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clark, P.L. Protein folding in the cell: Reshaping the folding funnel. Trends Biochem. Sci. 2004, 29, 527–534. [Google Scholar] [CrossRef]
- Jahn, T.R.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008, 469, 100–117. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Seong, B.L. A social distancing measure governing the whole proteome. Curr. Opin. Struct. Biol. 2021, 66, 104–111. [Google Scholar] [CrossRef]
- Lyubarev, A.E.; Kurganov, B.I. Modeling of irreversible thermal protein denaturation at varying temperature. II. The complete kinetic model of Lumry and Eyring. Biochemistry 1999, 64, 832–838. [Google Scholar] [PubMed]
- Valastyan, J.S.; Lindquist, S. Mechanisms of protein-folding diseases at a glance. Dis. Model Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Choi, S.I. A simple principle for understanding the combined cellular protein folding and aggregation. Curr. Protein Pept. Sci. 2020, 21, 3–21. [Google Scholar] [CrossRef]
- Chiti, F.; Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Dobson, C.M. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. USA 2002, 99, 16419–16426. [Google Scholar] [CrossRef]
- Zhou, H.X.; Pang, X. Electrostatic interactions in protein structure, folding, binding, and condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Kurnik, M.; Hedberg, L.; Danielsson, J.; Oliveberg, M. Folding without charges. Proc. Natl. Acad. Sci. USA 2012, 109, 5705–5710. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Phillips, K.J.; Liu, D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 2007, 129, 10110–10112. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Han, K.S.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Choi, S.I.; Seong, B.L. N-terminal domains of native multidomain proteins have the potential to assist de novo folding of their downstream domains in vivo by acting as solubility enhancers. Protein Sci. 2007, 16, 635–643. [Google Scholar] [CrossRef]
- Ryu, K.; Kim, C.W.; Kim, B.H.; Han, K.S.; Kim, K.H.; Choi, S.I.; Seong, B.L. Assessment of substrate-stabilizing factors for DnaK on the folding of aggregation-prone proteins. Biochem. Biophys. Res. Commun. 2008, 373, 74–79. [Google Scholar] [CrossRef]
- Choi, S.I.; Han, K.S.; Kim, C.W.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Kim, S.I.; Kang, T.H.; Shin, H.C.; Lim, K.H.; et al. Protein solubility and folding enhancement by interaction with RNA. PLoS ONE 2008, 3, e2677. [Google Scholar] [CrossRef]
- Kwon, S.B.; Ryu, K.; Son, A.; Jeong, H.; Lim, K.H.; Kim, K.H.; Seong, B.L.; Choi, S.I. Conversion of a soluble protein into a potent chaperone in vivo. Sci. Rep. 2019, 9, 2735. [Google Scholar] [CrossRef] [PubMed]
- Wennerstrom, H.; Vallina Estrada, E.; Danielsson, J.; Oliveberg, M. Colloidal stability of the living cell. Proc. Natl. Acad. Sci. USA 2020, 117, 10113–10121. [Google Scholar] [CrossRef]
- Fenton, W.A.; Kashi, Y.; Furtak, K.; Horwich, A.L. Residues in chaperonin GroEL required for polypeptide binding and release. Nature 1994, 371, 614–619. [Google Scholar] [CrossRef]
- Rüdiger, S.; Germeroth, L.; Schneider-Mergener, J.; Bukau, B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997, 16, 1501–1507. [Google Scholar] [CrossRef]
- Pappenberger, G.; Wilsher, J.A.; Roe, S.M.; Counsell, D.J.; Willison, K.R.; Pearl, L.H. Crystal structure of the CCTgamma apical domain: Implications for substrate binding to the eukaryotic cytosolic chaperonin. J. Mol. Biol. 2002, 318, 1367–1379. [Google Scholar] [CrossRef]
- Koldewey, P.; Stull, F.; Horowitz, S.; Martin, R.; Bardwell, J.C.A. Forces driving chaperone action. Cell 2016, 166, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Choi, S.I.; Kwon, S.; Son, A.; Jeong, H.; Kim, K.H.; Seong, B.L. Protein folding in vivo revisited. Curr. Protein Pept. Sci. 2013, 14, 721–733. [Google Scholar] [CrossRef]
- Olsen, S.N.; Andersen, K.B.; Randolph, T.W.; Carpenter, J.F.; Westh, P. Role of electrostatic repulsion on colloidal stability of Bacillus halmapalus alpha-amylase. Biochim. Biophys. Acta. 2009, 1794, 1058–1065. [Google Scholar] [CrossRef]
- Ortega-Vinuesa, J.L.; Martín-Rodríguez, A.; Hidalgo-Álvarez, R. Colloidal stability of polymer colloids with different interfacial properties: Mechanisms. J. Colloid Interface Sci. 1996, 184, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Taketomi, H.; Ueda, Y.; Gō, N. Studies on protein folding, unfolding and fluctuations by computer simulation. I. The effect of specific amino acid sequence represented by specific inter-unit interactions. Int. J. Pept. Protein Res. 1975, 7, 445–459. [Google Scholar] [CrossRef]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins 1995, 21, 167–195. [Google Scholar] [CrossRef]
- Best, R.B.; Hummer, G.; Eaton, W.A. Native contacts determine protein folding mechanisms in atomistic simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 17874–17879. [Google Scholar] [CrossRef]
- Rose, G.D. Protein folding—seeing is deceiving. Protein Sci. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Gupta, A.J.; Haldar, S.; Miličić, G.; Hartl, F.U.; Hayer-Hartl, M. Active cage mechanism of chaperonin-assisted protein folding demonstrated at single-molecule level. J. Mol. Biol. 2014, 426, 2739–2754. [Google Scholar] [CrossRef] [PubMed]
- Korobko, I.; Mazal, H.; Haran, G.; Horovitz, A. Measuring protein stability in the GroEL chaperonin cage reveals massive destabilization. ELife 2020, 9, e56511. [Google Scholar] [CrossRef] [PubMed]
- Apetri, A.C.; Horwich, A.L. Chaperonin chamber accelerates protein folding through passive action of preventing aggregation. Proc. Natl. Acad. Sci. USA 2008, 105, 17351–17355. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Molecular chaperones: Inside and outside the Anfinsen cage. Curr. Biol. 2001, 11, R1038–R1040. [Google Scholar] [CrossRef][Green Version]
- Cohen, S.I.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Adamcik, J.; Mezzenga, R. Amyloid polymorphism in the protein folding and aggregation energy landscape. Angew. Chem. Int. Ed. Engl. 2018, 57, 8370–8382. [Google Scholar] [CrossRef]
- Libich, D.S.; Tugarinov, V.; Clore, G.M. Intrinsic unfoldase/foldase activity of the chaperonin GroEL directly demonstrated using multinuclear relaxation-based NMR. Proc. Natl. Acad. Sci. USA 2015, 112, 8817–8823. [Google Scholar] [CrossRef]
- Singh, A.K.; Balchin, D.; Imamoglu, R.; Hayer-Hartl, M.; Hartl, F.U. Efficient catalysis of protein folding by GroEL/ES of the obligate chaperonin substrate MetF. J. Mol. Biol. 2020, 432, 2304–2318. [Google Scholar] [CrossRef]
- Imamoglu, R.; Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. Bacterial Hsp70 resolves misfolded states and accelerates productive folding of a multi-domain protein. Nat. Commun. 2020, 11, 365. [Google Scholar] [CrossRef]
- Agashe, V.R.; Guha, S.; Chang, H.C.; Genevaux, P.; Hayer-Hartl, M.; Stemp, M.; Georgopoulos, C.; Hartl, F.U.; Barral, J.M. Function of trigger factor and DnaK in multidomain protein folding: Increase in yield at the expense of folding speed. Cell 2004, 117, 199–209. [Google Scholar] [CrossRef]
- Horwich, A.L.; Apetri, A.C.; Fenton, W.A. The GroEL/GroES cis cavity as a passive anti-aggregation device. FEBS Lett. 2009, 583, 2654–2662. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.P.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Trigger factor slows co-translational folding through kinetic trapping while sterically protecting the nascent chain from aberrant cytosolic interactions. J. Am. Chem. Soc. 2012, 134, 10920–10932. [Google Scholar] [CrossRef]
- Marchenko, N.Y.; Marchenkov, V.V.; Semisotnov, G.V.; Finkelstein, A.V. Strict experimental evidence that apo-chaperonin GroEL does not accelerate protein folding, although it does accelerate one of its steps. Proc. Natl. Acad. Sci. USA 2015, 112, E6831–E6832. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Rossi, P.; Saio, T.; Kalodimos, C.G. Structural basis for the antifolding activity of a molecular chaperone. Nature 2016, 537, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Stull, F.; Koldewey, P.; Humes, J.R.; Radford, S.E.; Bardwell, J.C.A. Substrate protein folds while it is bound to the ATP- independent chaperone Spy. Nat. Struct. Mol. Biol. 2016, 23, 53–58. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Marinelli, P.; Navarro, S.; Baño-Polo, M.; Morel, B.; Graña-Montes, R.; Sabe, A.; Canals, F.; Fernandez, M.R.; Conejero-Lara, F.; Ventura, S. Global Protein Stabilization Does Not Suffice to Prevent Amyloid Fibril Formation. ACS Chem. Biol. 2018, 13, 2094–2105. [Google Scholar] [CrossRef]
- Gazit, E. The “Correctly Folded” state of proteins: Is it a metastable state? Angew. Chem. Int. Ed. Engl. 2002, 41, 257–259. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Thirumalai, D.; Reddy, G. Protein thermodynamics: Are native proteins metastable? Nat. Chem. 2011, 3, 910–911. [Google Scholar] [CrossRef]
- Varela, A.E.; Lang, J.F.; Wu, Y.; Dalphin, M.D.; Stangl, A.J.; Okuno, Y.; Cavagnero, S. Kinetic trapping of folded proteins relative to aggregates under physiologically relevant conditions. J. Phys. Chem. B. 2018, 122, 7682–7698. [Google Scholar] [CrossRef]
- Ciryam, P.; Tartaglia, G.G.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013, 5, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B.; Haber, E. Studies on the reduction and re-formation of protein disulfide bonds. J. Biol. Chem. 1961, 236, 1361–1363. [Google Scholar] [CrossRef]
- Zhou, A.Q.; O’Hern, C.S.; Regan, L. Revisiting the Ramachandranplot from a new angle. Protein Sci. 2011, 20, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Radley, T.L.; Markowska, A.I.; Bettinger, B.T.; Ha, J.H.; Loh, S.N. Allosteric switching by mutually exclusive folding of protein domains. J. Mol. Biol. 2003, 332, 529–536. [Google Scholar] [CrossRef]
- Zhou, H.X.; Rivas, G.; Minton, A.P. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008, 37, 375–397. [Google Scholar] [CrossRef]
- Honig, B.H.; Hubbell, W.L. Stability of “salt bridges” in membrane proteins. Proc. Natl. Acad. Sci. USA 1984, 81, 5412–5416. [Google Scholar] [CrossRef]
- Honig, B.H.; Hubbell, W.L.; Flewelling, R.F. Electrostatic interactions in membranes and proteins. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 163–193. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.L.; Grimsley, G.R.; Yakovlev, G.I.; Makarov, A.A.; Pace, C.N. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Sci. 2001, 10, 1206–1215. [Google Scholar] [CrossRef]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, E.; Nordlund, A.; Andersen, P.M.; Marklund, S.S.; Oliveberg, M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J. Biol. Chem. 2007, 282, 21230–21236. [Google Scholar] [CrossRef]
- Speed, M.A.; Wang, D.I.; King, J. Specific aggregation of partially folded polypeptide chains: The molecular basis of inclusion body composition. Nat. Biotechnol. 1996, 14, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Bravo, R.; Espargaro, A.; Sisquella, X.; Aviles, F.X.; Fernandez- Busquets, X.; Ventura, S. Inclusion bodies: Specificity in their aggregation process and amyloid-like structure. Biochim. Biophys. Acta. 2008, 1783, 1815–1825. [Google Scholar] [CrossRef]
- Mu, X.; Choi, S.; Lang, L.; Mowray, D.; Dokholyan, N.V.; Danielsson, J.; Oliveberg, M. Physicochemical code for quinary protein interactions in Escherichia coli. Proc. Natl. Acad. Sci. USA 2017, 114, E4556–E4563. [Google Scholar] [CrossRef]
- De Los Rios, P.; Ben-Zvi, A.; Slutsky, O.; Azem, A.; Goloubinoff, P. Hsp70 chaperones accelerate protein translocation and the unfolding of stable protein aggregates by entropic pulling. Proc. Natl. Acad. Sci. USA 2006, 103, 6166–6171. [Google Scholar] [CrossRef]
- Kellner, R.; Hofmann, H.; Barducci, A.; Wunderlich, B.; Nettels, D.; Schuler, B. Single-molecule spectroscopy reveals chaperone-mediated expansion of substrate protein. Proc. Natl. Acad. Sci. USA 2014, 111, 13355–13360. [Google Scholar] [CrossRef] [PubMed]
- Wayne, N.; Bolon, D.N. Charge-rich regions modulate the anti- aggregation activity of Hsp90. J. Mol. Biol. 2010, 401, 931–939. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Chothia, C.; Gough, J.; Vogel, C.; Teichmann, S.A. Evolution of the protein repertoire. Science 2003, 300, 1701–1703. [Google Scholar] [CrossRef]
- Ellis, R.J.; Hartl, F.U. Principles of protein folding in the cellular environment. Curr. Opin. Struct. Biol. 1999, 9, 102–110. [Google Scholar] [CrossRef]
- Maier, T.; Ferbitz, L.; Deuerling, E.; Ban, N. A cradle for new proteins: Trigger factor at the ribosome. Curr. Opin. Struct. Biol. 2005, 15, 204–212. [Google Scholar] [CrossRef]
- Schimmele, B.; Gräfe, N.; Plückthun, A. Ribosome display of mammalian receptor domains. Protein Eng. Des. Sel. 2005, 18, 285–294. [Google Scholar] [CrossRef]
- Sørensen, H.P.; Kristensen, J.E.; Sperling-Petersen, H.U.; Mortensen, K.K. Soluble expression of aggregating proteins by covalent coupling to the ribosome. Biochem. Biophys. Res. Commun. 2004, 319, 715–719. [Google Scholar] [CrossRef]
- Tillotson, B.J.; Cho, Y.K.; Shusta, E.V. Cells and cell lysates: A direct approach for engineering antibodies against membrane proteins using yeast surface display. Methods 2013, 60, 27–37. [Google Scholar] [CrossRef]
- Waugh, D.S. Making the most of affinity tags. Trends Biotechnol. 2005, 23, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, V.; Falcone, F.H. Polyionic Tags as Enhancers of Protein Solubility in Recombinant Protein Expression. Microorganisms 2018, 6, 47. [Google Scholar] [CrossRef]
- Kaldmäe, M.; Leppert, A.; Chen, G.; Sarr, M.; Sahin, C.; Nordling, K.; Kronqvist, N.; Gonzalvo-Ulla, M.; Fritz, N.; Abelein, A.; et al. High intracellular stability of the spidroin N-terminal domain in spite of abundant amyloidogenic segments revealed by in-cell hydrogen/deuterium exchange mass spectrometry. FEBS J. 2020, 287, 2823–2833. [Google Scholar] [CrossRef]
- Butt, T.R.; Jonnalagadda, S.; Monia, B.P.; Sternberg, E.J.; Marsh, J.A.; Stadel, J.M.; Ecker, D.J.; Crooke, S.T. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 2540–2544. [Google Scholar] [CrossRef] [PubMed]
- Butt, T.R.; Edavettal, S.C.; Hall, J.P.; Mattern, M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 2005, 43, 1–9. [Google Scholar] [CrossRef]
- Santner, A.A.; Croy, C.H.; Vasanwala, F.H.; Uversky, V.N.; Van, Y.Y.; Dunker, A.K. Sweeping away protein aggregation with entropic bristles: Intrinsically disordered protein fusions enhance soluble expression. Biochemistry 2012, 51, 7250–7262. [Google Scholar] [CrossRef]
- Choi, S.I.; Ryu, K.; Seong, B.L. RNA-mediated chaperone type for de novo protein folding. RNA Biol. 2009, 6, 21–24. [Google Scholar] [CrossRef]
- Wu, K.; Stull, F.; Lee, C.; Bardwell, J.C.A. Protein folding while chaperone bound is dependent on weak interactions. Nat. Commun. 2019, 10, 4833. [Google Scholar] [CrossRef]
- Kyratsous, C.A.; Panagiotidis, C.A. Heat-shock protein fusion vectors for improved expression of soluble recombinant proteins in Escherichia coli. Methods Mol. Biol. 2012, 824, 109–129. [Google Scholar]
- Basters, A.; Ketscher, L.; Deuerling, E.; Arkona, C.; Rademann, J.; Knobeloch, K.P.; Fritz, G. High yield expression of catalytically active USP18 (UBP43) using a Trigger Factor fusion system. BMC Biotechnol. 2012, 12, 56. [Google Scholar] [CrossRef]
- Ruan, A.; Ren, C.; Quan, S. Conversion of the molecular chaperone Spy into a novel fusion tag to enhance recombinant protein expression. J. Biotechnol. 2020, 307, 131–138. [Google Scholar] [CrossRef]
- Kajino, T.; Ohto, C.; Muramatsu, M.; Obata, S.; Udaka, S.; Yamada, Y.; Takahashi, H. A protein disulfide isomerase gene fusion expression system that increases the extracellular productivity of Bacillus brevis. Appl. Environ. Microbiol. 2000, 66, 638–642. [Google Scholar] [CrossRef]
- Ideno, A.; Furutani, M.; Iwabuchi, T.; Iida, T.; Iba, Y.; Kurosawa, Y.; Sakuraba, H.; Ohshima, T.; Kawarabayashi, Y.; Maruyama, T. Expression of foreign proteins in Escherichia coli by fusing with an archaeal FK506 binding protein. Appl. Microbiol. Biotechnol. 2004, 64, 99–105. [Google Scholar] [CrossRef]
- Das, D.; Das, A.; Samanta, D.; Ghosh, J.; Dasgupta, S.; Bhattacharya, A.; Basu, A.; Sanyal, S.; Das Gupta, C. Role of the ribosome in protein folding. Biotechnol. J. 2008, 3, 999–1009. [Google Scholar] [CrossRef]
- Viennet, T.; Wördehoff, M.M.; Uluca, B.; Poojari, C.; Shaykhalishahi, H.; Willbold, D.; Strodel, B.; Heise, H.; Buell, A.K.; Hoyer, W.; et al. Structural insights from lipid-bilayer nanodiscs link α-Synuclein membrane-binding modes to amyloid fibril formation. Commun. Biol. 2018, 1, 44. [Google Scholar] [CrossRef]
- Guha, S.; Manna, T.K.; Das, K.P.; Bhattacharyya, B. Chaperone-like activity of tubulin. J. Biol. Chem. 1998, 273, 30077–30080. [Google Scholar] [CrossRef]
- Zarzecka, U.; Harrer, A.; Zawilak-Pawlik, A.; Skorko-Glonek, J.; Backert, S. Chaperone activity of serine protease HtrA of Helicobacter pylori as a crucial survival factor under stress conditions. Cell Commun. Signal. 2019, 17, 161. [Google Scholar] [CrossRef]
- Begeman, A.; Son, A.; Litberg, T.J.; Wroblewski, T.H.; Gehring, T.; Huizar Cabral, V.; Bourne, J.; Xuan, Z.; Horowitz, S. G-Quadruplexes act as sequence-dependent protein chaperones. EMBO Rep. 2020, 21, e49735. [Google Scholar] [CrossRef] [PubMed]
- Litberg, T.J.; Docter, B.; Hughes, M.P.; Bourne, J.; Horowitz, S. DNA facilitates oligomerization and prevents aggregation via DNA networks. Biophys. J. 2020, 118, 162–171. [Google Scholar] [CrossRef]
- Xie, L.; Jakob, U. Inorganic polyphosphate, a multifunctional polyanionic protein scaffold. J. Biol. Chem. 2019, 294, 2180–2190. [Google Scholar] [CrossRef] [PubMed]
- Gestwicki, J.E.; Crabtree, G.R.; Graef, I.A. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science 2004, 306, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Southwell, A.L.; Patterson, P.H. Antibody therapy in neurodegenerative disease. Rev. Neurosci. 2010, 21, 273–287. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.I.; Seong, B.L. A Conceptual Framework for Integrating Cellular Protein Folding, Misfolding and Aggregation. Life 2021, 11, 605. https://doi.org/10.3390/life11070605
Choi SI, Seong BL. A Conceptual Framework for Integrating Cellular Protein Folding, Misfolding and Aggregation. Life. 2021; 11(7):605. https://doi.org/10.3390/life11070605
Chicago/Turabian StyleChoi, Seong Il, and Baik L. Seong. 2021. "A Conceptual Framework for Integrating Cellular Protein Folding, Misfolding and Aggregation" Life 11, no. 7: 605. https://doi.org/10.3390/life11070605
APA StyleChoi, S. I., & Seong, B. L. (2021). A Conceptual Framework for Integrating Cellular Protein Folding, Misfolding and Aggregation. Life, 11(7), 605. https://doi.org/10.3390/life11070605