
BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML?




Abstract
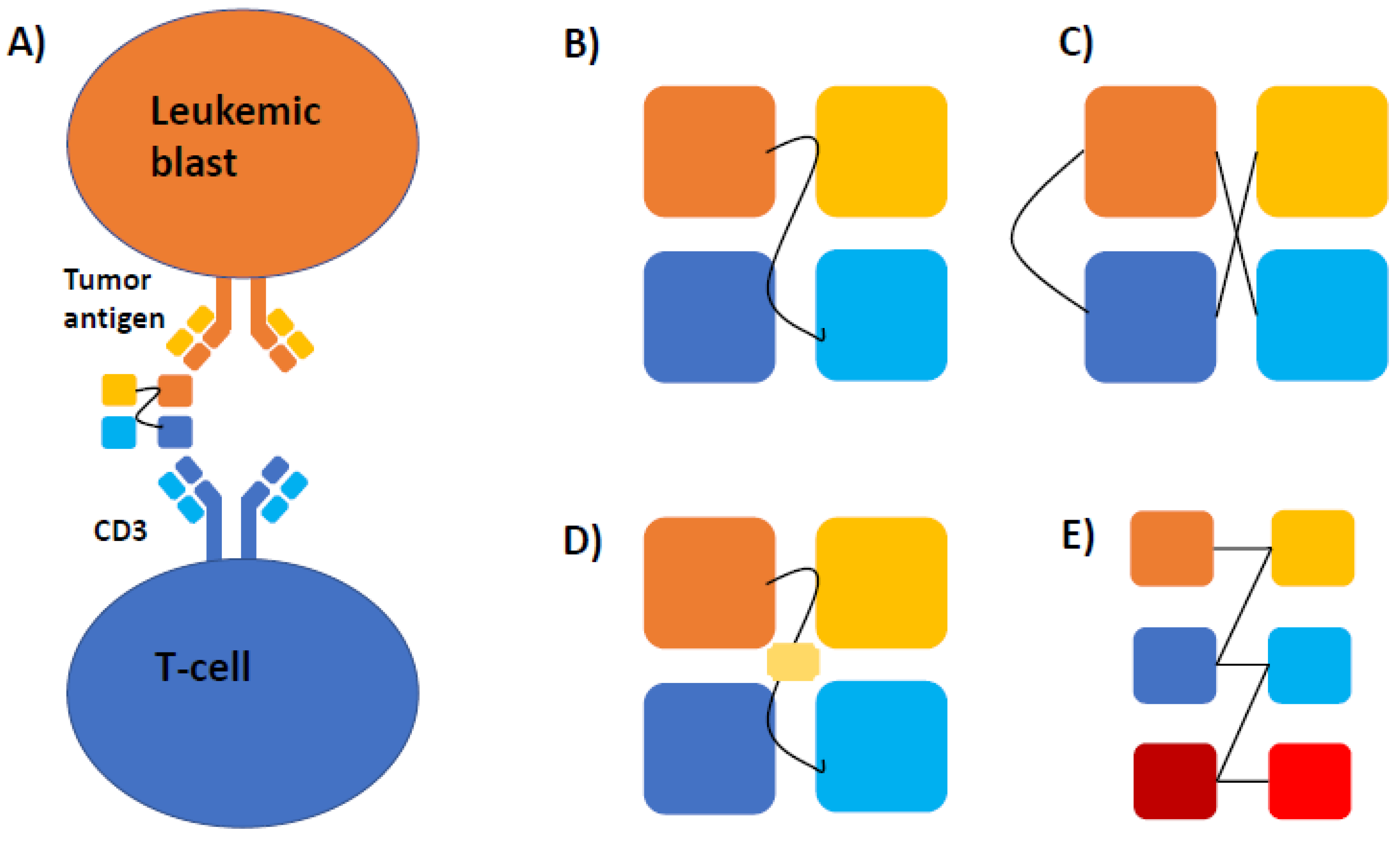
1. Introduction
2. Historical Overview and Safety Considerations
3. Mechanism and Structure
4. Antigen Targets
4.1. CD33
4.2. CD123
4.3. CLL-1/CLEC12A
4.4. FLT3
5. Cytokine Release Syndrome and Other Toxicities
6. Overcoming Limitations of Bispecific Antibodies and Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Carraway, H.E.; Chandhok, N.S.; Prebet, T. Advances in non-intensive chemotherapy treatment options for adults diagnosed with acute myeloid leukemia. Leuk. Res. 2020, 91, 106339. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Guyatt, G.; Abel, G.; Alibhai, S.; Altman, J.K.; Buckstein, R.; Choe, H.; Desai, P.; Erba, H.; Hourigan, C.S.; et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020, 4, 3528–3549. [Google Scholar] [CrossRef] [PubMed]
- Juliusson, G.; Hagberg, O.; Lazarevic, V.L.; Ölander, E.; Antunovic, P.; Cammenga, J.; Wennström, L.; Möllgård, L.; Brune, M.; Jädersten, M.; et al. Improved survival of men 50 to 75 years old with acute myeloid leukemia over a 20-year period. Blood 2019, 134, 1558–1561. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute–Surveillance, Epidemiology, and End Results Program, Cancer Stat Facts: Leukemia—Acute Myeloid Leukemia (AML) [cited 2021]. Available online: https://seer.cancer.gov/statfacts/html/amyl.html (accessed on 2 January 2021).
- DiNardo, C.D.; Wei, A.H. How I treat acute myeloid leukemia in the era of new drugs. Blood 2020, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- DeWolf, S.; Tallman, M.S. How I treat relapsed or refractory AML. Blood 2020, 136, 1023–1032. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Are we witnessing the start of a therapeutic revolution in acute myeloid leukemia? Leuk. Lymphoma 2019, 60, 1354–1369. [Google Scholar] [CrossRef]
- Liu, Y.; Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? Blood Rev. 2019, 34, 67–83. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.-A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res. 2009, 69, 4941. [Google Scholar] [CrossRef]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Sun, Z.-Y.J.; Kim, K.S.; Wagner, G.; Reinherz, E.L. Mechanisms Contributing to T Cell Receptor Signaling and Assembly Revealed by the Solution Structure of an Ectodomain Fragment of the CD3ϵγ Heterodimer. Cell 2001, 105, 913–923. [Google Scholar] [CrossRef]
- Tedder, T.F.; Inaoki, M.; Sato, S. The CD19–CD21 Complex Regulates Signal Transduction Thresholds Governing Humoral Immunity and Autoimmunity. Immunity 1997, 6, 107–118. [Google Scholar] [CrossRef]
- Hoseini, S.S.; Cheung, N.K. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer J. 2017, 7, e522. [Google Scholar] [CrossRef]
- Slade, M.J.; Uy, G.L. CD123 bi-specific antibodies in development in AML: What do we know so far? Best Pract. Res. Clin. Haematol. 2020, 33, 101219. [Google Scholar] [CrossRef] [PubMed]
- Kubasch, A.S.; Schulze, F.; Giagounidis, A.; Götze, K.S.; Krönke, J.; Sockel, K.; Middeke, J.M.; Chermat, F.; Gloaguen, S.; Puttrich, M.; et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia 2020, 34, 1182–1186. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Brischwein, K.; Parr, L.; Pflanz, S.; Volkland, J.; Lumsden, J.; Klinger, M.; Locher, M.; Hammond, S.A.; Kiener, P.; Kufer, P.; et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J. Immunother. 2007, 30, 798–807. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef]
- Zhu, M.; Wu, B.; Brandl, C.; Johnson, J.; Wolf, A.; Chow, A.; Doshi, S. Blinatumomab, a Bispecific T-cell Engager (BiTE(®)) for CD-19 Targeted Cancer Immunotherapy: Clinical Pharmacology and Its Implications. Clin. Pharm. 2016, 55, 1271–1288. [Google Scholar] [CrossRef]
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balazs, M.; Rock, D.; Boyle, M.C.; Goldstein, R.; Coxon, A.; et al. Generation of a Half-Life Extended Anti-CD19 BiTE® Antibody Construct Compatible with Once-Weekly Dosing for Treatment of CD19-Positive Malignancies. Blood 2017, 130 (Suppl. 1), 2815. [Google Scholar]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134 (Suppl. 1), 833. [Google Scholar] [CrossRef]
- Rader, C. DARTs take aim at BiTEs. Blood 2011, 117, 4403–4404. [Google Scholar] [CrossRef] [PubMed]
- Anegón, I.; Cuturi, M.C.; Trinchieri, G.; Perussia, B. Interaction of Fc receptor (CD16) ligands induces transcription of interleukin 2 receptor (CD25) and lymphokine genes and expression of their products in human natural killer cells. J. Exp. Med. 1988, 167, 452–472. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and Trispecific Killer Cell Engagers Directly Activate Human NK Cells through CD16 Signaling and Induce Cytotoxicity and Cytokine Production. Mol. Cancer Ther. 2012, 11, 2674. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting Natural Killer Cells to Acute Myeloid Leukemia In Vitro with a CD16 × 33 Bispecific Killer Cell Engager and ADAM17 Inhibition. Clin. Cancer Res. 2013, 19, 3844. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Arvindam, U.S.; van Hauten, P.; Hallstrom, C.; Vallera, D.A.; Dolstra, H.; Miller, J.S.; Felices, M. CD16-IL15-CLEC12A Trispecific Killer Engager (TriKE) Drives NK Cell Expansion, Activation, and Antigen Specific Killing of Cancer Stem Cells in Acute Myeloid Leukemia. Blood 2018, 132 (Suppl. 1), 1454. [Google Scholar] [CrossRef]
- Arvindam, U.S.; van Hauten, P.M.M.; Schirm, D.; Schaap, N.; Hobo, W.; Blazar, B.R.; Vallera, D.A.; Dolstra, H.; Felices, M.; Miller, J.S. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia 2020. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]
- Clark, M.C.; Stein, A. CD33 directed bispecific antibodies in acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2020, 33, 101224. [Google Scholar] [CrossRef]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef]
- Jen, E.Y.; Ko, C.-W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly Diagnosed CD33-Positive Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 3242. [Google Scholar] [CrossRef] [PubMed]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.H.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell–engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, G.S.; Gudgeon, C.J.; Harrington, K.H.; Dell’Aringa, J.; Newhall, K.J.; Means, G.D.; Sinclair, A.M.; Kischel, R.; Frankel, S.R.; Walter, R.B. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014, 123, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.H.; Gudgeon, C.J.; Laszlo, G.S.; Newhall, K.J.; Sinclair, A.M.; Frankel, S.R.; Kischel, R.; Chen, G.; Walter, R.B. The Broad Anti-AML Activity of the CD33/CD3 BiTE Antibody Construct, AMG 330, Is Impacted by Disease Stage and Risk. PLoS ONE 2015, 10, e0135945. [Google Scholar]
- Laszlo, G.S.; Gudgeon, C.J.; Harrington, K.H.; Walter, R.B. T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood Cancer J. 2015, 5, e340. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Kohnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef]
- Ravandi, F.; Stein, A.S.; Kantarjian, H.M.; Walter, R.B.; Paschka, P.; Jongen-Lavrencic, M.; Ossenkoppele, G.J.; Yang, Z.; Mehta, B.; Subklewe, M. A Phase 1 First-in-Human Study of AMG 330, an Anti-CD33 Bispecific T-Cell Engager (BiTE®) Antibody Construct, in Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). Blood 2018, 132 (Suppl. 1), 25. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38 (Suppl. 15), 7508. [Google Scholar] [CrossRef]
- Eissenberg, L.G.; Ritchey, J.; Rettig, M.P.; Fox, J.A.; Guenot, J.; DiPersio, J.F. AMV564, a Bivalent Bispecific (2 × 2) CD33/CD3 T-Cell Engager, Is Active and Improves Survival in a Mouse Model of Acute Myeloid Leukemia. Blood 2018, 132 (Suppl. 1), 2727. [Google Scholar] [CrossRef]
- Westervelt, P.; Roboz, G.J.; Cortes, J.E.; Kantarjian, H.M.; Lee, S.; Rettig, M.P.; Han, T.H.; Guenot, J.; Feldman, E.J.; DiPersio, J.F. Phase 1 First-in-Human Trial of AMV564, a Bivalent Bispecific (2 × 2) CD33/CD3 T-Cell Engager, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2018, 132 (Suppl. 1), 1455. [Google Scholar] [CrossRef]
- Westervelt, P.; Cortes, J.E.; Altman, J.K.; Long, M.; Oehler, V.G.; Gojo, I.; Guenot, J.; Chun, P.; Roboz, G.J. Phase 1 First-in-Human Trial of AMV564, a Bivalent Bispecific (2:2) CD33/CD3 T-Cell Engager, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2019, 134 (Suppl. 1), 834. [Google Scholar] [CrossRef]
- Smith, V.; Eckard, S.; Rettig, M.P.; Gehrs, L.N.; Guenot, J.; Wei, S.; DiPersio, J.F. Abstract 5699: AMV564, a bivalent, bispecific T-cell engager, depletes myeloid-derived suppressor cells and activates T cells in cancer patients. Cancer Res. 2020, 80 (Suppl. 16), 5699. [Google Scholar]
- Nair-Gupta, P.; Diem, M.; Reeves, D.; Wang, W.; Schulingkamp, R.; Sproesser, K.; Mattson, B.; Heidrich, B.; Mendonça, M.; Joseph, J.; et al. A novel C2 domain binding CD33xCD3 bispecific antibody with potent T-cell redirection activity against acute myeloid leukemia. Blood Adv. 2020, 4, 906–919. [Google Scholar] [CrossRef]
- Carson, W.E.; Giri, J.G.; Lindemann, M.J.; Linett, M.L.; Ahdieh, M.; Paxton, R.; Anderson, D.; Eisenmann, J.; Grabstein, K.; Caligiuri, M.A. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 1994, 180, 1395–1403. [Google Scholar] [CrossRef]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef]
- Chu, S.Y.; Pong, E.; Chen, H.; Phung, S.; Chan, E.W.; Endo, N.A.; Rashid, R.; Bonzon, C.; Leung, I.W.L.; Muchhal, U.S.; et al. Immunotherapy with Long-Lived Anti-CD123 × Anti-CD3 Bispecific Antibodies Stimulates Potent T Cell-Mediated Killing of Human AML Cell Lines and of CD123+ Cells in Monkeys: A Potential Therapy for Acute Myelogenous Leukemia. Blood 2014, 124, 2316. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, J.M.; Mawad, R.; Egan, D.; Blum, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 × CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136 (Suppl. 1), 4–5. [Google Scholar] [CrossRef]
- Gaudet, F.; Nemeth, J.F.; McDaid, R.; Li, Y.; Harman, B.; Millar, H.; .Teplyakov, A.; Wheeler, J.; Luo, J.; Tam, S.; et al. Development of a CD123xCD3 Bispecific Antibody (JNJ-63709178) for the Treatment of Acute Myeloid Leukemia (AML). Blood 2016, 128, 2824. [Google Scholar] [CrossRef]
- Comeau, M.R.; Gottschalk, R.; Daugherty, M.; Sewell, T.; Misher, L.; Jeannette, B.; Johnson, S.; Parr, L.; Kumer, J.; Jablonski, D.; et al. Abstract LB-199: APVO436, a bispecific anti-CD123 × anti-CD3 ADAPTIR™ molecule for redirected T-cell cytotoxicity with limited cytokine release, is well tolerated in repeat dose toxicology studies in cynomolgus macaques. Cancer Res. 2019, 13 (Suppl. 13), LB-199-LB. [Google Scholar]
- Watts, J.M.; Lin, T.; Wang, E.S.; Mims, A.S.; Cull, E.H.; Patel, P.A.; Shami, P.J.; Walter, R.B.; Cogle, C.R.; Chenault, R.A.; et al. Preliminary Results from a Phase 1 Study of APVO436, a Novel Anti-CD123 × Anti-CD3 Bispecific Molecule, in Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2020, 136 (Suppl. 1), 11–12. [Google Scholar] [CrossRef]
- Chichili, G.R.; Huang, L.; Li, H.; Burke, S.; He, L.; Tang, Q.; Jin, L.; Gorlatov, S.; Ciccarone, V.; Chen, F.; et al. A CD3 × CD123 bispecific DART for redirecting host T cells to myelogenous leukemia: Preclinical activity and safety in nonhuman primates. Sci. Transl. Med. 2015, 7, 289ra82. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R.; et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood 2016, 127, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Davidson-Moncada, J.K.; Uy, G.L.; Rizzieri, D.; Khoury, H.J.; Foster, M.C.; Godwin, J.E.; Topp, M.S.; Martinelli, G.; Ciceri, F.; et al. A phase I, first-in-human study of MGD006/S80880 (CD123 × CD3 DART) in AML/MDS. J. Clin. Oncol. 2017, 35 (Suppl. 15), TPS7070-TPS. [Google Scholar] [CrossRef]
- Uy, G.L.; Rettig, M.P.; Vey, N.; Godwin, J.; Foster, M.C.; Rizzieri, D.A.; Arellano, M.L.; Topp, M.S.; Huls, G.; Jongen-Lavrencic, M.; et al. Phase 1 Cohort Expansion of Flotetuzumab, a CD123×CD3 Bispecific Dart® Protein in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2018, 132 (Suppl. 1), 764. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Wei, A.H.; Fong, C.Y.; Montesinos, P.; Calbacho, M.; Gil, J.S.; Perez De Oteyza, J.; Rowe, J.M.; Wolach, O.; Ofran, Y.; Moshe, Y.; et al. A Phase 1 Study of Flotetuzumab, a CD123 × CD3 DART® Protein, Combined with MGA012, an Anti-PD-1 Antibody, in Patients with Relapsed or Refractory Acute Myeloid Leukemia. Blood 2019, 134 (Suppl. 1), 2662. [Google Scholar] [CrossRef]
- Montesinos, P.; Roboz, G.J.; Bulabois, C.E.; Subklewe, M.; Platzbecker, U.; Ofran, Y.; Papayannidis, C.; Wierzbowska, A.; Shin, H.J.; Doronin, V.; et al. Safety and efficacy of talacotuzumab plus decitabine or decitabine alone in patients with acute myeloid leukemia not eligible for chemotherapy: Results from a multicenter, randomized, phase 2/3 study. Leukemia 2021, 35, 62–74. [Google Scholar] [CrossRef]
- van Rhenen, A.; van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Stigter-van Walsum, M.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef]
- Van Loo, P.F.; Doornbos, R.; Dolstra, H.; Shamsili, S.; Bakker, L. Preclinical Evaluation of MCLA117, a CLEC12AxCD3 Bispecific Antibody Efficiently Targeting a Novel Leukemic Stem Cell Associated Antigen in AML. Blood 2015, 126, 325. [Google Scholar] [CrossRef]
- Willier, S.; Rothämel, P.; Hastreiter, M.; Wilhelm, J.; Stenger, D.; Blaeschke, F.; Rohlfs, M.; Kaeuferle, T.; Schmid, I.; Albert, M.H.; et al. CLEC12A and CD33 coexpression as a preferential target for pediatric AML combinatorial immunotherapy. Blood 2021, 137, 1037–1049. [Google Scholar] [CrossRef]
- Noordhuis, P.; Terwijn, M.; Rutten, A.P.; Smit, L.; Ossenkoppele, G.J.; Schuurhuis, G.J. Targeting of CLEC12A In Acute Myeloid Leukemia by Antibody-Drug-Conjugates and Bispecific CLL-1×CD3 BiTE Antibody. Blood 2010, 116, 2890. [Google Scholar] [CrossRef]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti–CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef]
- van Loo, P.F.; Hangalapura, B.N.; Thordardottir, S.; Gibbins, J.D.; Veninga, H.; Hendriks, L.J.A.; Kramer, A.; Roovers, R.C.; Leenders, M.; de Kruif, J.; et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukaemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert Opin. Biol. Ther. 2019, 19, 721–733. [Google Scholar] [CrossRef]
- Krawczyk, E.; Zolov, S.N.; Huang, K.; Bonifant, C.L. T-cell Activity against AML Improved by Dual-Targeted T Cells Stimulated through T-cell and IL7 Receptors. Cancer Immunol. Res. 2019, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Durben, M.; Schmiedel, D.; Hofmann, M.; Vogt, F.; Nübling, T.; Pyz, E.; Bühring, H.J.; Rammensee, H.G.; Salih, H.R.; Große-Hovest, L.; et al. Characterization of a bispecific FLT3 × CD3 antibody in an improved, recombinant format for the treatment of leukemia. Mol. Ther. 2015, 23, 648–655. [Google Scholar] [CrossRef]
- Brauchle, B.; Goldstein, R.L.; Karbowski, C.M.; Henn, A.; Li, C.M.; Bücklein, V.L.; Krupka, C.; Boyle, M.C.; Koppikar, P.; Haubner, S.; et al. Characterization of a Novel FLT3 BiTE Molecule for the Treatment of Acute Myeloid Leukemia. Mol. Cancer Ther. 2020, 19, 1875–1888. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Krishnamoorthy, V.; Dettling, D.; Sommer, C.; Poulsen, K.; Ni, I.; Pham, A.; Chen, W.; Liao-Chan, S.; Lindquist, K.; et al. An Optimized Full-Length FLT3/CD3 Bispecific Antibody Demonstrates Potent Anti-leukemia Activity and Reversible Hematological Toxicity. Mol. Ther. 2020, 28, 889–900. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Brandl, C.; Haas, C.; d’Argouges, S.; Fisch, T.; Kufer, P.; Brischwein, K.; Prang, N.; Bargou, R.; Suzich, J.; Baeuerle, P.A.; et al. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol. Immunother. 2007, 56, 1551–1563. [Google Scholar] [CrossRef]
- Subklewe, M. BiTEs better than CAR T cells. Blood Adv. 2021, 5, 607–612. [Google Scholar] [CrossRef]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef]
- Stein, A.S.; Schiller, G.; Benjamin, R.; Jia, C.; Zhang, A.; Zhu, M.; Zimmerman, Z.; Topp, M.S. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: Management and mitigating factors. Ann. Hematol. 2019, 98, 159–167. [Google Scholar] [CrossRef]
- Khadka, R.H.; Sakemura, R.; Kenderian, S.S.; Johnson, A.J. Management of cytokine release syndrome: An update on emerging antigen-specific T cell engaging immunotherapies. Immunotherapy 2019, 11, 851–857. [Google Scholar] [CrossRef]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.; Zhukovsky, E.A.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef]
- Hoseini, S.S.; Guo, H.; Wu, Z.; Hatano, M.N.; Cheung, N.-K.V. A potent tetravalent T-cell–engaging bispecific antibody against CD33 in acute myeloid leukemia. Blood Adv. 2018, 2, 1250–1258. [Google Scholar] [CrossRef]
- Cheng, P.; Eksioglu, E.; Chen, X.; Wei, M.; Guenot, J.; Fox, J.; List, A.F.; Wei, S. Immunodepletion of MDSC By AMV564, a Novel Tetravalent Bispecific CD33/CD3 T Cell Engager Restores Immune Homeostasis in MDS in Vitro. Blood 2017, 130 (Suppl. 1), 51. [Google Scholar]
- Herrmann, M.; Krupka, C.; Deiser, K.; Brauchle, B.; Marcinek, A.; Ogrinc Wagner, A.; Rataj, F.; Mocikat, R.; Metzeler, K.H.; Spiekermann, K.; et al. Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 2018, 132, 2484–2494. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Reeder, S.; Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci. Transl. Med. 2020, 12, eaaz0463. [Google Scholar] [CrossRef]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef]
- Mu-Mosley, H.; Ostermann, L.B.; Zhao, R.; Bonifant, C.L.; Gottschalk, S.; Velasquez, M.P.; Andreeff, M. Venetoclax Enhances Anti-Leukemia Activity of CD123-Specific BiTE-Secreting T-Cells in AML. Blood 2020, 136 (Suppl. 1), 12–13. [Google Scholar] [CrossRef]
- Upadhyay, R.; Boiarsky, J.A.; Pantsulaia, G.; Svensson-Arvelund, J.; Lin, M.J.; Wroblewska, A.; Bhalla, S.; Scholler, N.; Bot, A.; Rossi, J.M.; et al. A Critical Role for Fas-Mediated Off-Target Tumor Killing in T-cell Immunotherapy. Cancer Discov. 2021, 11, 599–613. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Shallis, R.M.; Boddu, P.C.; Wood, B.; Radich, J.; Halene, S.; Zeidan, A.M. The minimal that kills: Why defining and targeting measurable residual disease is the “Sine Qua Non” for further progress in management of acute myeloid leukemia. Blood Rev. 2019, 43, 100650. [Google Scholar] [CrossRef]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C.; Hambach, L.; Stadler, M.; Koenecke, C.; Flintrop, M.; et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef]
- Terwijn, M.; van Putten, W.L.; Kelder, A.; van der Velden, V.H.; Brooimans, R.A.; Pabst, T.; Maertens, J.; Boeckx, N.; de Greef, G.E.; Valk, P.J.; et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: Data from the HOVON/SAKK AML 42A study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef]
- Ehninger, A.; Kramer, M.; Rollig, C.; Thiede, C.; Bornhauser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.-J.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 bispecific antibody–induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019, 11, eaax8861. [Google Scholar] [CrossRef]
- Uy, G.L.; Rettig, M.P.; Christ, S.; Aldoss, I.; Byrne, M.T.; Erba, H.P.; Arellano, M.L.; Foster, M.C.; Godwin, J.E.; Ravandi, F.; et al. Prophylactic Ruxolitinib for Cytokine Release Syndrome (CRS) in Relapse/Refractory (R/R) AML Patients Treated with Flotetuzumab. Blood 2020, 136 (Suppl. 1), 19–21. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef]


{kind=link}
| Construct | Components |
|---|---|
| Bispecific Antibody | 2 scFVs: CD3 of T cells and antigen target |
| DARTs | 2 scFVs: CD3 of T cells and antigen target, with addition of disulfide bridge |
| BiKES | 2 scFVs: CD16 of NK cells and antigen target |
| Trikes | 2 scFVs: CD16 of NK cells and antigen target and IL-15 crosslinker |
| Author | Drug (Construct) | Patient Population | Outcomes |
|---|---|---|---|
| Uy [27] | Flotetuzumab (anti CD3 × CD123 DART) | 92 R/R-AML patients | Primary induction failure or early relapse cohort (n = 30): Efficacy: 27% with CR/CRh; median OS 10.2 months among responders Safety: 100% CRS (3% ≥ grade) |
| Ravandi [28] | AMG 330 (anti-CD3 × CD33 BiTE) | 55 patients with R/R-AML | Efficacy: 19% ORR (7% CR) Safety: 60% CRS |
| Subklewe [39] | AMG 673 (Half-Life Extended Anti-CD3 × CD33 BiTE) | 30 patients with R/R-AML | Efficacy: 44% with bone marrow blast reduction Safety: 50% CRS (13% ≥ grade 3) |
| Ravandi [30] | Vibecotamab (XmAb14045; anti CD3 × CD123 BiTE) | 104 R/R-AML, 1 B-cell ALL, and 1 CML | Efficacy: 14% ORR (4% CR); 71% SD Safety: 59% CRS (15% ≥ grade 3) |
| Watts [31] | APVO436 (anti CD3 × CD123 BiTE) | 22 R/R-AML and 6 R/R-MDS | Efficacy: 2 patients with blast reduction Safety: edema (32%), febrile neutropenia (29%), infusion reaction (21%), CRS (18%) |
| Drug | NCT | Patient Population | Target | Phase |
|---|---|---|---|---|
| AMG330 | NCT02520427 | Relapsed/Refractory AML, Minimal Residual Disease Positive AML, MDS | CD3 × CD33 bispecific antibody | 1 |
| AMV564 | NCT03144245 | Relapsed/Refractory AML | CD3 × CD33 bispecific antibody | 1 |
| AMV564 | NCT03516591 | MDS | CD3 × CD33 bispecific antibody | 1 |
| AMG673 | NCT03224819 | Relapsed/Refractory AML | CD3 × CD33 bispecific antibody | 1 |
| GEM333 | NCT03516760 | Relapsed/Refractory AML | CD3 × CD33 bispecific antibody | 1 |
| JNJ-67571244 | NCT03915379 | Relapsed/Refractory AML, MDS | CD3 × CD33 bispecific antibody | 1 |
| GTB-3550 | NCT03214666 | Relapsed/Refractory AML, MDS, Advanced Systemic Mastocytosis | CD16/IL-15/CD33 TriKE | 1/2 |
| APVO436 | NCT03647800 | Relapsed/Refractory AML, MDS | CD3 × CD123 bispecific antibody | 1 |
| XmAb14045 | NCT02730312 | CD123 Expressing hematologic malignancies | CD3 × CD123 bispecific antibody | 1 |
| JNJ-63709178 | NCT02715011 | Relapsed/Refractory AML | CD3 × CD123 bispecific antibody | 1 |
| SAR440234 | NCT03594955 | Relapsed/Refractory AML, MDS, B-ALL | CD3 × CD123 bispecific antibody | 1/2 |
| MGD006 | NCT02152956 | Relapsed/Refractory AML, MDS | CD3 × CD123 DART | 1/2 |
| MCLA-117 | NCT03038230 | Relapsed/Refractory AML and newly diagnosed elderly AML | CD3 × CLEC12A bispecific antibody | 1 |
| AMG427 | NCT03541369 | Relapsed/Refractory AML | CD3 × CD135(FLT3) bispecific antibody | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, C.; Zeidan, A.M.; Bewersdorf, J.P. BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML? Life 2021, 11, 465. https://doi.org/10.3390/life11060465
Allen C, Zeidan AM, Bewersdorf JP. BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML? Life. 2021; 11(6):465. https://doi.org/10.3390/life11060465
Chicago/Turabian StyleAllen, Cecily, Amer M. Zeidan, and Jan Philipp Bewersdorf. 2021. "BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML?" Life 11, no. 6: 465. https://doi.org/10.3390/life11060465
APA StyleAllen, C., Zeidan, A. M., & Bewersdorf, J. P. (2021). BiTEs, DARTS, BiKEs and TriKEs—Are Antibody Based Therapies Changing the Future Treatment of AML? Life, 11(6), 465. https://doi.org/10.3390/life11060465