
Evidence of the Cellular Senescence Stress Response in Mitotically Active Brain Cells—Implications for Cancer and Neurodegeneration

 ,
, 
 , , and
, , and 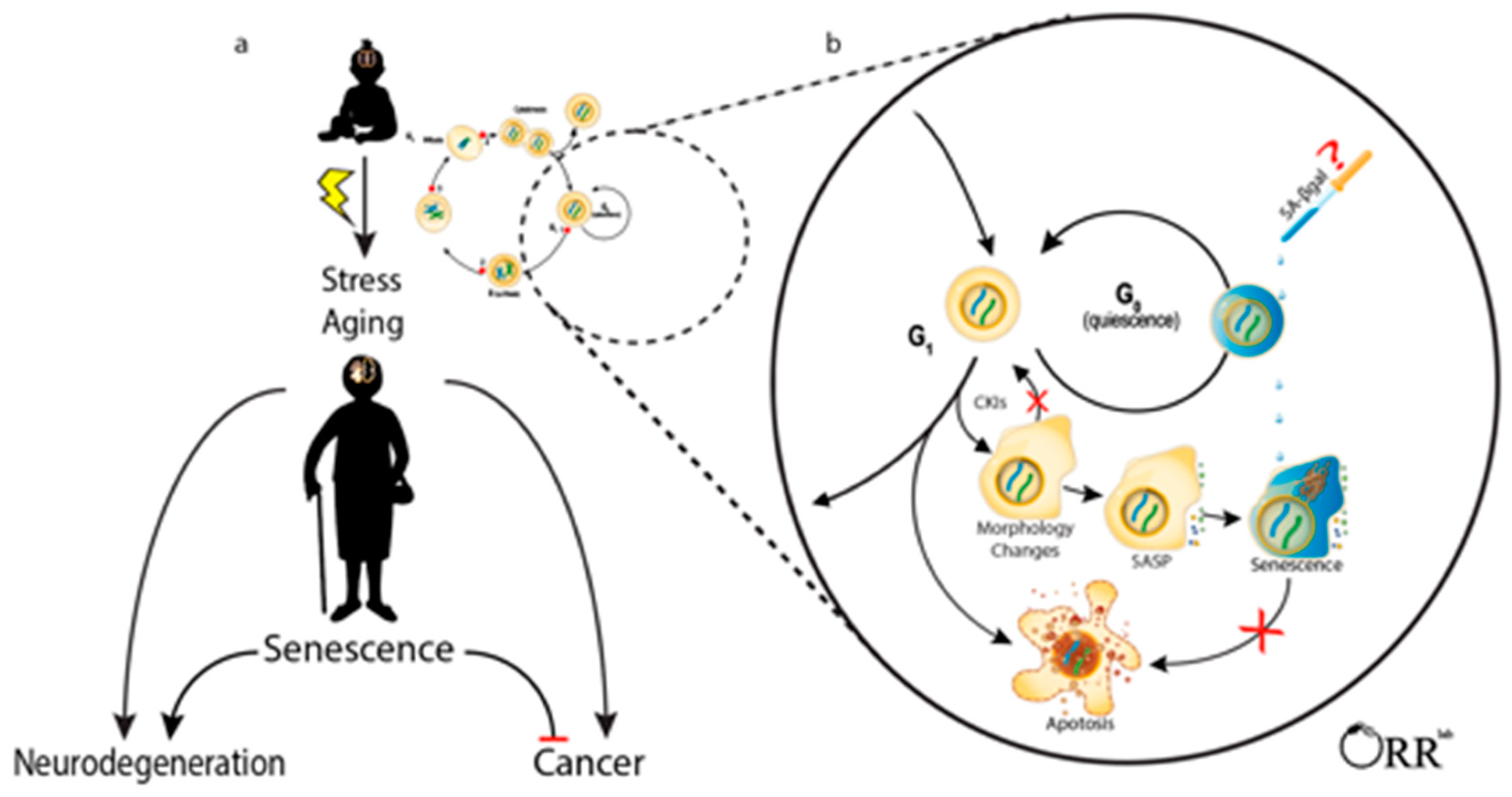{kind=link}
Abstract
1. Introduction
2. Identifying Senescent Brain Cells
2.1. Absence of Proliferation/Stable Cell Cycle Arrest
2.2. Apoptosis Resistance
2.3. Secretory Phenotye
2.4. Senescence-Associated β-Galactosidase
2.5. Concluding Remarks on Identifying Senescent Cells

3. Neuronal Precursor Cells
Concluding Remarks
4. Oligodendrocyte Precursor Cells
Concluding Remarks
5. Microglia
Concluding Remarks
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.M.; Fitzpatrick, A.L.; Xiong, C.; Sieh, W.; Kuller, L.; Miller, J.P.; Williams, M.M.; Kopan, R.; Behrens, M.I.; Morris, J.C. Cancer linked to Alzheimer disease but not vascular dementia. Neurology 2009, 74, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Tabarés-Seisdedos, R.; Dumont, N.; Baudot, A.; Valderas, J.M.; Climent, J.; Valencia, A.; Crespo-Facorro, B.; Vieta, E.; Gómez-Beneyto, M.; Martínez, S.; et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol. 2011, 12, 604–608. [Google Scholar] [CrossRef]
- Driver, J.A.; Logroscino, G.; Buring, J.E.; Gaziano, J.M.; Kurth, T. A prospective cohort study of cancer incidence following the diagnosis of Parkinson’s disease. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1260–1265. [Google Scholar] [CrossRef]
- Shi, H.B.; Tang, B.; Liu, Y.W.; Wang, X.F.; Chen, G.J. Alzheimer disease and cancer risk: A meta-analysis. J. Cancer Res. Clin. Oncol. 2015, 141, 485–494. [Google Scholar] [CrossRef]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads Between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2019, 11, 77–89. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. (Lond.) 2018, 38, 40. [Google Scholar] [CrossRef]
- Irizar, P.A.; Schäuble, S.; Esser, D.; Groth, M.; Frahm, C.; Priebe, S.; Baumgart, M.; Hartmann, N.; Marthandan, S.; Menzel, U.; et al. Transcriptomic alterations during ageing reflect the shift from cancer to degenerative diseases in the elderly. Nat. Commun. 2018, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Congrains, A.; Kamide, K.; Ohishi, M.; Rakugi, H. ANRIL: Molecular Mechanisms and Implications in Human Health. Int. J. Mol. Sci. 2013, 14, 1278–1292. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2011, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2017, 217, 65–77. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- Justice, J.N.; Gregory, M.H.; Tchkonia, T.; Lebrasseur, N.K.; Kirkland, J.L.; Kritchevsky, S.B.; Nicklas, B.J. Cellular Senescence Biomarker p16INK4a+ Cell Burden in Thigh Adipose is Associated With Poor Physical Function in Older Women. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2017, 73, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Uyar, B.; Palmer, D.; Kowald, A.; Escobar, H.M.; Barrantes, I.; Möller, S.; Akalin, A.; Fuellen, G. Single-cell analyses of aging, inflammation and senescence. Ageing Res. Rev. 2020, 64, 101156. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nat. Cell Biol. 2008, 455, 58–63. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; DeMaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Matjusaitis, M.; Chin, G.; Sarnoski, E.A.; Stolzing, A. Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 2016, 29, 1–12. [Google Scholar] [CrossRef]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 483–497. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef]
- Roy, A.L.; Sierra, F.; Howcroft, K.; Singer, D.S.; Sharpless, N.; Hodes, R.J.; Wilder, E.L.; Anderson, J.M. A Blueprint for Characterizing Senescence. Cell 2020, 183, 1143–1146. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Homem, C.C.F.; Repic, M.; Knoblich, J.A. Proliferation control in neural stem and progenitor cells. Nat. Rev. Neurosci. 2015, 16, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.-I.; Aida, J.; Izumiyama-Shimomura, N.; Nakamura, K.-I.; Ishikawa, N.; Matsuda, Y.; Arai, T.; Ishiwata, T.; Kumasaka, T.; Takahashi-Fujigasaki, J.; et al. Changes in telomere length with aging in human neurons and glial cells revealed by quantitative fluorescence in situ hybridization analysis. Geriatr. Gerontol. Int. 2018, 18, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomeres in endothelial dysfunction. J. Cardiol. 2003, 41, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nat. Cell Biol. 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 Induces p21-Dependent Cellular Senescence in Post-mitotic Dopaminergic Neurons. Cell Stem Cell 2019, 25, 514–530.e8. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.-M.; Shi, M.; Cheng, A.; Gao, Y.; Chen, G.; Song, X.; So, R.W.L.; Zhang, J.; Herrup, K. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 2019, 22, 1806–1819. [Google Scholar] [CrossRef]
- Turner, K.J.; Vasu, V.; Griffin, D.K. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, X.; Ding, X.; Wang, F.; Geng, X. Telomere and its role in the aging pathways: Telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.-P.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160451. [Google Scholar] [CrossRef]
- Lai, T.-P.; Zhang, N.; Noh, J.; Mender, I.; Tedone, E.; Huang, E.; Wright, W.E.; Danuser, G.; Shay, J.W. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Zhang, X.; Lou, X.; Xia, F. Advances in the detection of telomerase activity using isothermal amplification. Theranostics 2017, 7, 1847–1862. [Google Scholar] [CrossRef]
- Zheng, K.-W.; Liu, C.; Meng, Q.; Hao, Y.-H.; Zheng, J.-P.; Li, W.; Tan, Z. One-Step High-Throughput Telomerase Activity Measurement of Cell Populations, Single Cells, and Single-Enzyme Complexes. ACS Omega 2020, 5, 24666–24673. [Google Scholar] [CrossRef]
- Liu, Y.; Li, S.; Zhang, L.; Zhao, Q.; Li, N.; Wu, Y. Catalytic Hairpin Assembly-Assisted Rolling Circle Amplification for High-Sensitive Telomerase Activity Detection. ACS Omega 2020, 5, 11836–11841. [Google Scholar] [CrossRef]
- Díaz-Cartagena, D.C.; Hernández-Cancel, G.; Bracho-Rincón, D.P.; González-Feliciano, J.A.; Cunci, L.; González, C.I.; Cabrera, C.R. Label-Free Telomerase Activity Detection via Electrochemical Impedance Spectroscopy. ACS Omega 2019, 4, 16724–16732. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.-I.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; De Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.M.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Fan, Y.; Cheng, J.; Zeng, H.; Shao, L. Senescent Cell Depletion through Targeting BCL-Family Proteins and Mitochondria. Front. Physiol. 2020, 11, 593630. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 2019, 18, e12841. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; DeMaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Haak, A.J.; Tschumperlin, D.J.; Lebrasseur, N.K. Targeting Senescent Cells in Fibrosis: Pathology, Paradox, and Practical Considerations. Curr. Rheumatol. Rep. 2018, 20, 3. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Fujimaki, K.; Li, R.; Chen, H.; Della Croce, K.; Zhang, H.H.; Xing, J.; Bai, F.; Yao, G. Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl. Acad. Sci. USA 2019, 116, 22624–22634. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of senescent cells by beta-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Ke, Z.; Gorbunova, V.; Seluanov, A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging 2012, 4, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Hammelrath, L.; Škokić, S.; Khmelinskii, A.; Hess, A.; Van Der Knaap, N.; Staring, M.; Lelieveldt, B.P.; Wiedermann, D.; Hoehn, M. Morphological maturation of the mouse brain: An in vivo MRI and histology investigation. NeuroImage 2016, 125, 144–152. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106-107, 1–16. [Google Scholar] [CrossRef]
- Altman, J. Are new neurons formed in the brains of adult mammals? Science 1962, 135, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Tramontin, A.D.; Quiñones-Hinojosa, A.; Barbaro, N.M.; Gupta, N.; Kunwar, S.; Lawton, M.T.; McDermott, M.W.; Parsa, A.T.; Verdugo, J.M.-G.; et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nat. Cell Biol. 2004, 427, 740–744. [Google Scholar] [CrossRef]
- Temple, S. Division and differentiation of isolated CNS blast cells in microculture. Nat. Cell Biol. 1989, 340, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef]
- Gonzalez-Perez, O.; Quiñones-Hinojosa, A. Astrocytes as neural stem cells in the adult brain. J. Stem Cells 2012, 7, 181–188. [Google Scholar]
- Doetsch, F.; Caillé, I.; Lim, D.A.; García-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular Zone Astrocytes Are Neural Stem Cells in the Adult Mammalian Brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef]
- Knoth, R.; Singec, I.; Ditter, M.; Pantazis, G.; Capetian, P.; Meyer, R.P.; Horvat, V.; Volk, B.; Kempermann, G. Murine Features of Neurogenesis in the Human Hippocampus across the Lifespan from 0 to 100 Years. PLoS ONE 2010, 5, e8809. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of Hippocampal Neurogenesis in Adult Humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.-M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e3. [Google Scholar] [CrossRef]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Höglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Evercooren, A.B.-V. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef] [PubMed]
- Donega, V.; Burm, S.M.; Van Strien, M.E.; Van Bodegraven, E.J.; Paliukhovich, I.; Geut, H.; Van De Berg, W.D.J.; Li, K.W.; Smit, A.B.; Basak, O.; et al. Transcriptome and proteome profiling of neural stem cells from the human subventricular zone in Parkinson’s disease. Acta Neuropathol. Commun. 2019, 7, 84. [Google Scholar] [CrossRef]
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 2018, 22, 589–599.e5. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Dennis, C.V.; Suh, L.S.; Rodriguez, M.L.; Kril, J.J.; Sutherland, G.T. Human adult neurogenesis across the ages: An immunohistochemical study. Neuropathol. Appl. Neurobiol. 2016, 42, 621–638. [Google Scholar] [CrossRef]
- Sanai, N.; Nguyen, T.; Ihrie, R.A.; Mirzadeh, Z.; Tsai, H.-H.; Wong, M.; Gupta, N.; Berger, M.S.; Huang, E.; Garcia-Verdugo, J.-M.; et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nat. Cell Biol. 2011, 478, 382–386. [Google Scholar] [CrossRef]
- Mathews, K.J.; Allen, K.M.; Boerrigter, D.; Ball, H.; Weickert, C.S.; Double, K.L. Evidence for reduced neurogenesis in the aging human hippocampus despite stable stem cell markers. Aging Cell 2017, 16, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Morshead, C.M.; Reynolds, B.A.; Craig, C.G.; McBurney, M.W.; Staines, W.A.; Morassutti, D.; Weiss, S.; Van Der Kooy, D. Neural stem cells in the adult mammalian forebrain: A relatively quiescent subpopulation of subependymal cells. Neuron 1994, 13, 1071–1082. [Google Scholar] [CrossRef]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Urbán, N.; Blomfield, I.M.; Guillemot, F. Quiescence of Adult Mammalian Neural Stem Cells: A Highly Regulated Rest. Neuron 2019, 104, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Obernier, K.; Alvarez-Buylla, A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146, dev156059. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Willis, C.M.; Crocker, S.J.; Pluchino, S. Stem Cells of the Aging Brain. Front. Aging Neurosci. 2020, 12, 247. [Google Scholar] [CrossRef]
- Patel, A.P.; Fisher, J.L.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.; Agius, D.; Alahdab, F.; Alam, T.; et al. Global, regional, and national burden of brain and other CNS cancer, 1990-2016: A systematic analysis for the Global Burden of Disease Study. Lancet Neurol. 2019, 18, 376–393. [Google Scholar] [CrossRef]
- Azzarelli, R.; Simons, B.D.; Philpott, A. The developmental origin of brain tumours: A cellular and molecular framework. Development 2018, 145, dev162693. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nat. Cell Biol. 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; Van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Raabe, E.H.; Lim, K.S.; Kim, J.M.; Meeker, A.; Mao, X.-G.; Nikkhah, G.; Maciaczyk, J.; Kahlert, U.; Jain, D.; Bar, E.; et al. BRAF Activation Induces Transformation and Then Senescence in Human Neural Stem Cells: A Pilocytic Astrocytoma Model. Clin. Cancer Res. 2011, 17, 3590–3599. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.M.; Lan, M.L.; Kan, V.H.; Patel, N.H.; Giedzinski, E.; Tseng, B.P.; Limoli, C.L. Consequences of ionizing radiation-induced damage in human neural stem cells. Free. Radic. Biol. Med. 2010, 49, 1846–1855. [Google Scholar] [CrossRef]
- Monje, M.L.; Mizumatsu, S.; Fike, J.R.; Palmer, T.D. Irradiation induces neural precursor-cell dysfunction. Nat. Med. 2002, 8, 955–962. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, N.; Ellerby, L.M.; Davalos, A.R.; Zeng, X.; Campisi, J.; Desprez, P.-Y. Responses of human embryonic stem cells and their differentiated progeny to ionizing radiation. Biochem. Biophys. Res. Commun. 2012, 426, 100–105. [Google Scholar] [CrossRef]
- Schneider, L.; Pellegatta, S.; Favaro, R.; Pisati, F.; Roncaglia, P.; Testa, G.; Nicolis, S.K.; Finocchiaro, G.; Di Fagagna, F.D. DNA Damage in Mammalian Neural Stem Cells Leads to Astrocytic Differentiation Mediated by BMP2 Signaling through JAK-STAT. Stem Cell Rep. 2013, 1, 123–138. [Google Scholar] [CrossRef]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef]
- Dong, C.-M.; Wang, X.-L.; Wang, G.-M.; Zhang, W.-J.; Zhu, L.; Gao, S.; Yang, D.-J.; Qin, Y.; Liang, Q.-J.; Chen, Y.-L.; et al. A stress-induced cellular aging model with postnatal neural stem cells. Cell Death Dis. 2014, 5, e1116. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Da Pozzo, E.; Iofrida, C.; Martini, C. Human Neural Stem Cell Aging Is Counteracted by α-Glycerylphosphorylethanolamine. ACS Chem. Neurosci. 2016, 7, 952–963. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Houben, S.; Leroy, K.; Ando, K.; Yilmaz, Z.; Widomski, C.; Buée, L.; Brion, J.-P. Genetic ablation of tau in postnatal neurons rescues decreased adult hippocampal neurogenesis in a tauopathy model. Neurobiol. Dis. 2019, 127, 131–141. [Google Scholar] [CrossRef]
- Komuro, Y.; Xu, G.; Bhaskar, K.; Lamb, B.T. Human tau expression reduces adult neurogenesis in a mouse model of tauopathy. Neurobiol. Aging 2015, 36, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- He, N.; Jin, W.L.; Lok, K.H.; Wang, Y.; Yin, M.; Wang, Z.J. Amyloid-beta(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor. Cell Death Dis. 2013, 4, e924. [Google Scholar] [CrossRef]
- Orr, M.E.; Pitstick, R.; Canine, B.; Ashe, K.H.; Carlson, G.A. Genotype-Specific Differences between Mouse CNS Stem Cell Lines Expressing Frontotemporal Dementia Mutant or Wild Type Human Tau. PLoS ONE 2012, 7, e39328. [Google Scholar] [CrossRef]
- Voisin, J.; Farina, F.; Naphade, S.; Fontaine, M.; Tshilenge, K.T.; Aguirre, C.G.; Lopez-Ramirez, A.; Dancourt, J.; Ginisty, A.; Nair, S.S.; et al. FOXO3 targets are reprogrammed as Huntington’s disease neural cells and striatal neurons face senescence with p16(INK4a) increase. Aging Cell 2020, 19, e13226. [Google Scholar] [CrossRef]
- Orr, M.E.; Sullivan, A.C.; Frost, B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 637–648. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nat. Cell Biol. 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Kao, A.W.; Karydas, A.; Onanuga, K.; Martinez, R.; Argouarch, A.; Wang, C.; Huang, C.; Sohn, P.D.; Bowles, K.R.; et al. A Comprehensive Resource for Induced Pluripotent Stem Cells from Patients with Primary Tauopathies. Stem Cell Rep. 2019, 13, 939–955. [Google Scholar] [CrossRef]
- Liang, K.X.; Kristiansen, C.K.; Mostafavi, S.; Vatne, G.H.; Zantingh, G.A.; Kianian, A.; Tzoulis, C.; Høyland, L.E.; Ziegler, M.; Perez, R.M.; et al. Disease-specific phenotypes in iPSC -derived neural stem cells with POLG mutations. EMBO Mol. Med. 2020, 12, 12146. [Google Scholar] [CrossRef]
- Gao, J.; Wu, Y.; He, D.; Zhu, X.; Li, H.; Liu, H.; Liu, H. Anti-aging effects of Ribes meyeri anthocyanins on neural stem cells and aging mice. Aging 2020, 12, 17738–17753. [Google Scholar] [CrossRef]
- Hu, G.; Xia, Y.; Chen, B.; Zhang, J.; Gong, L.; Chen, Y.; Li, Q.; Wang, Y.; Deng, Z. ESC-sEVs Rejuvenate Aging Hippocampal NSCs by Transferring SMADs to Regulate the MYT1-Egln3-Sirt1 Axis. Mol. Ther. 2021, 29, 103–120. [Google Scholar] [CrossRef]
- Xiao, Y.Z.; Yang, M.; Xiao, Y.; Guo, Q.; Huang, Y.; Li, C.J.; Cai, D.; Luo, X.H. Reducing Hypothalamic Stem Cell Senescence Protects against Aging-Associated Physiological Decline. Cell Metab. 2020, 31, 534–548.e5. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.-K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nat. Cell Biol. 2003, 425, 962–967. [Google Scholar] [CrossRef]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005, 19, 1432–1437. [Google Scholar] [CrossRef]
- Bruggeman, S.W.; Valk-Lingbeek, M.E.; Van Der Stoop, P.P.; Jacobs, J.J.; Kieboom, K.; Tanger, E.; Hulsman, D.; Leung, C.; Arsenijevic, Y.; Marino, S.; et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef]
- Micheli, L.; D’Andrea, G.; Ceccarelli, M.; Ferri, A.; Scardigli, R.; Tirone, F. p16Ink4a Prevents the Activation of Aged Quiescent Dentate Gyrus Stem Cells by Physical Exercise. Front. Cell. Neurosci. 2019, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nat. Cell Biol. 2006, 443, 448–452. [Google Scholar] [CrossRef]
- Kippin, T.E.; Martens, D.J.; Van Der Kooy, D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005, 19, 756–767. [Google Scholar] [CrossRef]
- Qiu, J.; Takagi, Y.; Harada, J.; Rodrigues, N.; Moskowitz, M.A.; Scadden, D.T.; Cheng, T. Regenerative Response in Ischemic Brain Restricted by p21cip1/waf1. J. Exp. Med. 2004, 199, 937–945. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef]
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Anwar, T.; Sen, B.; Aggarwal, S.; Nath, R.; Pathak, N.; Katoch, A.; Aiyaz, M.; Trehanpati, N.; Khosla, S.; Ramakrishna, G. Differentially regulated gene expression in quiescence versus senescence and identification of ARID5A as a quiescence associated marker. J. Cell. Physiol. 2018, 233, 3695–3712. [Google Scholar] [CrossRef] [PubMed]
- Salmenperä, P.; Karhemo, P.-R.; Räsänen, K.; Laakkonen, P.; Vaheri, A. Fibroblast spheroids as a model to study sustained fibroblast quiescence and their crosstalk with tumor cells. Exp. Cell Res. 2016, 345, 17–24. [Google Scholar] [CrossRef]
- Tomé, M.; Tchorz, J.; Gassmann, M.; Bettler, B. Constitutive activation of Notch2 signalling confers chemoresistance to neural stem cells via transactivation of fibroblast growth factor receptor-1. Stem Cell Res. 2019, 35, 101390. [Google Scholar] [CrossRef]
- Yadirgi, G.; Leinster, V.; Acquati, S.; Bhagat, H.; Shakhova, O.; Marino, S. Conditional Activation of Bmi1 Expression Regulates Self-renewal, Apoptosis, and Differentiation of Neural Stem/Progenitor Cells In Vitro and In Vivo. Stem Cells 2011, 29, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.M.; Reynolds, R.; Fawcett, J.W. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001, 24, 39–47. [Google Scholar] [CrossRef]
- Rowitch, D.H. Glial specification in the vertebrate neural tube. Nat. Rev. Neurosci. 2004, 5, 409–419. [Google Scholar] [CrossRef]
- Gallo, V.; Deneen, B. Glial Development: The Crossroads of Regeneration and Repair in the CNS. Neuron 2014, 83, 283–308. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, E.; de Theije, C.G.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Galvao, R.P.; Kasina, A.; McNeill, R.S.; Harbin, J.E.; Foreman, O.; Verhaak, R.G.W.; Nishiyama, A.; Miller, C.R.; Zong, H. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc. Natl. Acad. Sci. USA 2014, 111, E4214–E4223. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, N.; Kastemar, M.; Olofsson, T.; Smits, A.; Uhrbom, L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 2009, 28, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, N.; Jiang, Y.; Xie, Y.; Bolouri, H.; Kastemar, M.; Olofsson, T.; Holland, E.C.; Uhrbom, L. Oncogenic Signaling Is Dominant to Cell of Origin and Dictates Astrocytic or Oligodendroglial Tumor Development from Oligodendrocyte Precursor Cells. J. Neurosci. 2014, 34, 14644–14651. [Google Scholar] [CrossRef]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.-J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte Dynamics in the Healthy Adult CNS: Evidence for Myelin Remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.B.; Jackiewicz, M.; McKenzie, I.A.; Kougioumtzidou, E.; Grist, M.; Richardson, W.D. Remarkable Stability of Myelinating Oligodendrocytes in Mice. Cell Rep. 2017, 21, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Orthmann-Murphy, J.L.; Langseth, A.J.; Bergles, D.E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 2018, 21, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.A.; Li, A.M.; Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 2018, 21, 683–695. [Google Scholar] [CrossRef]
- Mitew, S.; Gobius, I.; Fenlon, L.R.; McDougall, S.J.; Hawkes, D.; Xing, Y.L.; Bujalka, H.; Gundlach, A.L.; Richards, L.J.; Kilpatrick, T.J.; et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal Activity Promotes Oligodendrogenesis and Adaptive Myelination in the Mammalian Brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-Resident Glial Progenitor/Stem Cells Produce Schwann Cells as well as Oligodendrocytes during Repair of CNS Demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef]
- Tang, D.G.; Tokumoto, Y.M.; Apperly, J.A.; Lloyd, A.C.; Raff, M.C. Lack of Replicative Senescence in Cultured Rat Oligodendrocyte Precursor Cells. Science 2001, 291, 868–871. [Google Scholar] [CrossRef]
- Zezula, J.; Casaccia-Bonnefil, P.; Ezhevsky, S.A.; Osterhout, D.J.; Levine, J.M.; Dowdy, S.F.; Chao, M.V.; Koff, A. p21 cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2001, 2, 27–34. [Google Scholar] [CrossRef]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; Van Wijngaarden, P.; et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell 2019, 25, 473–485.e8. [Google Scholar] [CrossRef] [PubMed]
- Kujuro, Y.; Suzuki, N.; Kondo, T. Esophageal cancer-related gene 4 is a secreted inducer of cell senescence expressed by aged CNS precursor cells. Proc. Natl. Acad. Sci. USA 2010, 107, 8259–8264. [Google Scholar] [CrossRef] [PubMed]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Fernandez-Castaneda, A.; Gaultier, A. Adult oligodendrocyte progenitor cells—Multifaceted regulators of the CNS in health and disease. Brain Behav. Immun. 2016, 57, 1–7. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.M.; Gorospe, M.; et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J.M. The Age-Related Decrease in CNS Remyelination Efficiency Is Attributable to an Impairment of Both Oligodendrocyte Progenitor Recruitment and Differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating Oligodendrocytes in Chronic Lesions of Multiple Sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cuo, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef]
- Woodruff, R.H.; Franklin, R.J.M. Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: A comparative study. Glia 1999, 25, 216–228. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Vanzulli, I.; Papanikolaou, M.; De-La-Rocha, I.C.; Pieropan, F.; Rivera, A.D.; Gomez-Nicola, D.; Verkhratsky, A.; Rodríguez, J.J.; Butt, A.M. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 94, 130–139. [Google Scholar] [CrossRef]
- Ossola, B.; Zhao, C.; Compston, A.; Pluchino, S.; Franklin, R.J.M.; Spillantini, M.G. Neuronal expression of pathological tau accelerates oligodendrocyte progenitor cell differentiation. Glia 2015, 64, 457–471. [Google Scholar] [CrossRef]
- Dunckley, T.; Beach, T.G.; Ramsey, K.E.; Grover, A.; Mastroeni, D.; Walker, D.G.; LaFleur, B.J.; Coon, K.D.; Brown, K.M.; Caselli, R.; et al. Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48, 405–415. [Google Scholar] [CrossRef]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef]
- Ginhoux, F.; Prinz, M. Origin of Microglia: Current Concepts and Past Controversies. Cold Spring Harb. Perspect. Biol. 2015, 7, a020537. [Google Scholar] [CrossRef]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-Stimulating Factor 1 Receptor Signaling Is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef]
- Nomura, K.; Vilalta, A.; Allendorf, D.H.; Hornik, T.C.; Brown, G.C. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 2017, 198, 4792–4801. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Liu, W.; Hu, X.; Hanna, M.; Caravaca, A.; Paul, S.M. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci. Rep. 2015, 5, 11161. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Kaur, C.; Rathnasamy, G.; Ling, E.-A. Biology of Microglia in the Developing Brain. J. Neuropathol. Exp. Neurol. 2017, 76, 736–753. [Google Scholar] [CrossRef]
- Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef]
- Afridi, R.; Lee, W.-H.; Suk, K. Microglia Gone Awry: Linking Immunometabolism to Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Watabe, K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. (Landmark Ed.) 2017, 22, 1805–1829. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Abels, E.R.; Maas, S.L.; Nieland, L.; Wei, Z.; Cheah, P.S.; Tai, E.; Kolsteeg, C.-J.; Dusoswa, S.A.; Ting, D.T.; Hickman, S.; et al. Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep. 2019, 28, 3105–3119.e7. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.N.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflammation 2020, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, L.; Heinen, Y.; Van Dam, A.-M.; Lucassen, P.J.; Korosi, A. Microglial Priming and Alzheimer’s Disease: A Possible Role for (Early) Immune Challenges and Epigenetics? Front. Hum. Neurosci. 2016, 10, 398. [Google Scholar] [CrossRef]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflammation 2015, 12, 114. [Google Scholar] [CrossRef]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial Activation and Increased Microglial Density Observed in the Dorsolateral Prefrontal Cortex in Autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef]
- Hendrickx, D.A.; Van Eden, C.G.; Schuurman, K.G.; Hamann, J.; Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 2017, 309, 12–22. [Google Scholar] [CrossRef]
- Thrupp, N.; Frigerio, C.S.; Wolfs, L.; Skene, N.G.; Fattorelli, N.; Poovathingal, S.; Fourne, Y.; Matthews, P.M.; Theys, T.; Mancuso, R.; et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020, 32, 108189. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, R. Detection of Senescence-Associated Heterochromatin Foci (SAHF). Methods Mol. Biol. 2013, 965, 185–196. [Google Scholar] [CrossRef]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef]
- Trias, E.; Beilby, P.R.; Kovacs, M.; Ibarburu, S.; Varela, V.; Barreto-Núñez, R.; Bradford, S.C.; Beckman, J.S.; Barbeito, L. Emergence of Microglia Bearing Senescence Markers During Paralysis Progression in a Rat Model of Inherited ALS. Front. Aging Neurosci. 2019, 11, 42. [Google Scholar] [CrossRef]
- Chen, N.C.; Partridge, A.T.; Sell, C.; Torres, C.; Martín-García, J. Fate of microglia during HIV-1 infection: From activation to senescence? Glia 2017, 65, 431–446. [Google Scholar] [CrossRef]
- Caldeira, C.; Oliveira, A.F.; Ecunha, C.; Vaz, A.R.; Falcão, A.S.; Efernandes, A.; Ebrites, D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell. Neurosci. 2014, 8, 152. [Google Scholar] [CrossRef]
- Kronenberg, G.; Uhlemann, R.; Schöner, J.; Wegner, S.; Boujon, V.; Deigendesch, N.; Endres, M.; Gertz, K. Repression of telomere-associated genes by microglia activation in neuropsychiatric disease. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 473–477. [Google Scholar] [CrossRef]
- Yu, H.-M.; Zhao, Y.-M.; Luo, X.-G.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.-Y.; Chen, S.-D.; Wang, X.-Y. Repeated Lipopolysaccharide Stimulation Induces Cellular Senescence in BV2 Cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence That Aging And Amyloid Promote Microglial Cell Senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef]
- Nikodemova, M.; Small, A.L.; Kimyon, R.S.; Watters, J.J. Age-dependent differences in microglial responses to systemic inflammation are evident as early as middle age. Physiol. Genom. 2016, 48, 336–344. [Google Scholar] [CrossRef]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia Priming with Aging and Stress. Neuropsychopharmacology 2016, 42, 318–333. [Google Scholar] [CrossRef]
- Stojiljkovic, M.R.; Ain, Q.; Bondeva, T.; Heller, R.; Schmeer, C.; Witte, O.W. Phenotypic and functional differences between senescent and aged murine microglia. Neurobiol. Aging 2019, 74, 56–69. [Google Scholar] [CrossRef]
- Scheffold, A.; Holtman, I.R.; Dieni, S.; Brouwer, N.; Katz, S.-F.; Jebaraj, B.M.C.; Kahle, P.J.; Hengerer, B.; Lechel, A.; Stilgenbauer, S.; et al. Telomere shortening leads to an acceleration of synucleinopathy and impaired microglia response in a genetic mouse model. Acta Neuropathol. Commun. 2016, 4, 87. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; DeMaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Liu, L.; Zeng, S. Senescence and Cancer. Cancer Transl. Med. 2018, 4, 70–74. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Huang, F.; Wang, M.; Liu, R.; Wang, J.Z.; Schadt, E.; Haroutunian, V.; Katsel, P.; Zhang, B.; Wang, X. CDT2-controlled cell cycle reentry regulates the pathogenesis of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, A.; Sekiya, M.; Beckmann, N.D.; Quan, X.; Schrode, N.; Fernando, M.B.; Yu, A.; Zhu, L.; Cao, J.; et al. Transformative Network Modeling of Multi-omics Data Reveals Detailed Circuits, Key Regulators, and Potential Therapeutics for Alzheimer’s Disease. Neuron 2021, 109, 257–272.e14. [Google Scholar] [CrossRef]
- Neff, R.A.; Wang, M.; Vatansever, S.; Guo, L.; Ming, C.; Wang, Q.; Wang, E.; Horgusluoglu-Moloch, E.; Song, W.-M.; Li, A.; et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv. 2021, 7, eabb5398. [Google Scholar] [CrossRef]
- Zhang, Y.; Kim, M.S.; Jia, B.; Yan, J.; Zuniga-Hertz, J.P.; Han, C.; Cai, D. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nat. Cell Biol. 2017, 548, 52–57. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillispie, G.J.; Sah, E.; Krishnamurthy, S.; Ahmidouch, M.Y.; Zhang, B.; Orr, M.E. Evidence of the Cellular Senescence Stress Response in Mitotically Active Brain Cells—Implications for Cancer and Neurodegeneration. Life 2021, 11, 153. https://doi.org/10.3390/life11020153
Gillispie GJ, Sah E, Krishnamurthy S, Ahmidouch MY, Zhang B, Orr ME. Evidence of the Cellular Senescence Stress Response in Mitotically Active Brain Cells—Implications for Cancer and Neurodegeneration. Life. 2021; 11(2):153. https://doi.org/10.3390/life11020153
Chicago/Turabian StyleGillispie, Gregory J., Eric Sah, Sudarshan Krishnamurthy, Mohamed Y. Ahmidouch, Bin Zhang, and Miranda E. Orr. 2021. "Evidence of the Cellular Senescence Stress Response in Mitotically Active Brain Cells—Implications for Cancer and Neurodegeneration" Life 11, no. 2: 153. https://doi.org/10.3390/life11020153
APA StyleGillispie, G. J., Sah, E., Krishnamurthy, S., Ahmidouch, M. Y., Zhang, B., & Orr, M. E. (2021). Evidence of the Cellular Senescence Stress Response in Mitotically Active Brain Cells—Implications for Cancer and Neurodegeneration. Life, 11(2), 153. https://doi.org/10.3390/life11020153