
Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Animal Characteristics
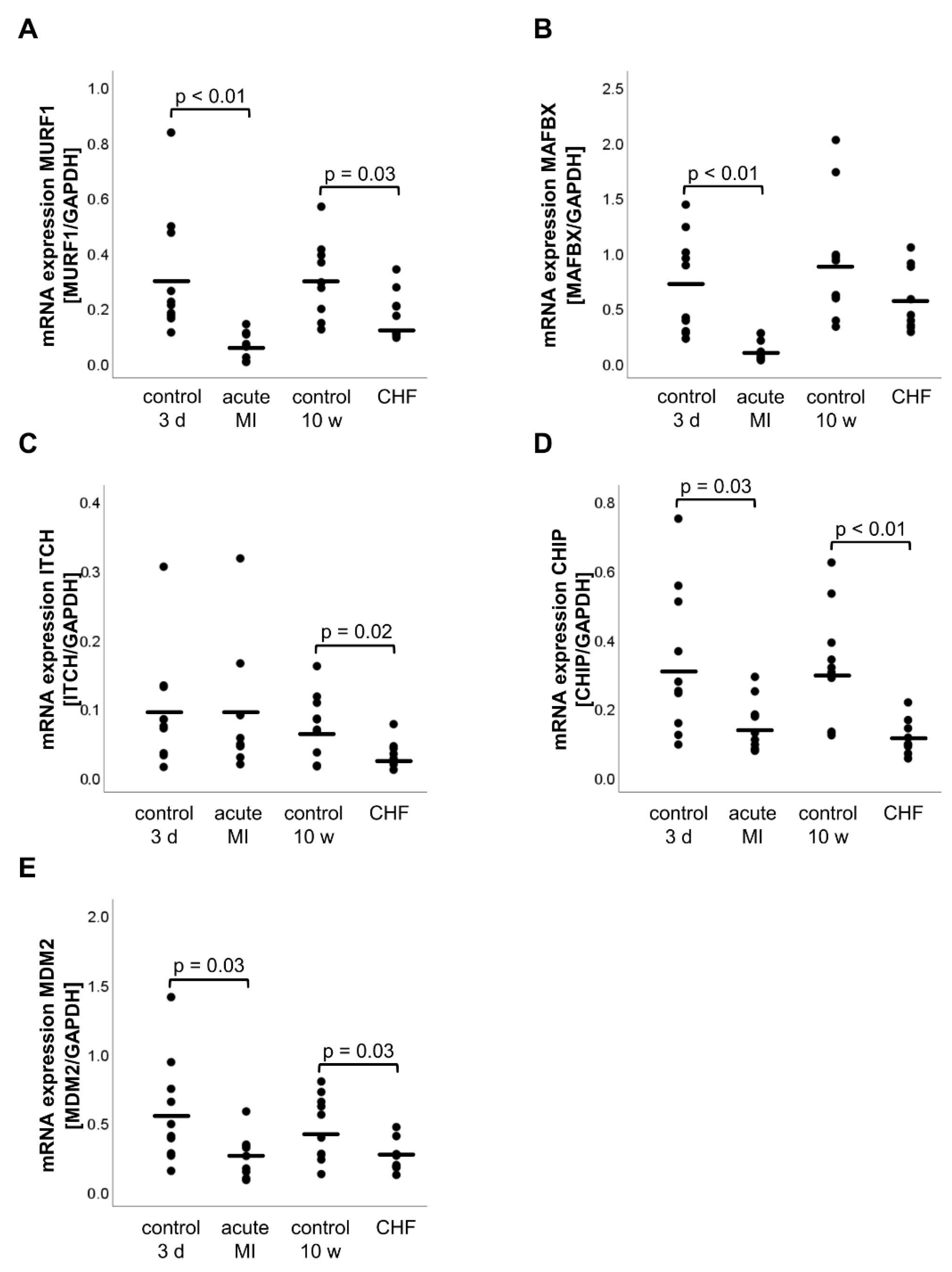
2.2. mRNA and Protein Expression of E3 Ligases
2.3. mRNA Expression of the CHIP-Interacting Molecule BAG3 (Bcl-2-Associated Athanogene 3)
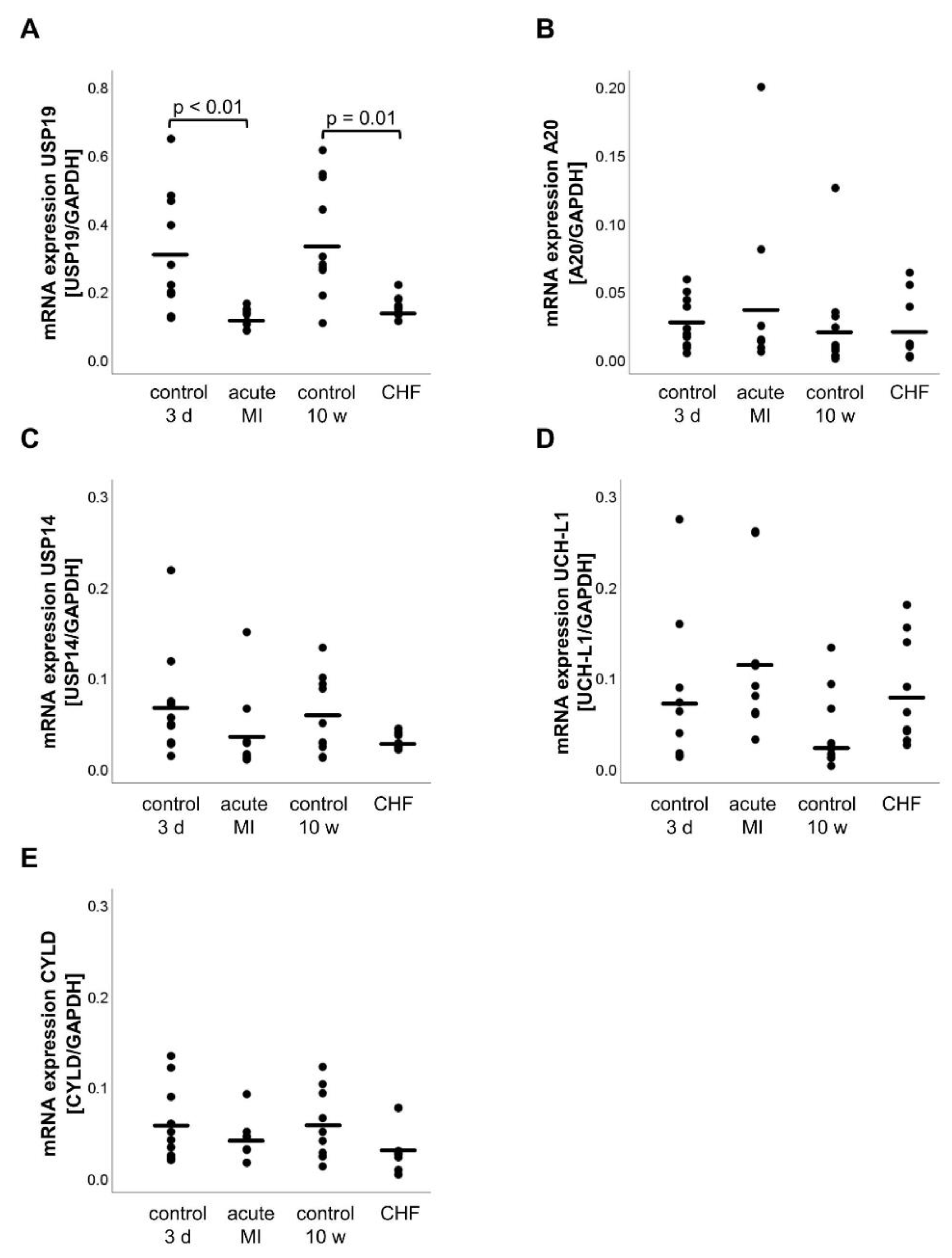
2.4. mRNA Expression of DUBs
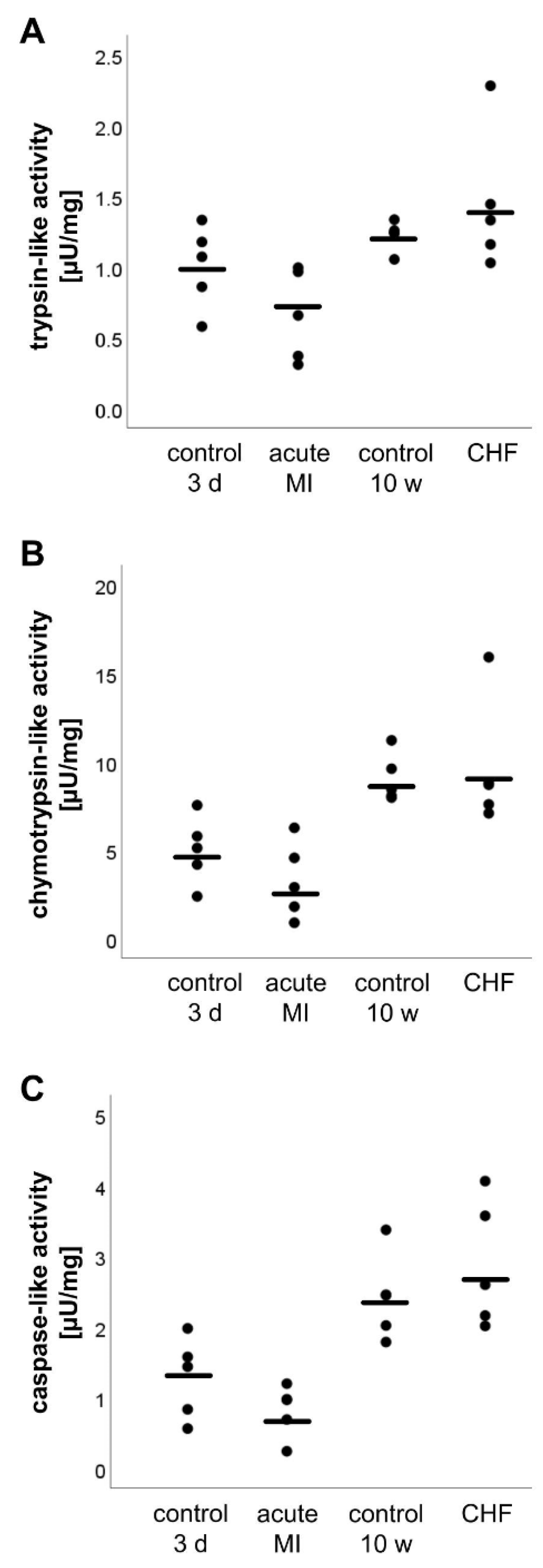
2.5. Analysis of the Proteasome Activity
3. Discussion
4. Materials and Methods
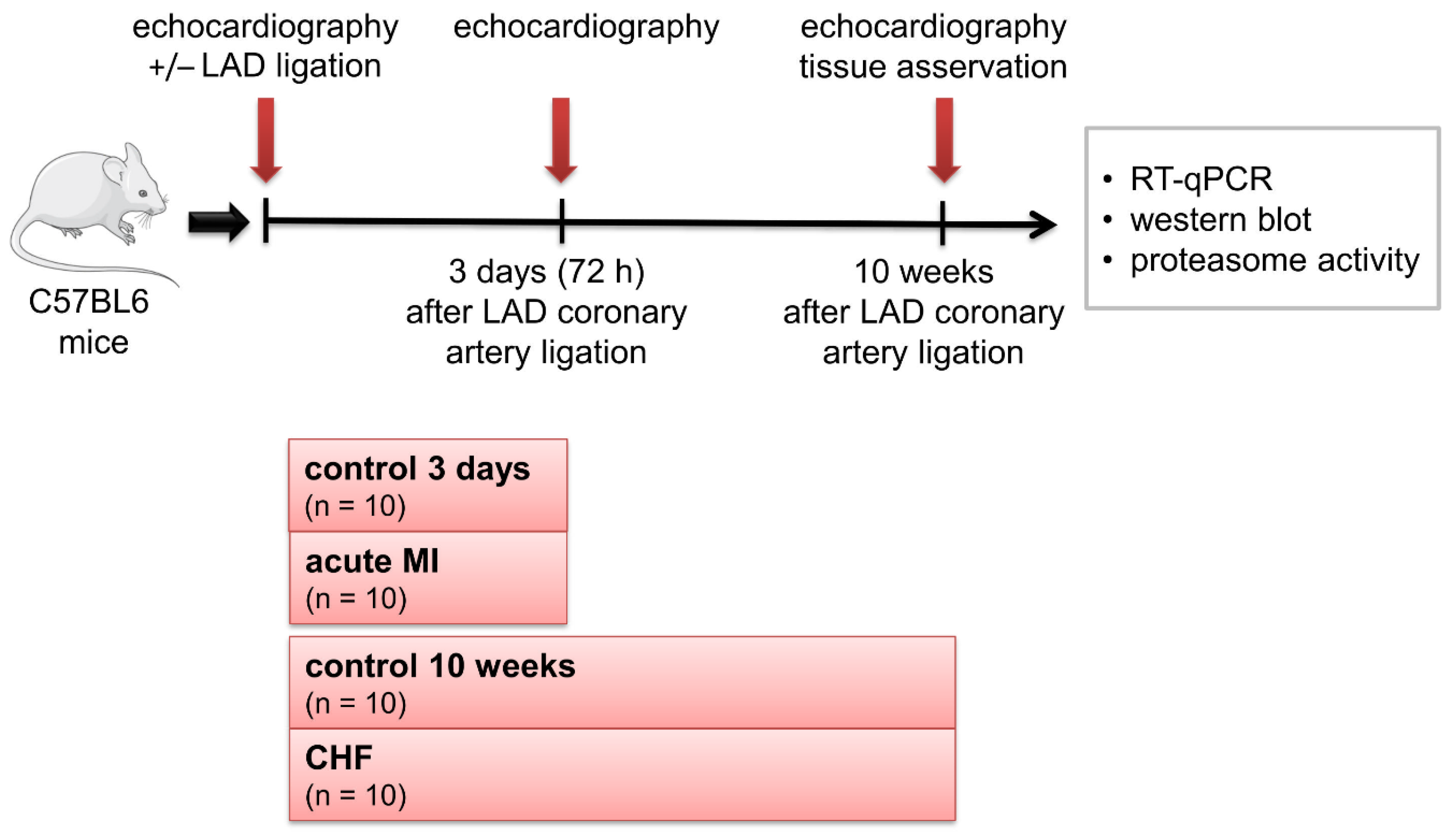
4.1. Study Design
4.2. Echocardiography Histology of the Heart
4.3. RNA Isolation
4.4. RT-qPCR Analysis
4.5. Proteasomal Activity
4.6. Immunoblot/Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis and Western Blotting Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, Q.Y.; Wang, H.X.; Liu, X.H.; Guo, C.X.; Hua, Q.; Yu, X.H.; Li, N.; Yang, Y.Z.; Du, J.; Xia, Y.L.; et al. Circulating E3 ligases are novel and sensitive biomarkers for diagnosis of acute myocardial infarction. Clin. Sci. 2015, 128, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Chai, R.; Liu, B.; Zhang, Z.; Zhang, S.; Zhang, J.; Liao, Y.; Cai, J.; Xia, X.; Li, A.; et al. Ubiquitin-specific protease 14 regulates cardiac hypertrophy progression by increasing GSK-3β phosphorylation. Biochem. Biophys. Res. Commun. 2016, 478, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Bevilacqua, A.; Pulinilkunnil, T.; Kienesberger, P.; Tannu, M.; Patterson, C. The role of ubiquitin ligases in cardiac disease. J. Mol. Cell Cardiol. 2014, 71, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Mearini, G.; Schlossarek, S.; Willis, M.S.; Carrier, L. The ubiquitin-proteasome system in cardiac dysfunction. Biochim. Biophys. Acta 2008, 1782, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Proteasome dysfunction in cardiomyopathies. J. Physiol. 2017, 595, 4051–4071. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Mangner, N.; Werner, S.; Glaser, S.; Kullnick, Y.; Schrepper, A.; Doenst, T.; Oberbach, A.; Linke, A.; Steil, L.; et al. Diaphragm muscle weakness in mice is early-onset post-myocardial infarction and associated with elevated protein oxidation. J. Appl Physiol. 2015, 118, 11–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parry, T.L.; Willis, M.S. Cardiac ubiquitin ligases: Their role in cardiac metabolism, autophagy, cardioprotection and therapeutic potential. Biochim. Biophys. Acta 2016, 1862, 2259–2269. [Google Scholar] [CrossRef]
- Zaglia, T.; Milan, G.; Ruhs, A.; Franzoso, M.; Bertaggia, E.; Pianca, N.; Carpi, A.; Carullo, P.; Pesce, P.; Sacerdoti, D.; et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Investig. 2014, 124, 2410–2424. [Google Scholar] [CrossRef]
- Gupta, I.; Varshney, N.K.; Khan, S. Emergence of Members of TRAF and DUB of Ubiquitin Proteasome System in the Regulation of Hypertrophic Cardiomyopathy. Front. Genet. 2018, 9, 336. [Google Scholar] [CrossRef]
- Adams, V.; Linke, A.; Wisloff, U.; Döring, C.; Erbs, S.; Kränkel, N.; Witt, C.C.; Labeit, S.; Müller-Werdan, U.; Schuler, G.; et al. Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: Effect on myocardial contractility. Cardiovasc. Res. 2007, 73, 120–129. [Google Scholar] [CrossRef]
- Adams, V.; Linke, A.; Gielen, S.; Erbs, S.; Hambrecht, R.; Schuler, G. Modulation of Murf-1 and MAFbx expression in the myocardium by physical exercise training. Eur. J. Cardiovasc. Prev. Rehabil. 2008, 15, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Schisler, J.C.; Li, L.; Rodríguez, J.E.; Hilliard, E.G.; Charles, P.C.; Patterson, C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ. Res. 2009, 105, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Galasso, G.; De Rosa, R.; Piscione, F.; Iaccarino, G.; Vosa, C.; Sorriento, D.; Piccolo, R.; Rapacciuolo, A.; Walsh, K.; Chiariello, M. Myocardial expression of FOXO3a-Atrogin-1 pathway in human heart failure. Eur. J. Heart Fail. 2010, 12, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Léger, B.; Senese, R.; Al-Khodairy, A.W.; Dériaz, O.; Gobelet, C.; Giacobino, J.P.; Russell, A.P. Atrogin-1, MuRF1, and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients. Muscle Nerve 2009, 40, 69–78. [Google Scholar] [CrossRef]
- Conraads, V.M.; Vrints, C.J.; Rodrigus, I.E.; Hoymans, V.Y.; Van Craenenbroeck, E.M.; Bosmans, J.; Claeys, M.J.; Van Herck, P.; Linke, A.; Schuler, G.; et al. Depressed expression of MuRF1 and MAFbx in areas remote of recent myocardial infarction: A mechanism contributing to myocardial remodeling? Basic Res. Cardiol. 2010, 105, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Spänig, S.; Kellermann, K.; Dieterlen, M.T.; Noack, T.; Lehmann, S.; Borger, M.A.; Garbade, J.; Barac, Y.D.; Emrich, F. The Ubiquitin Proteasome System in Ischemic and Dilated Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 6354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, C.; Gao, K.; Wang, D.; Mao, J.; An, J.; Xu, C.; Wu, D.; Yu, H.; Liu, J.O.; et al. The ubiquitin ligase itch regulates apoptosis by targeting thioredoxin-interacting protein for ubiquitin-dependent degradation. J. Biol. Chem. 2010, 285, 8869–8879. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, S.; Garbade, J.; Emrich, F.; Klaeske, K.; Borger, M.A.; Lehmann, S.; Jawad, K.; Dieterlen, M.T. Ischemic Cardiomyopathy Affects the Thioredoxin System in the Human Myocardium. J. Card Fail. 2019, 25, 204–212. [Google Scholar] [CrossRef]
- Portbury, A.L.; Ronnebaum, S.M.; Zungu, M.; Patterson, C.; Willis, M.S. Back to your heart: Ubiquitin proteasome system-regulated signal transduction. J. Mol. Cell Cardiol. 2012, 52, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.E.; Schisler, J.C.; Patterson, C.; Willis, M.S. Seek and destroy: The ubiquitin-proteasome system in cardiac disease. Curr. Hypertens. Rep. 2009, 11, 396–405. [Google Scholar] [CrossRef]
- Lam, B.; Roudier, E. Considering the Role of Murine Double Minute 2 in the Cardiovascular System? Front. Cell Dev. Biol. 2019, 7, 320. [Google Scholar] [CrossRef]
- Aiken, J.; Roudier, E.; Ciccone, J.; Genevieve, D.; Stromberg, A.; Vojnovic, J.; Olfert, I.M.; Haas, T.; Gustafsson, T.; Grenier, G.; et al. Phosphorylation of murine double minute-2 on Ser166 is downstream of VEGF-A in exercised skeletal muscle and regulates primary endothelial cell migration and FoxO gene expression. FASEB J. 2016, 30, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.D.; Semenchenko, K.; Wasylyk, B. Crosstalk between Mdm2, p53 and HIF1-α: Distinct responses to oxygen stress and implications for tumour hypoxia. Subcell Biochem. 2014, 85, 199–214. [Google Scholar] [CrossRef]
- Naito, A.T.; Okada, S.; Minamino, T.; Iwanaga, K.; Liu, M.L.; Sumida, T.; Nomura, S.; Sahara, N.; Mizoroki, T.; Takashima, A.; et al. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ. Res. 2010, 106, 1692–1702. [Google Scholar] [CrossRef]
- Stark, A.M.; Hugo, H.H.; Witzel, P.; Mihajlovic, Z.; Mehdorn, H.M. Age-related expression of p53, Mdm2, EGFR and Msh2 in glioblastoma multiforme. Zentralbl. Neurochir. 2003, 64, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tian, S.; Chen, Y.; Zhang, C.; Xie, W.; Xia, X.; Cui, J.; Wang, R.F. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. 2016, 35, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Yi, T.; Li, H.; Yan, Z.; Lv, Z.; Li, G.; Wang, Y. Ubiquitin C-terminal hydrolase L1 (UCHL1) regulates post-myocardial infarction cardiac fibrosis through glucose-regulated protein of 78 kDa (GRP78). Sci. Rep. 2020, 10, 10604. [Google Scholar] [CrossRef]
- Behl, C. BAG3 and friends: Co-chaperones in selective autophagy during aging and disease. Autophagy 2011, 7, 795–798. [Google Scholar] [CrossRef]
- Thomasova, D.; Mulay, S.R.; Bruns, H.; Anders, H.J. p53-independent roles of MDM2 in NF-κB signaling: Implications for cancer therapy, wound healing, and autoimmune diseases. Neoplasia 2012, 14, 1097–1101. [Google Scholar] [CrossRef]
- Ahmed, N.; Zeng, M.; Sinha, I.; Polin, L.; Wei, W.Z.; Rathinam, C.; Flavell, R.; Massoumi, R.; Venuprasad, K. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat. Immunol. 2011, 12, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Gilmore-Hebert, M.; Ramabhadran, R.; Stern, D.F. Interactions of ErbB4 and Kap1 connect the growth factor and DNA damage response pathways. Mol. Cancer Res. 2010, 8, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Léger, B.; Vergani, L.; Sorarù, G.; Hespel, P.; Derave, W.; Gobelet, C.; D‘Ascenzio, C.; Angelini, C.; Russell, A.P. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 2006, 20, 583–585. [Google Scholar] [CrossRef]
- Brattelid, T.; Winer, L.H.; Levy, F.O.; Liestøl, K.; Sejersted, O.M.; Andersson, K.B. Reference gene alternatives to Gapdh in rodent and human heart failure gene expression studies. BMC Mol. Biol. 2010, 11, 22. [Google Scholar] [CrossRef]
- Mangner, N.; Bowen, T.S.; Werner, S.; Fischer, T.; Kullnick, Y.; Oberbach, A.; Linke, A.; Steil, L.; Schuler, G.; Adams, V. Exercise Training Prevents Diaphragm Contractile Dysfunction in Heart Failure. Med. Sci. Sports Exerc. 2016, 48, 2118–2124. [Google Scholar] [CrossRef]
- Adams, V.; Bowen, S.T.; Werner, S.; Barthel, P.; Amberger, C.; Konzer, A.; Graumann, J.; Sehr, P.; Lewis, J.; Provaznik, J.; et al. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 2019, 10, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Primer3web. Available online: http://primer3.ut.ee (accessed on 12 December 2020).
- Feirer, N.; Dieterlen, M.T.; Klaeske, K.; Kiefer, P.; Oßmann, S.; Salameh, A.; Borger, M.A.; Hoyer, A. Impact of Custodiol-N cardioplegia on acute kidney injury after cardiopulmonary bypass. Clin. Exp. Pharmacol. Physiol. 2020, 47, 640–649. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control 3 d (n = 10) | Acute MI (n = 10) | p Value | Control 10 w (n = 10) | CHF (n = 10) | p Value | |
|---|---|---|---|---|---|---|
| Body weight [g] | 18.5 ± 0.7 | 16.4 ± 0.9 | <0.01 | 22.0 ± 1.0 | 21.9 ± 1.9 | 0.90 |
| Heart weight [mg] | 94.1 ± 7.9 | 116.9 ± 16.6 | <0.01 | 111.9 ± 15.8 | 175.3 ± 67.8 | <0.01 |
| Ejection fraction [%] | 60.5 ± 11.9 | 17.2 ± 17.6 | <0.01 | 59.3 ± 14.6 | 15.0 ± 4.5 | <0.01 |
| Fractional shortening [%] | 31.9 ± 8.1 | 11.5 ± 6.6 | <0.01 | 31.9 ± 10.7 | 6.8 ± 2.0 | <0.01 |
| Infarct size [%] | 0.0 ± 0.0 | 57.1 ± 3.3 | <0.01 | 0.0 ± 0.0 | 42.5 ± 13.1 | <0.01 |
| Gene | Sequence (5′-3′) | RT-qPCR Product Lenght (bp) | |
|---|---|---|---|
| MURF1 | forward | tgagtaactgcatctccatgct | 118 |
| reverse | tcacctggtggctattctcc | ||
| MAFBX | forward | tttgcaaacactgccacatt | 106 |
| reverse | cttgaggggaaagtgagacg | ||
| ITCH | forward | gggaggatttgctgacctta | 121 |
| reverse | tccaggcggttaaaacaagt | ||
| CHIP | forward | ccagctggagatggagagtt | 122 |
| reverse | gcgaagggcactaggaatatc | ||
| MDM2 | forward | taatctccgcttggaaggac | 104 |
| reverse | tctgtagcccttgatgaggaa | ||
| USP19 | forward | tatcgaaggttgtcccttgc | 111 |
| reverse | gcgtgtgggtattcacagatt | ||
| USP14 | forward | cccagtgagtaagtccttagatgtt | 100 |
| reverse | tcaaatcagaccagatcacga | ||
| A20 | forward | cacgactcacctgatcaacg | 105 |
| reverse | cgtgctgaacaagctcaaag | ||
| UCH-L1 | forward | gaccatcggaaactcctgtg | 134 |
| reverse | ggacagcttctccgtttcag | ||
| CYLD | forward | acgagtgcagggagtgctat | 116 |
| reverse | ttcaacctcctgggatgaag | ||
| BAG3 | forward | cagtggttgacaggcctca | 100 |
| reverse | ctgggcctggcttactttct | ||
| GAPDH | forward | aactttggcattgtggaagg | 132 |
| reverse | ggatgcagggatgatgttct |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaeske, K.; Dix, M.; Adams, V.; Jawad, K.; Eifert, S.; Etz, C.; Saeed, D.; Borger, M.A.; Dieterlen, M.-T. Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure. Life 2021, 11, 1430. https://doi.org/10.3390/life11121430
Klaeske K, Dix M, Adams V, Jawad K, Eifert S, Etz C, Saeed D, Borger MA, Dieterlen M-T. Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure. Life. 2021; 11(12):1430. https://doi.org/10.3390/life11121430
Chicago/Turabian StyleKlaeske, Kristin, Maria Dix, Volker Adams, Khalil Jawad, Sandra Eifert, Christian Etz, Diyar Saeed, Michael A. Borger, and Maja-Theresa Dieterlen. 2021. "Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure" Life 11, no. 12: 1430. https://doi.org/10.3390/life11121430
APA StyleKlaeske, K., Dix, M., Adams, V., Jawad, K., Eifert, S., Etz, C., Saeed, D., Borger, M. A., & Dieterlen, M.-T. (2021). Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure. Life, 11(12), 1430. https://doi.org/10.3390/life11121430