
Imaging of Intracellular and Plasma Membrane Pools of PI(4,5)P2 and PI4P in Human Platelets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Human Platelet Isolation and Platelet Spreading
2.3. HEK293T and BALB3T3 Cells
2.4. Immunostaining and Confocal Fluorescence Microscopy
2.5. Statistical Analysis
3. Results
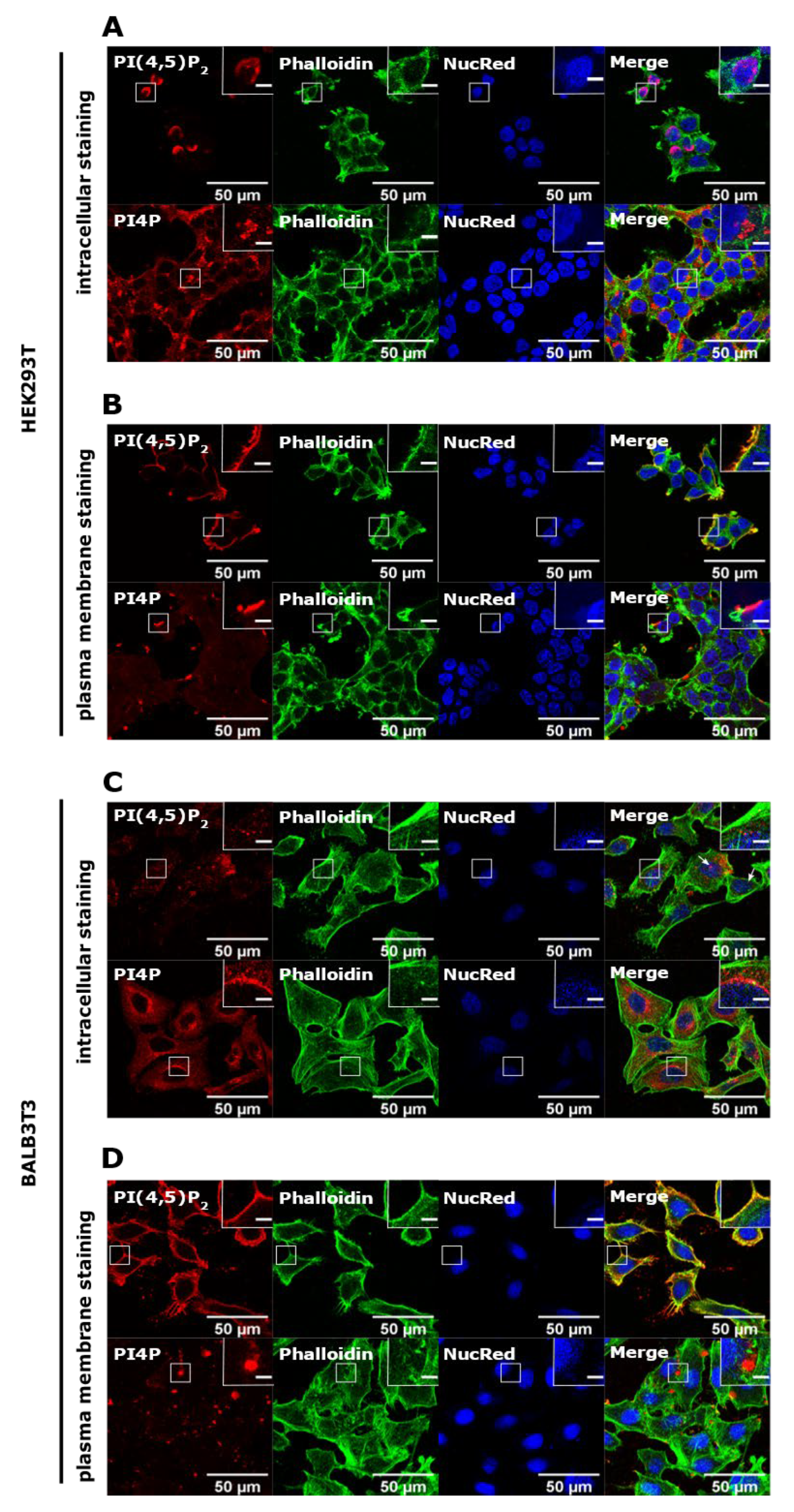
3.1. PI(4,5)P2 and PI4P Localize at Different Cellular Compartments in HEK293T and BALB3T3 Cells
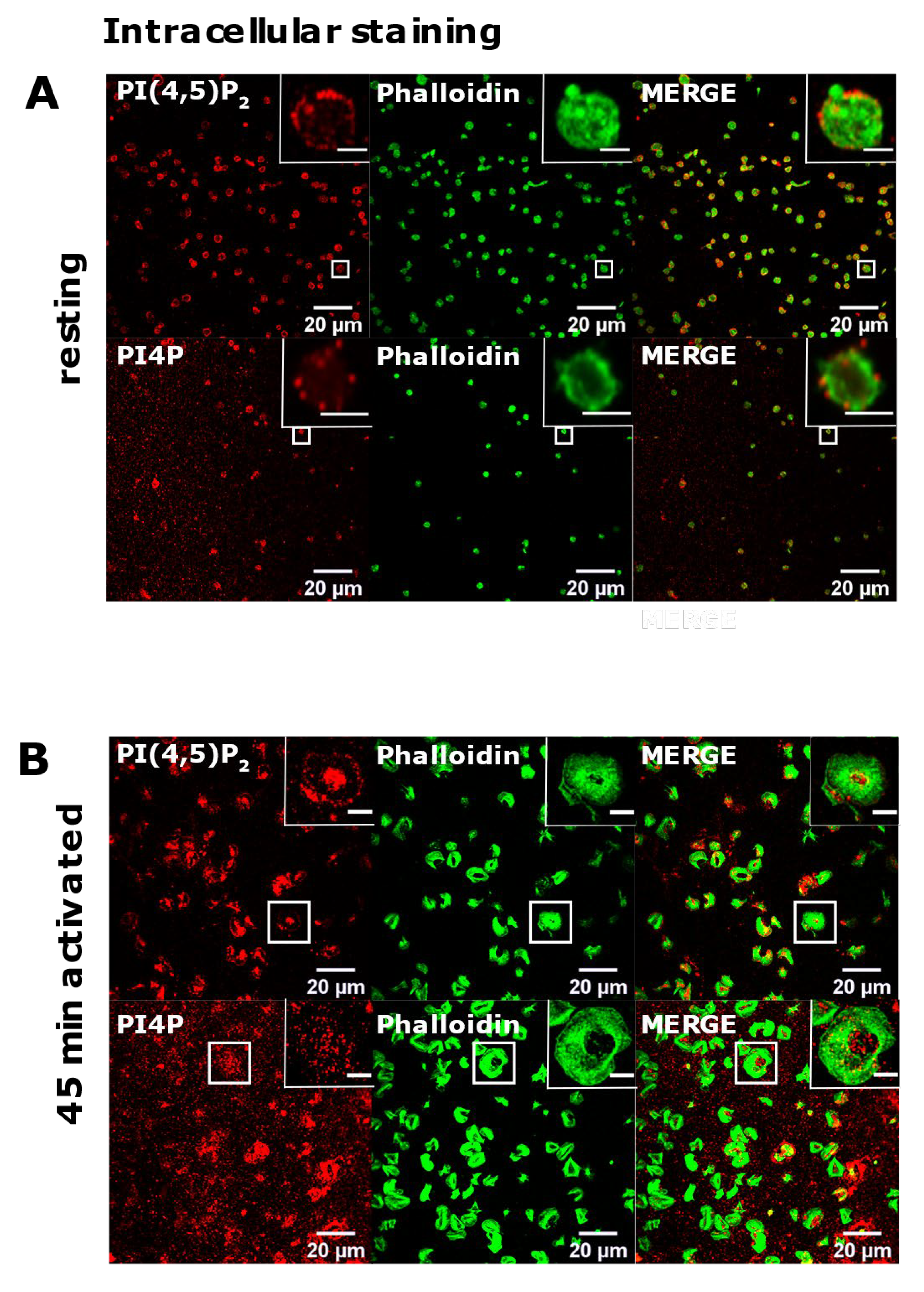
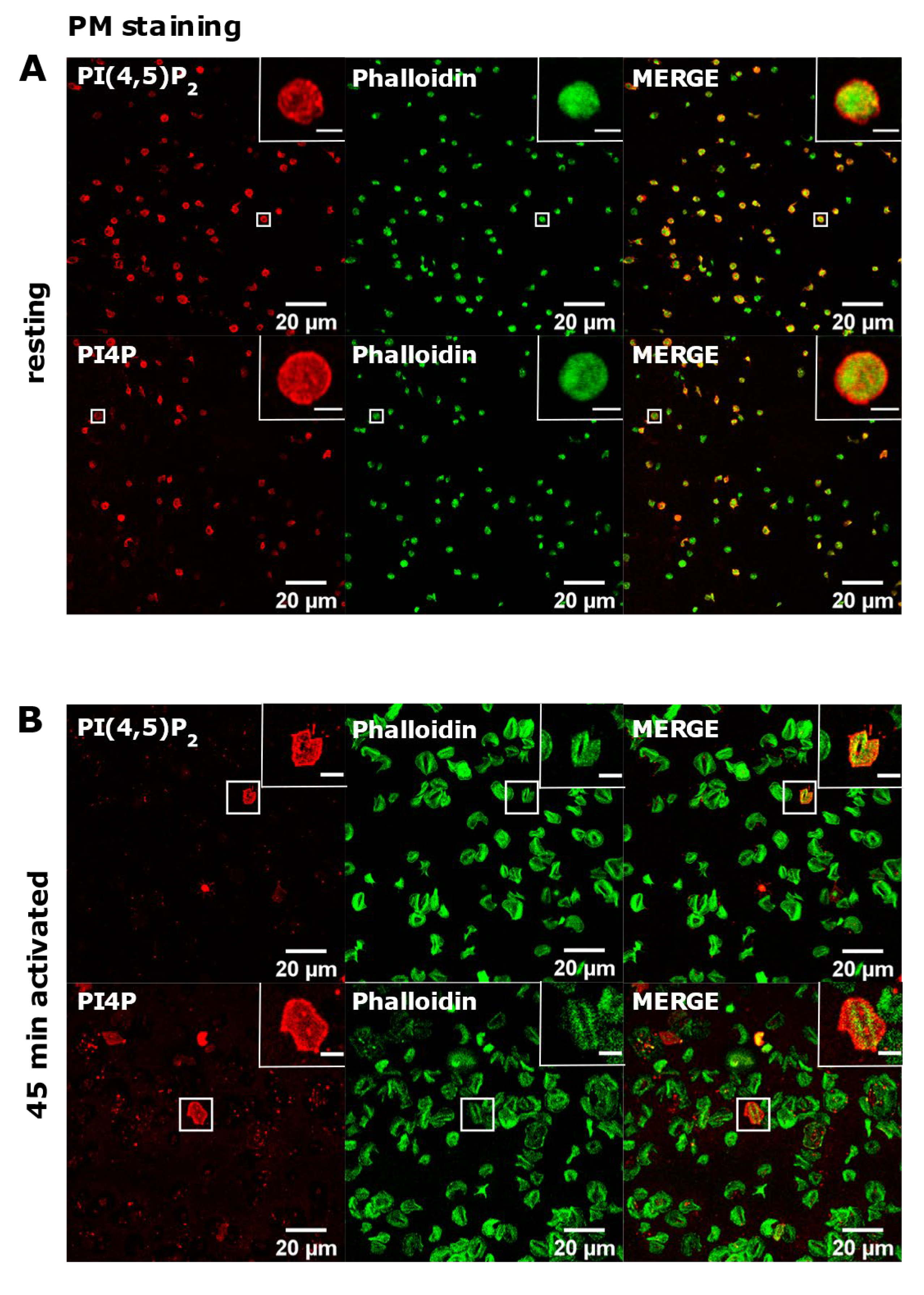
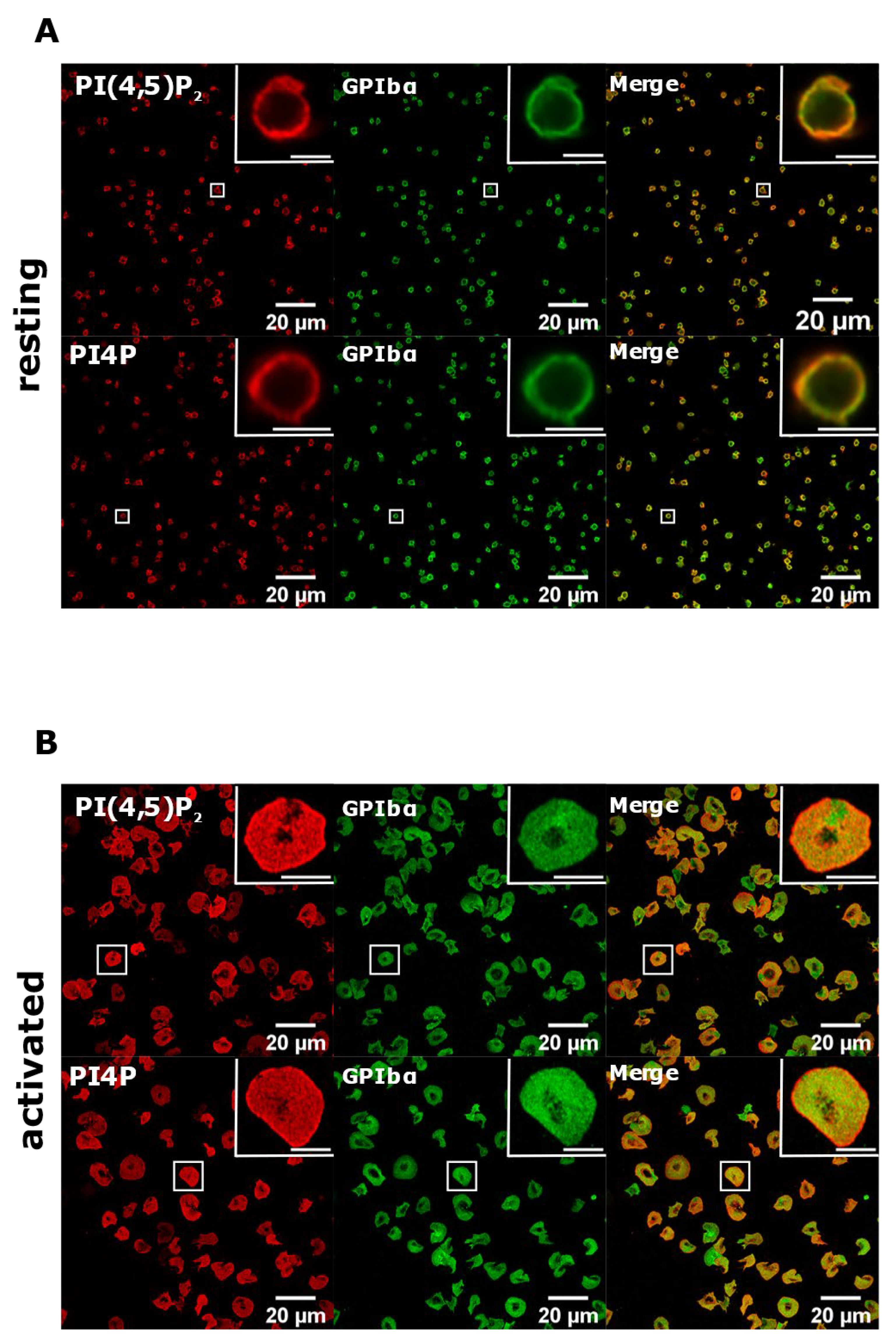
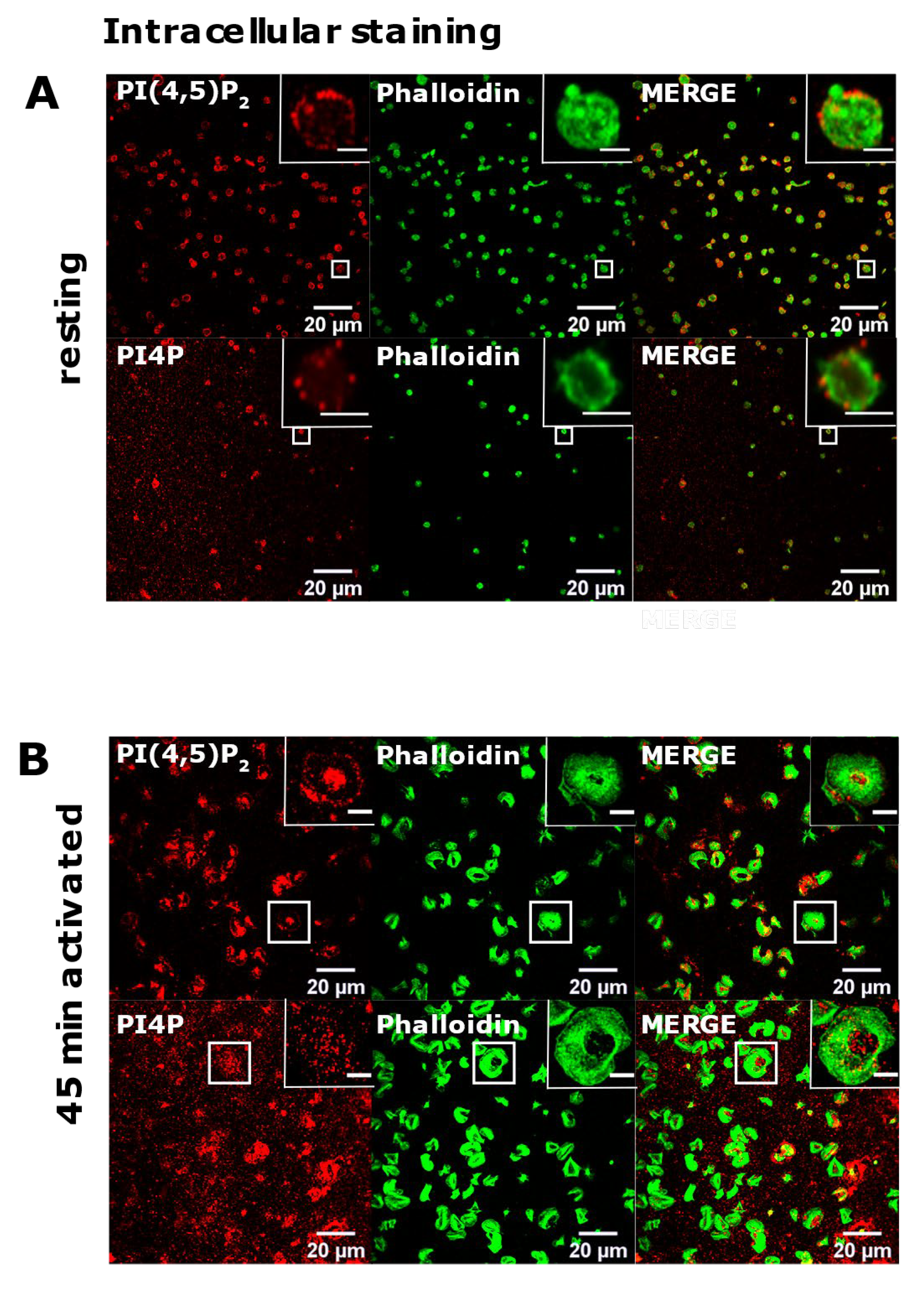
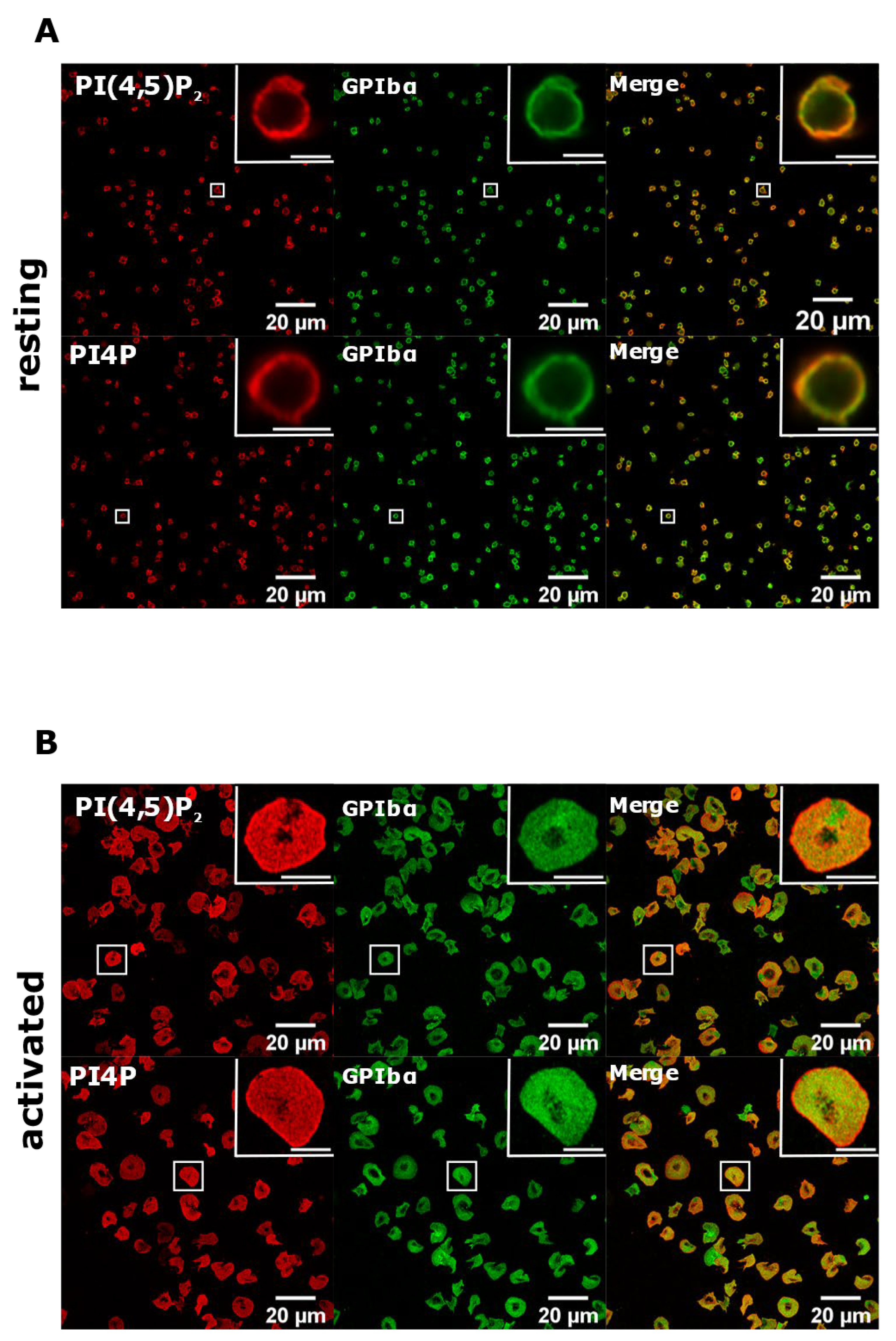
3.2. Resting and Activated Platelets Readily Stain for the Intracellular Pools but Not PM Pools of PI(4,5)P2 and PI4P
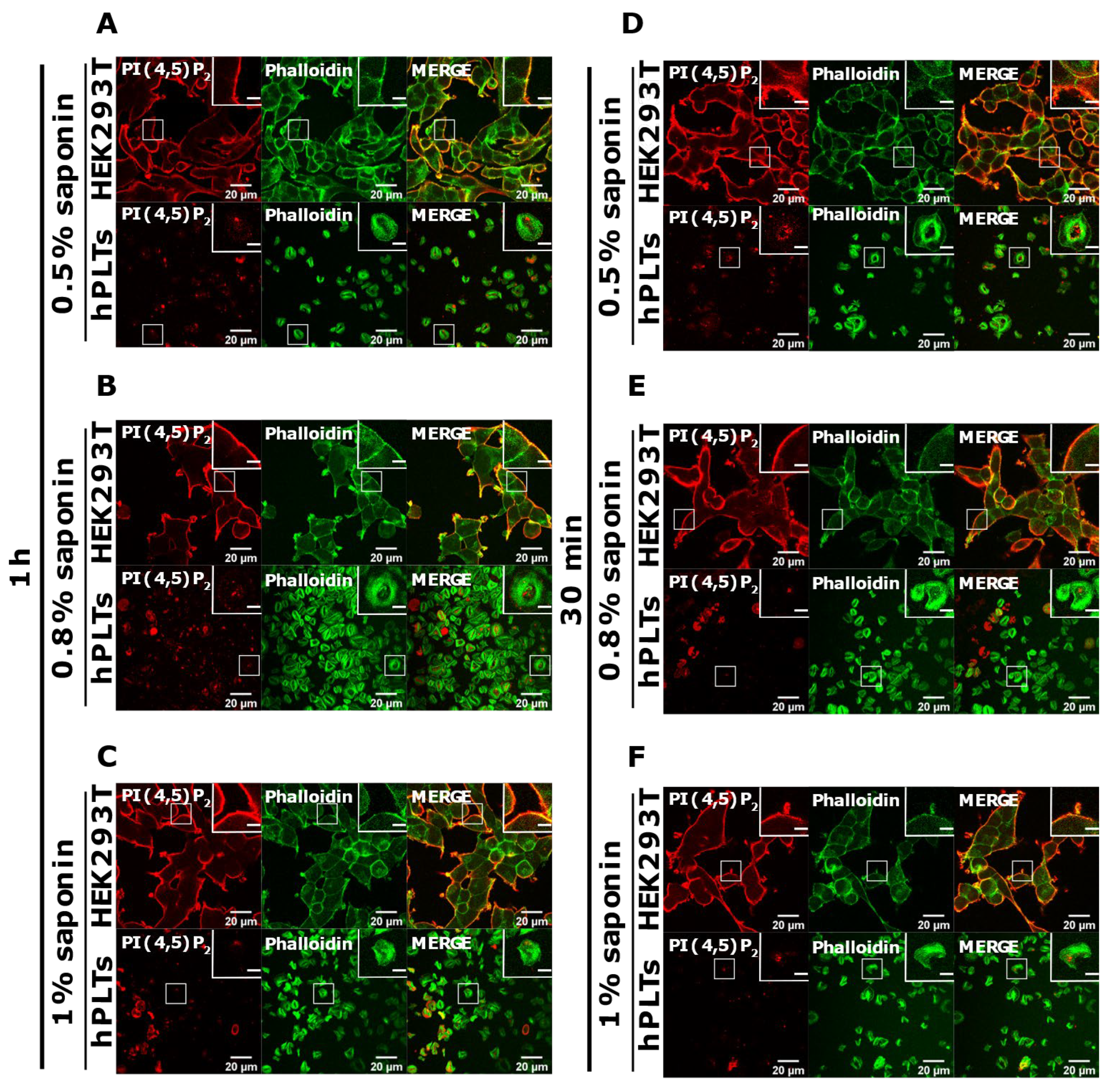
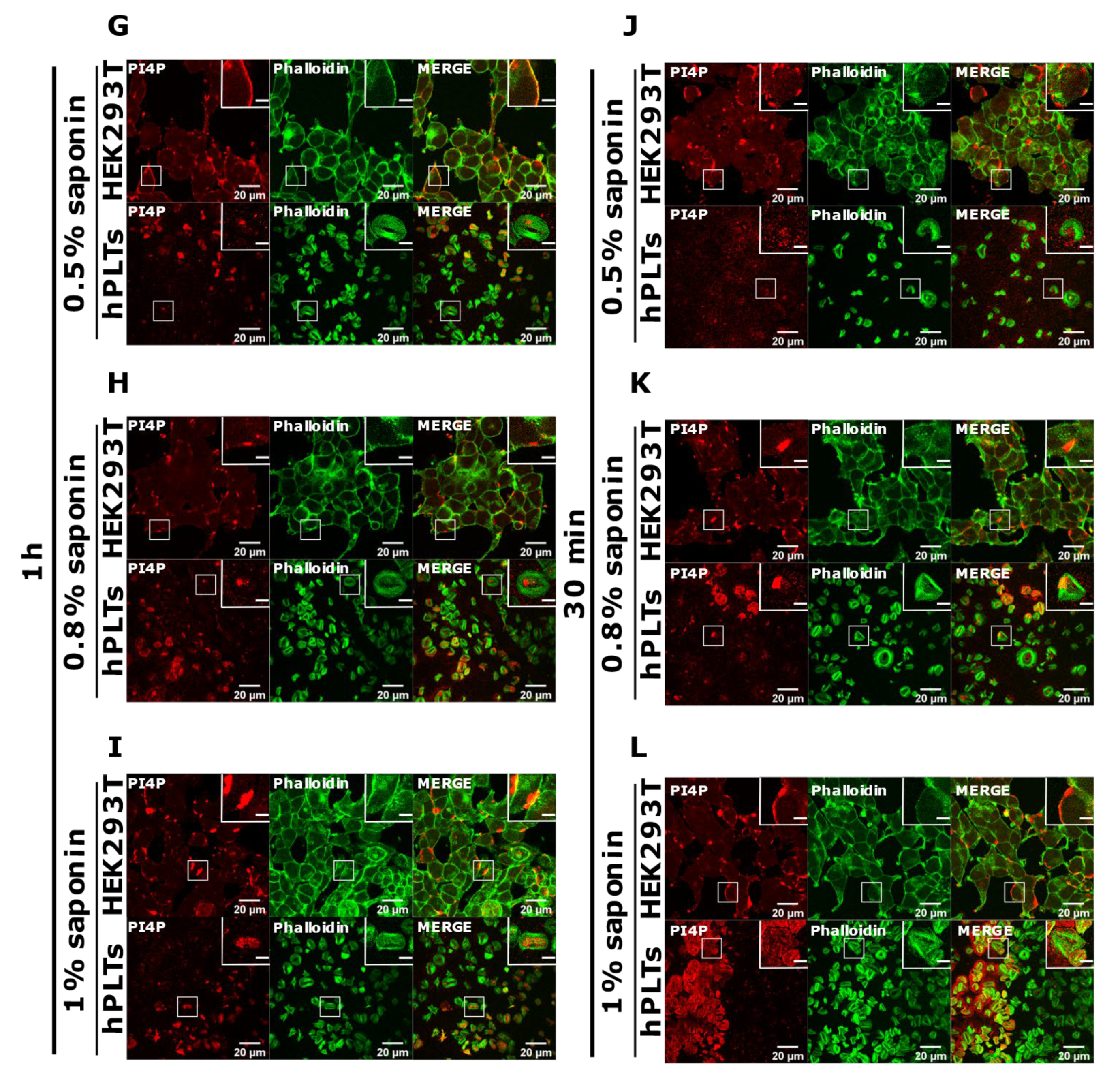
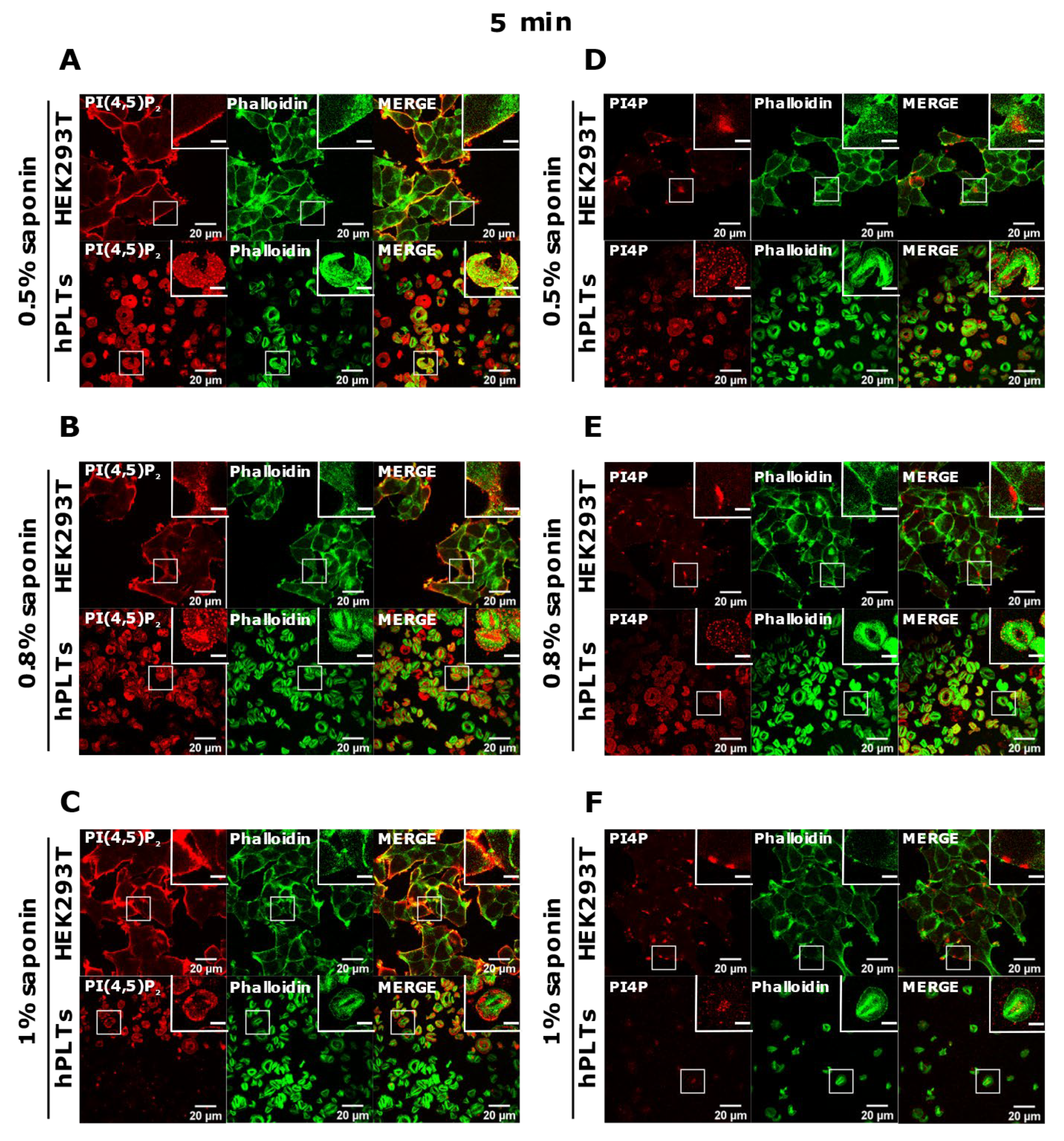
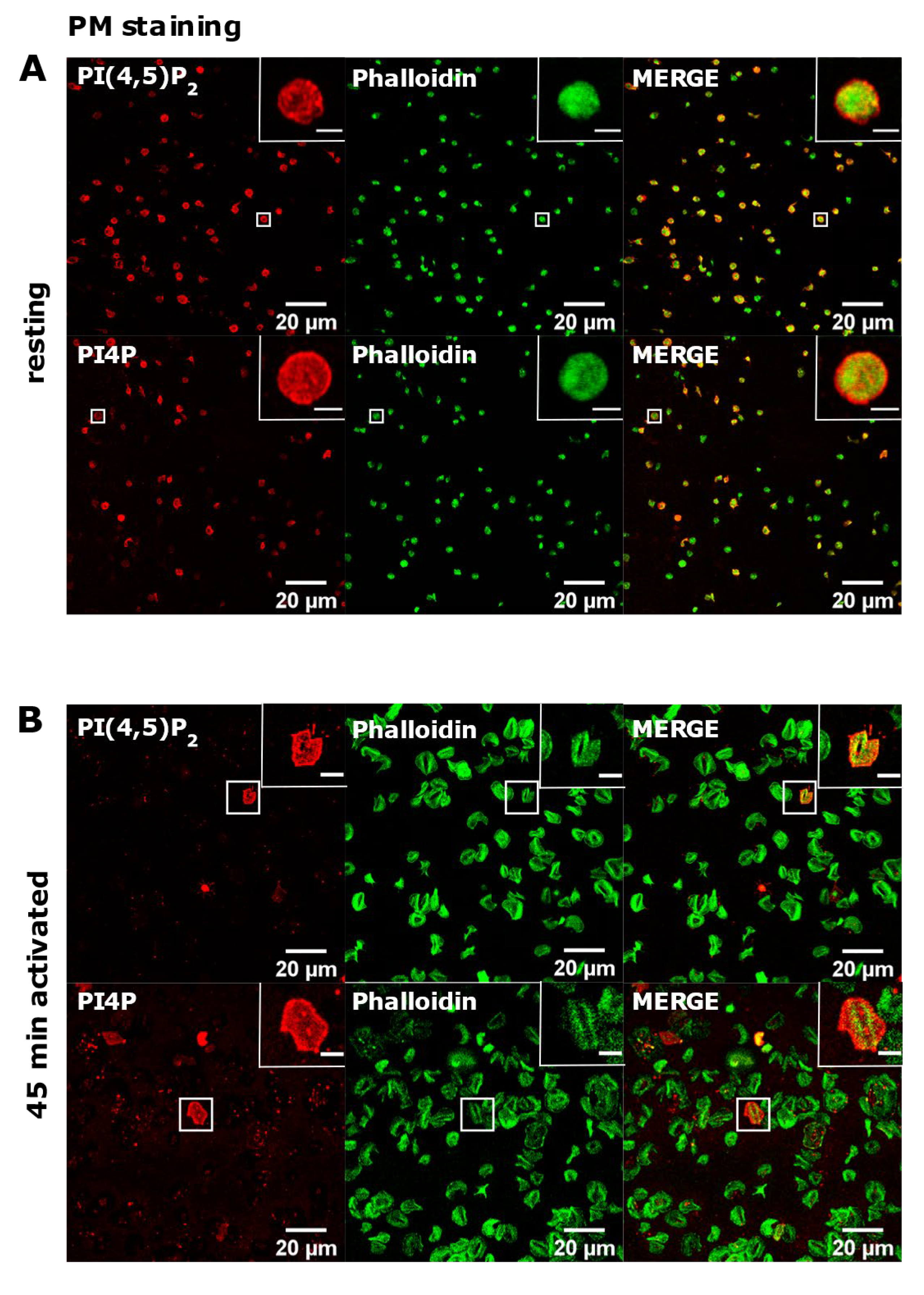
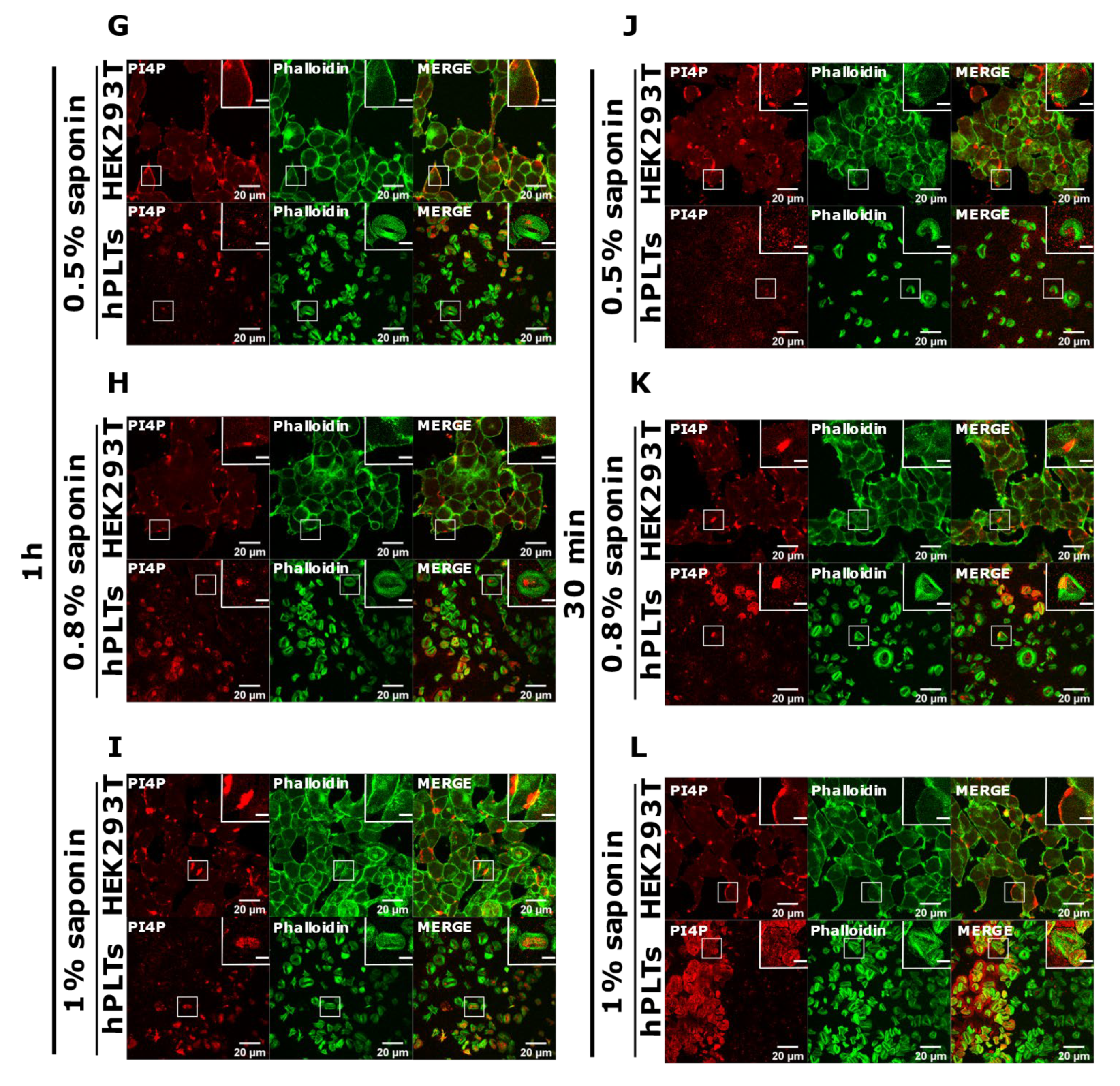
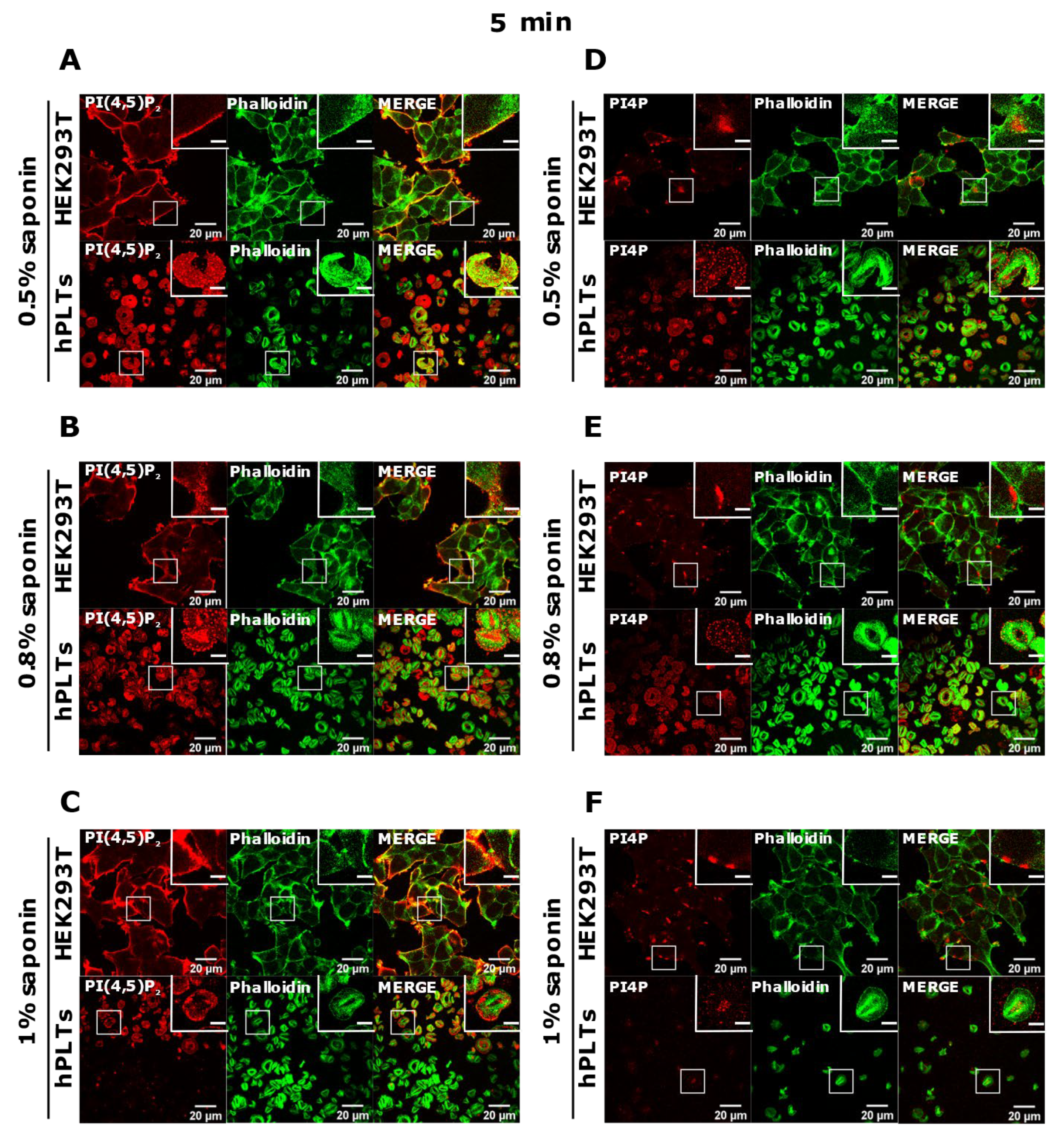
3.3. PM Staining of PI(4,5)P2 and PI4P Can Be Improved by Decreasing the Permeabilization Time in Activated Platelets
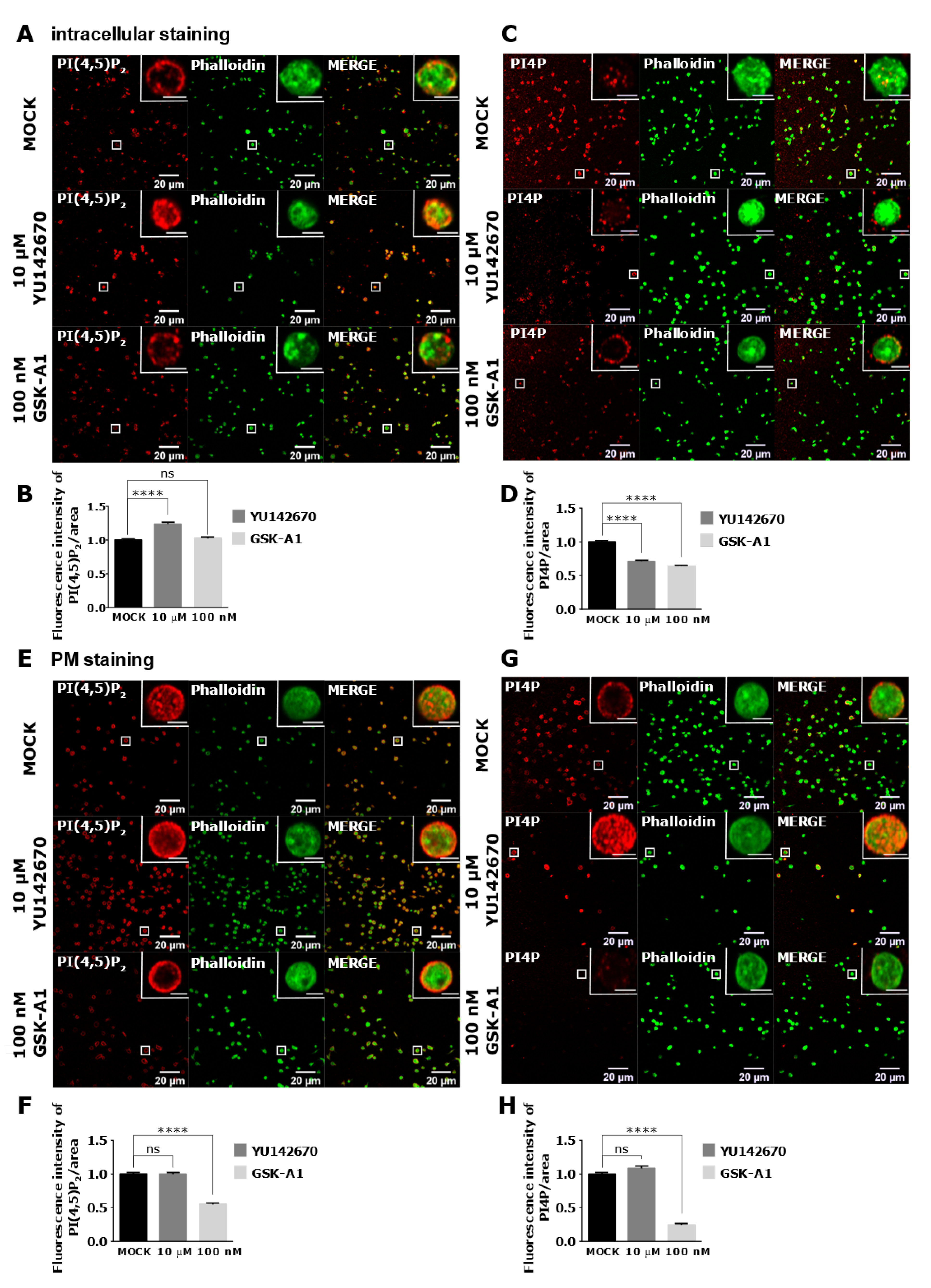
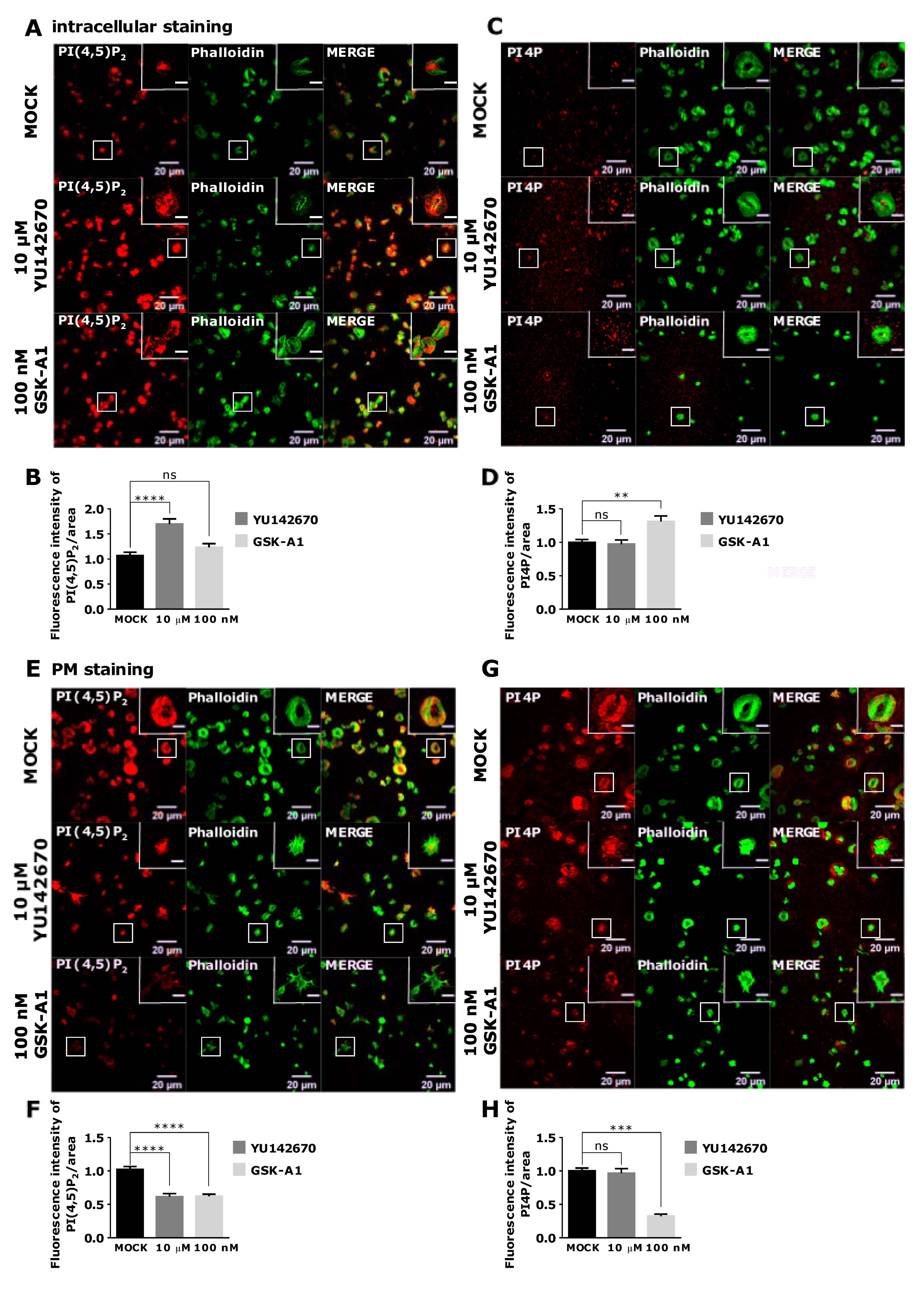
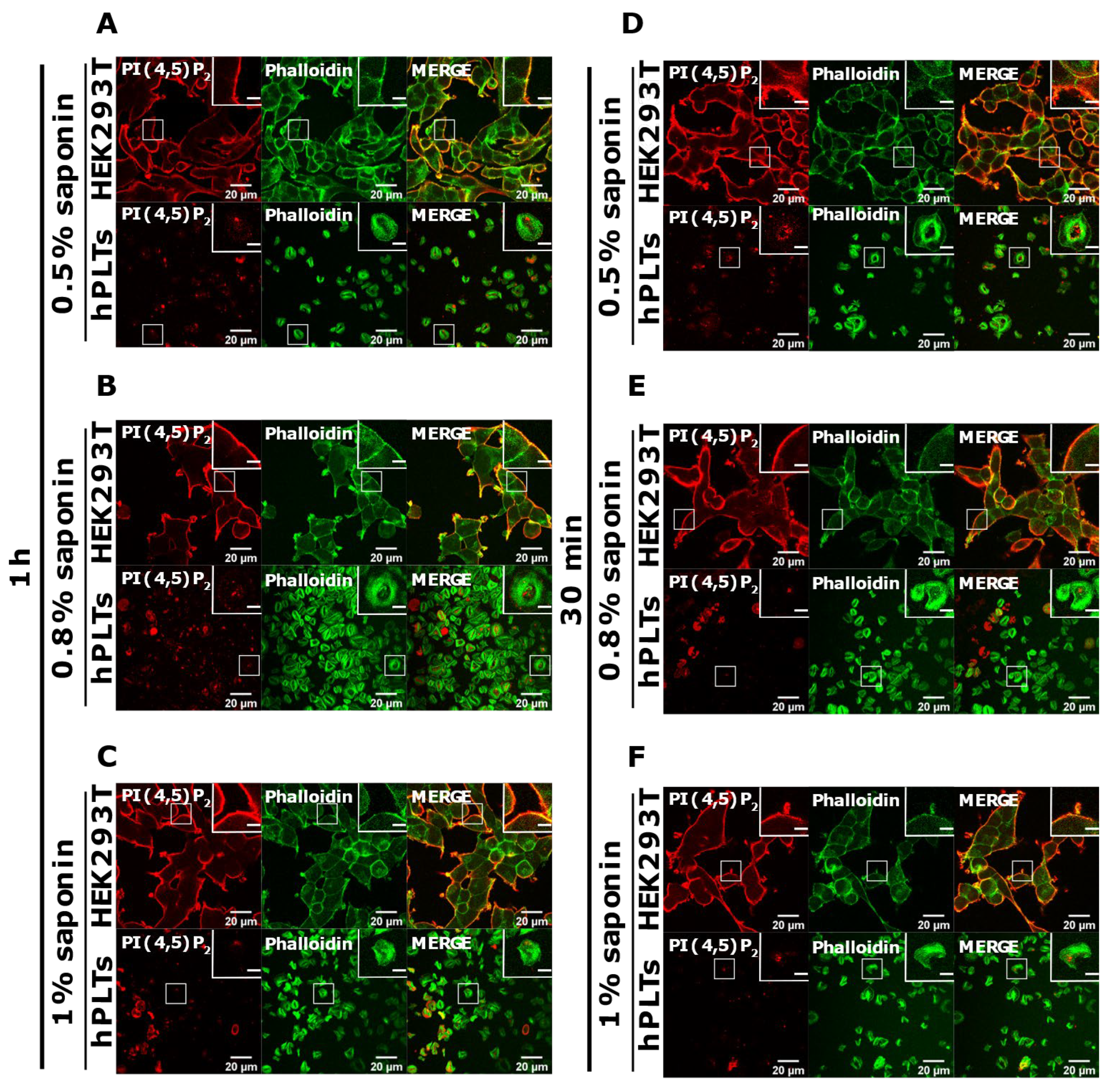
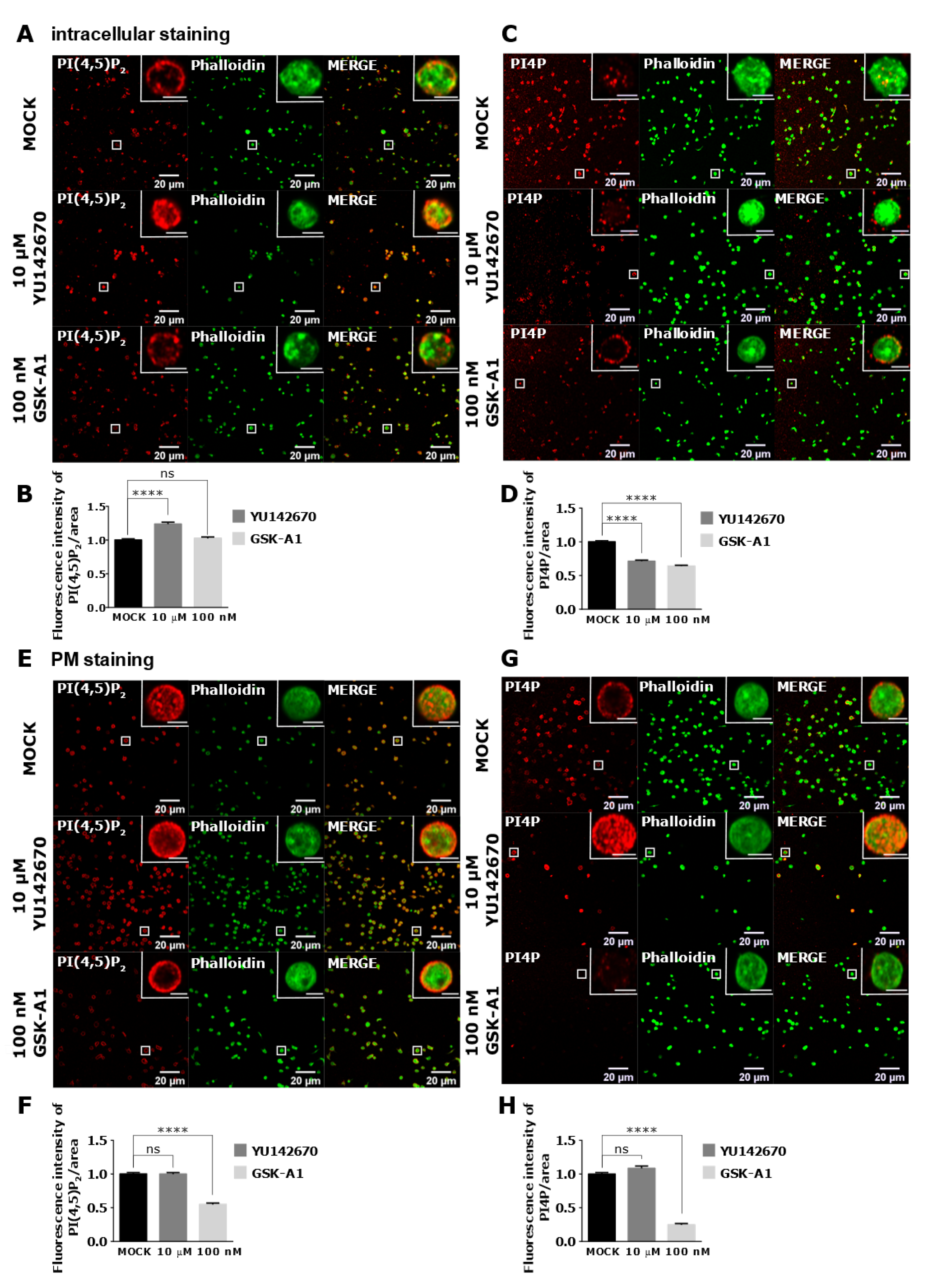
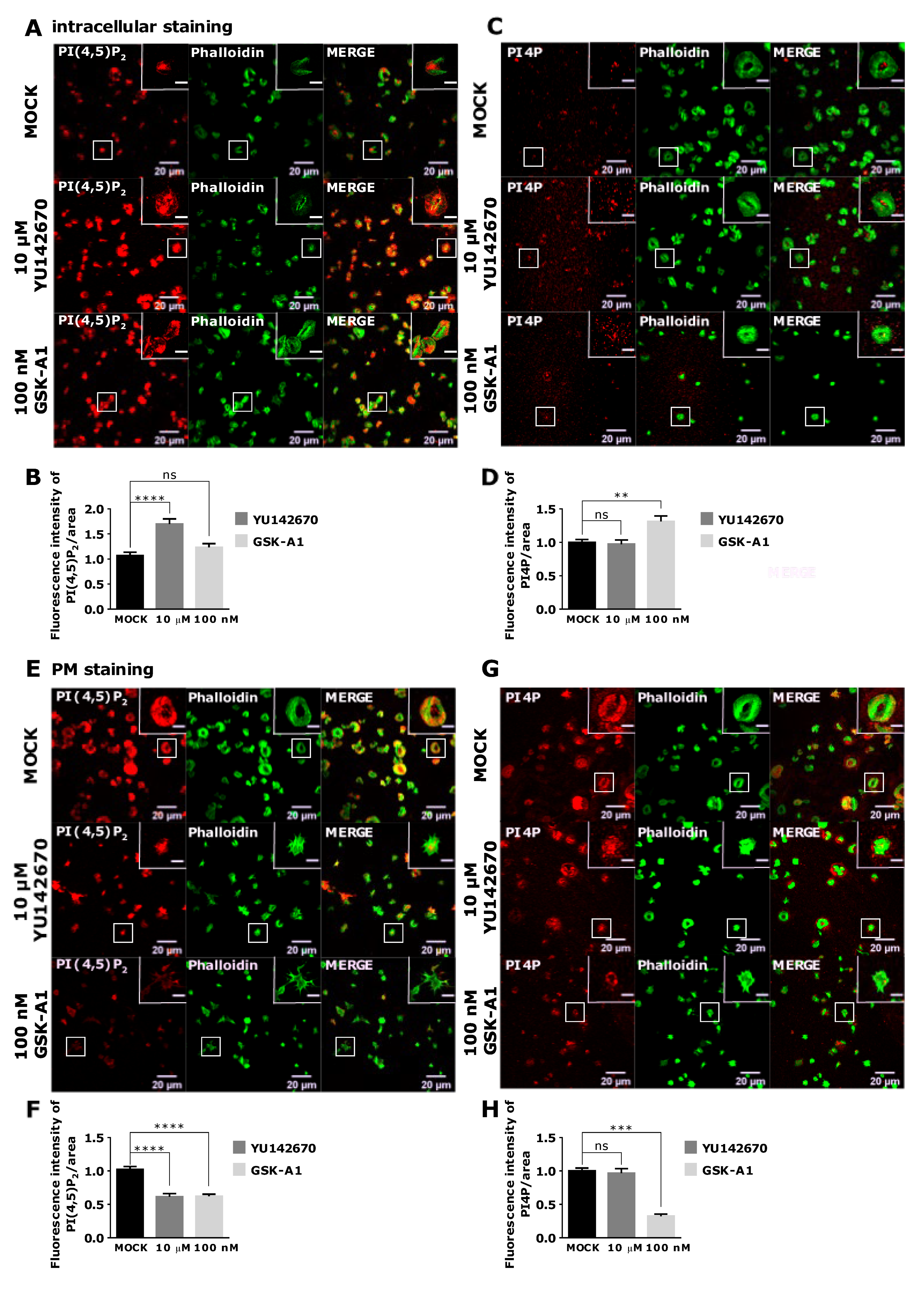
3.4. Intracellular and PM PI(4,5)P2 and PI4P Can Be Modulated by OCRL and PI4KIIIα Inhibitors in Resting and Activated Platelets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gresele, P.; Born, G.V.R.; Patrono, C.; Page, C.P. (Eds.) Antiplatelet Agents. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; Volume 210, ISBN 978-3-642-29422-8. [Google Scholar]
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and Biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankratz, S.; Bittner, S.; Kehrel, B.E.; Langer, H.F.; Kleinschnitz, C.; Meuth, S.G.; Göbel, K. The Inflammatory Role of Platelets: Translational Insights from Experimental Studies of Autoimmune Disorders. Int. J. Mol. Sci. 2016, 17, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelson, A.D. Platelets, 3rd ed.; Academic Press: Amsterdam, The Netherlands, 2013; ISBN 978-0-12-387837-3. [Google Scholar]
- Balla, T. Phosphoinositides: Tiny Lipids with Giant Impact on Cell Regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef]
- Gamper, N.; Shapiro, M.S. Target-specific PIP2signalling: How might it work? J. Physiol. 2007, 582, 967–975. [Google Scholar] [CrossRef]
- Suh, B.-C.; Hille, B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 2005, 15, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Katan, M.; Cockcroft, S. Phosphatidylinositol(4,5)bisphosphate: Diverse functions at the plasma membrane. Essays Biochem. 2020, 64, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Krauß, M.; Haucke, V. Phosphoinositides: Regulators of Membrane Traffic and Protein Function. FEBS Lett. 2007, 581, 2105–2111. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.; Machner, M.; Balla, T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 2014, 205, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Szentpetery, Z.; Varnai, P.; Balla, T. Acute manipulation of Golgi phosphoinositides to assess their importance in cellular trafficking and signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 8225–8230. [Google Scholar] [CrossRef] [Green Version]
- Cheong, F.Y.; Sharma, V.; Blagoveshchenskaya, A.; Oorschot, V.M.J.; Brankatschk, B.; Klumperman, J.; Freeze, H.H.; Mayinger, P. Spatial Regulation of Golgi Phosphatidylinositol-4-Phosphate Is Required for Enzyme Localization and Glycosylation Fidelity. Traffic 2010, 11, 1180–1190. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.V.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P2 Are Essential But Independent Lipid Determinants of Membrane Identity. Science 2012, 337, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.V.; Balla, T. Polyphosphoinositide Binding Domains: Key to Inositol Lipid Biology. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.V.; Schiavo, G.; Irvine, R.F. Immunocytochemical Techniques Reveal Multiple, Distinct Cellular Pools of PtdIns4P and PtdIns(4,5)P2. Biochem. J. 2009, 422, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.Y.; Heerklotz, H. Digitonin Does Not Flip across Cholesterol-Poor Membranes. J. Colloid Interface Sci. 2017, 504, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Oliver, C.; Jamur, M.C. (Eds.) Immunocytochemical Methods and Protocols. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 588, ISBN 978-1-58829-463-0. [Google Scholar]
- Gambaryan, S.; Kobsar, A.; Rukoyatkina, N.; Herterich, S.; Geiger, J.; Smolenski, A.; Lohmann, S.M.; Walter, U. Thrombin and Collagen Induce a Feedback Inhibitory Signaling Pathway in Platelets Involving Dissociation of the Catalytic Subunit of Protein Kinase A from an NFκB-IκB Complex. J. Biol. Chem. 2010, 285, 18352–18363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Shah, Z.H.; Jones, D.R.; Sommer, L.; Foulger, R.; Bultsma, Y.; D’Santos, C.; Divecha, N. Nuclear Phosphoinositides and Their Impact on Nuclear Functions. FEBS J. 2013, 280, 6295–6310. [Google Scholar] [CrossRef]
- Andrews, R.K.; Gardiner, E.E.; Shen, Y.; Whisstock, J.C.; Berndt, M.C. Glycoprotein Ib–IX–V. Int. J. Biochem. Cell Biol. 2003, 35, 1170–1174. [Google Scholar] [CrossRef]
- Nakatsu, F.; Baskin, J.M.; Chung, J.; Tanner, L.B.; Shui, G.; Lee, S.Y.; Pirruccello, M.; Hao, M.; Ingolia, N.T.; Wenk, M.R.; et al. PtdIns4P Synthesis by PI4KIIIα at the Plasma Membrane and Its Impact on Plasma Membrane Identity. J. Cell Biol. 2012, 199, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The First Comprehensive and Quantitative Analysis of Human Platelet Protein Composition Allows the Comparative Analysis of Structural and Functional Pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-Wide RNA-Seq Analysis of Human and Mouse Platelet Transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef] [Green Version]
- Roth, M.G. Phosphoinositides in Constitutive Membrane Traffic. Physiol. Rev. 2004, 84, 699–730. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Fairn, G.D. Molecular Probes to Visualize the Location, Organization and Dynamics of Lipids. J. Cell Sci. 2014, 127, 4801–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalasova, I.; Fáberová, V.; Kalendová, A.; Yildirim, S.; Uličná, L.; Venit, T.; Hozák, P. Tools for Visualization of Phosphoinositides in the Cell Nucleus. Histochem. Cell Biol. 2016, 145, 485–496. [Google Scholar] [CrossRef]
- Vicinanza, M.; Di Campli, A.; Polishchuk, E.; Santoro, M.; Di Tullio, G.; Godi, A.; Levtchenko, E.; De Leo, M.G.; Polishchuk, R.; Sandoval, L.; et al. OCRL Controls Trafficking through Early Endosomes via PtdIns4,5P 2 -Dependent Regulation of Endosomal Actin: OCRL Controls Endocytic Receptor Recycling. EMBO J. 2011, 30, 4970–4985. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, G.; Zini, N.; Rizzi, E.; Rizzoli, R.; Galanzi, A.; Ognibene, A.; Santi, S.; Matteucci, A.; Martelli, A.M.; Maraldi, N.M. Immunocytochemical Detection of Phosphatidylinositol 4,5-Bisphosphate Localization Sites within the Nucleus. J. Histochem. Cytochem. 1995, 43, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Gieni, R.S.; Hendzel, M.J. Actin Dynamics and Functions in the Interphase Nucleus: Moving toward an Understanding of Nuclear Polymeric Actin. Biochem. Cell Biol. 2009, 87, 283–306. [Google Scholar] [CrossRef]
- Mujalli, A.; Chicanne, G.; Bertrand-Michel, J.; Viars, F.; Stephens, L.; Hawkins, P.; Viaud, J.; Gaits-Iacovoni, F.; Severin, S.; Gratacap, M.; et al. Profiling of Phosphoinositide Molecular Species in Human and Mouse Platelets Identifies New Species Increasing Following Stimulation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1121–1131. [Google Scholar] [CrossRef]
- Cheung, H.Y.F.; Coman, C.; Westhoff, P.; Manke, M.; Sickmann, A.; Borst, O.; Gawaz, M.; Watson, S.P.; Heemskerk, J.W.M.; Ahrends, R. Targeted Phosphoinositides Analysis Using High-Performance Ion Chromatography-Coupled Selected Reaction Monitoring Mass Spectrometry. J. Proteome Res. 2021, 20, 3114–3123. [Google Scholar] [CrossRef]
- Mu, L.; Tu, Z.; Miao, L.; Ruan, H.; Kang, N.; Hei, Y.; Chen, J.; Wei, W.; Gong, F.; Wang, B.; et al. A Phosphatidylinositol 4,5-Bisphosphate Redistribution-Based Sensing Mechanism Initiates a Phagocytosis Programing. Nat. Commun. 2018, 9, 4259. [Google Scholar] [CrossRef] [Green Version]
- De Matteis, M.A.; Staiano, L.; Emma, F.; Devuyst, O. The 5-Phosphatase OCRL in Lowe Syndrome and Dent Disease 2. Nat. Rev. Nephrol. 2017, 13, 455–470. [Google Scholar] [CrossRef]
- Clayton, E.L.; Minogue, S.; Waugh, M.G. Mammalian Phosphatidylinositol 4-Kinases as Modulators of Membrane Trafficking and Lipid Signaling Networks. Prog. Lipid Res. 2013, 52, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Golebiewska, E.M.; Poole, A.W. Platelet Secretion: From Haemostasis to Wound Healing and Beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Wandall, H.H.; Rumjantseva, V.; Sørensen, A.L.T.; Patel-Hett, S.; Josefsson, E.C.; Bennett, E.P.; Italiano, J.E.; Clausen, H.; Hartwig, J.H.; Hoffmeister, K.M. The Origin and Function of Platelet Glycosyltransferases. Blood 2012, 120, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Williamson, J.K.; Aronova, M.A.; Prince, A.A.; Pokrovskaya, I.D.; Leapman, R.D.; Storrie, B. Golgi Proteins in Circulating Human Platelets Are Distributed across Non-Stacked, Scattered Structures. Platelets 2017, 28, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Storrie, B. The Cellular Basis of Platelet Secretion: Emerging Structure/Function Relationships. Platelets 2017, 28, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bura, A.; Jurak Begonja, A. Imaging of Intracellular and Plasma Membrane Pools of PI(4,5)P2 and PI4P in Human Platelets. Life 2021, 11, 1331. https://doi.org/10.3390/life11121331
Bura A, Jurak Begonja A. Imaging of Intracellular and Plasma Membrane Pools of PI(4,5)P2 and PI4P in Human Platelets. Life. 2021; 11(12):1331. https://doi.org/10.3390/life11121331
Chicago/Turabian StyleBura, Ana, and Antonija Jurak Begonja. 2021. "Imaging of Intracellular and Plasma Membrane Pools of PI(4,5)P2 and PI4P in Human Platelets" Life 11, no. 12: 1331. https://doi.org/10.3390/life11121331
APA StyleBura, A., & Jurak Begonja, A. (2021). Imaging of Intracellular and Plasma Membrane Pools of PI(4,5)P2 and PI4P in Human Platelets. Life, 11(12), 1331. https://doi.org/10.3390/life11121331