
Peripheral Neuropathy Presents Similar Symptoms and Pathological Changes in Both High-Fat Diet and Pharmacologically Induced Pre- and Diabetic Mouse Models

 , , , , and
, , , , and
Abstract
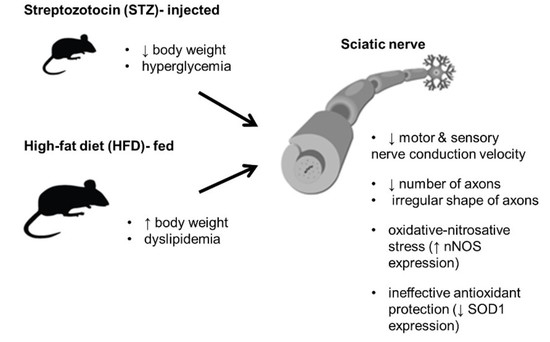
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Phenotypic Assays
2.3. Plasma Assays
2.4. Western Blot Analysis
2.5. Immunohistochemical (IHC) Staining
2.6. NCV Analysis
2.7. Morphometric Analysis
2.8. Statistical Analysis
3. Results
3.1. STZ Injections and HFD Administration Induce Diabetes-Related Phenotypic Characteristics in Mice
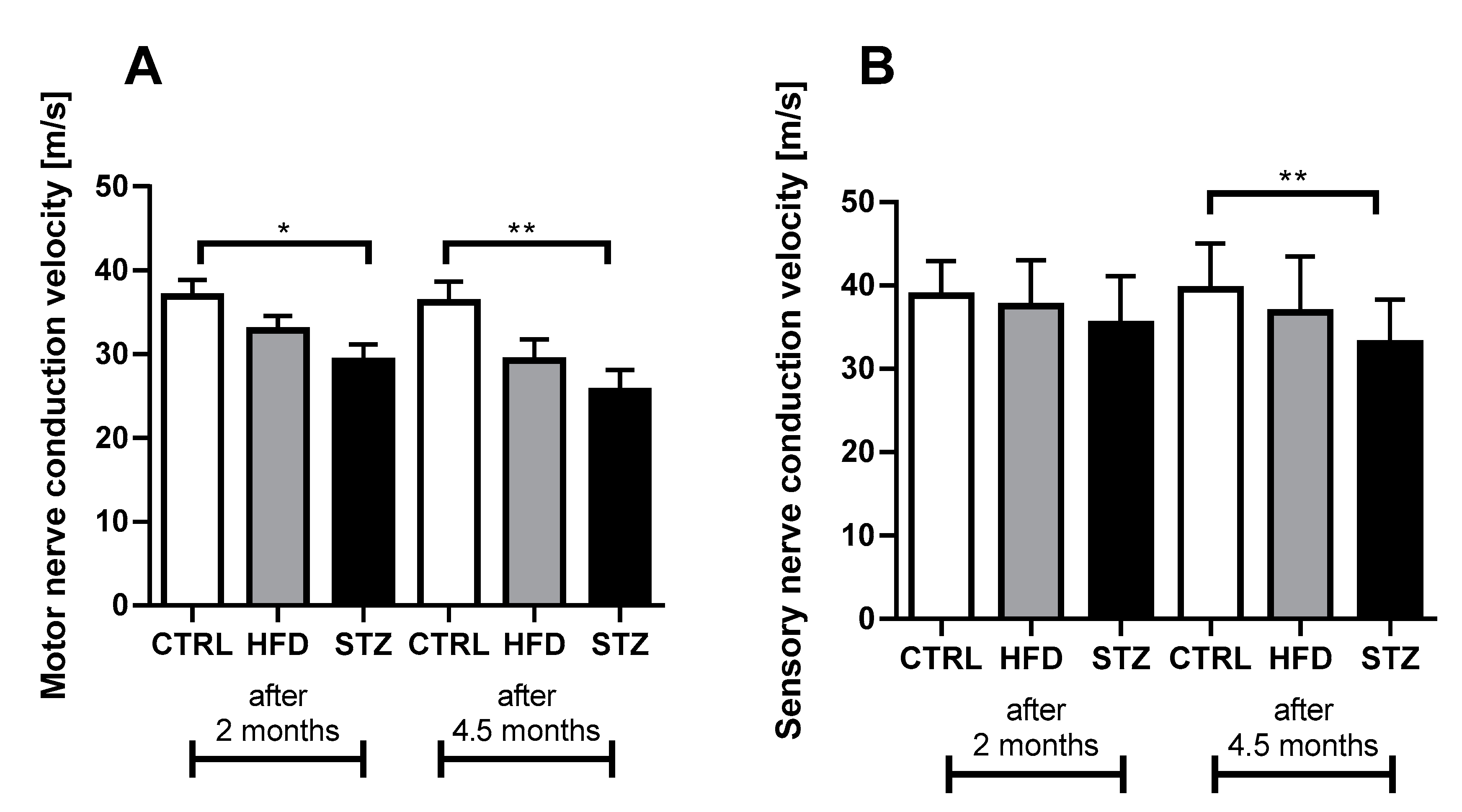
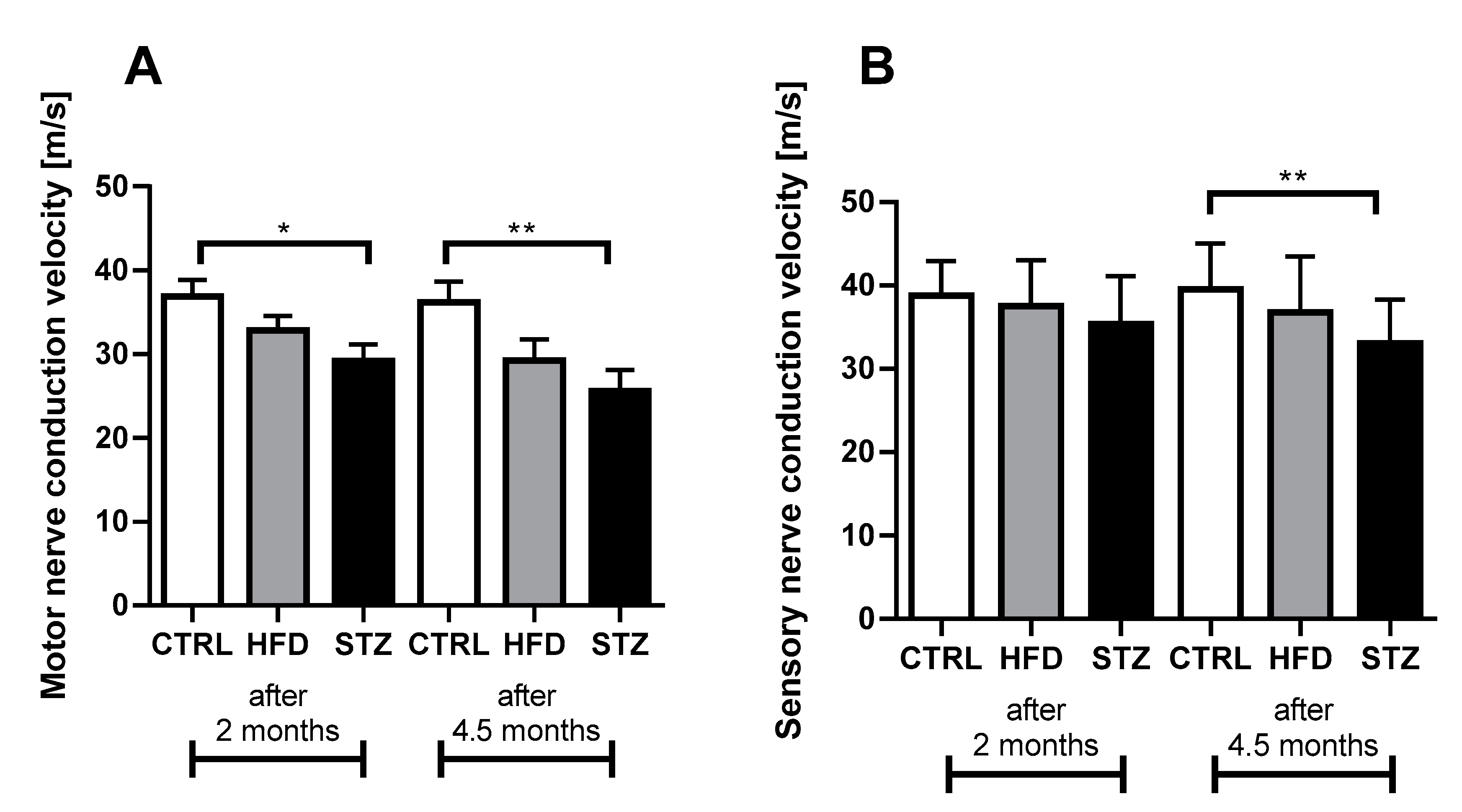
3.2. STZ-Treated Mice Exhibit Deficits in Both Sensory and Motor NCV after 4.5 Months of Diabetes
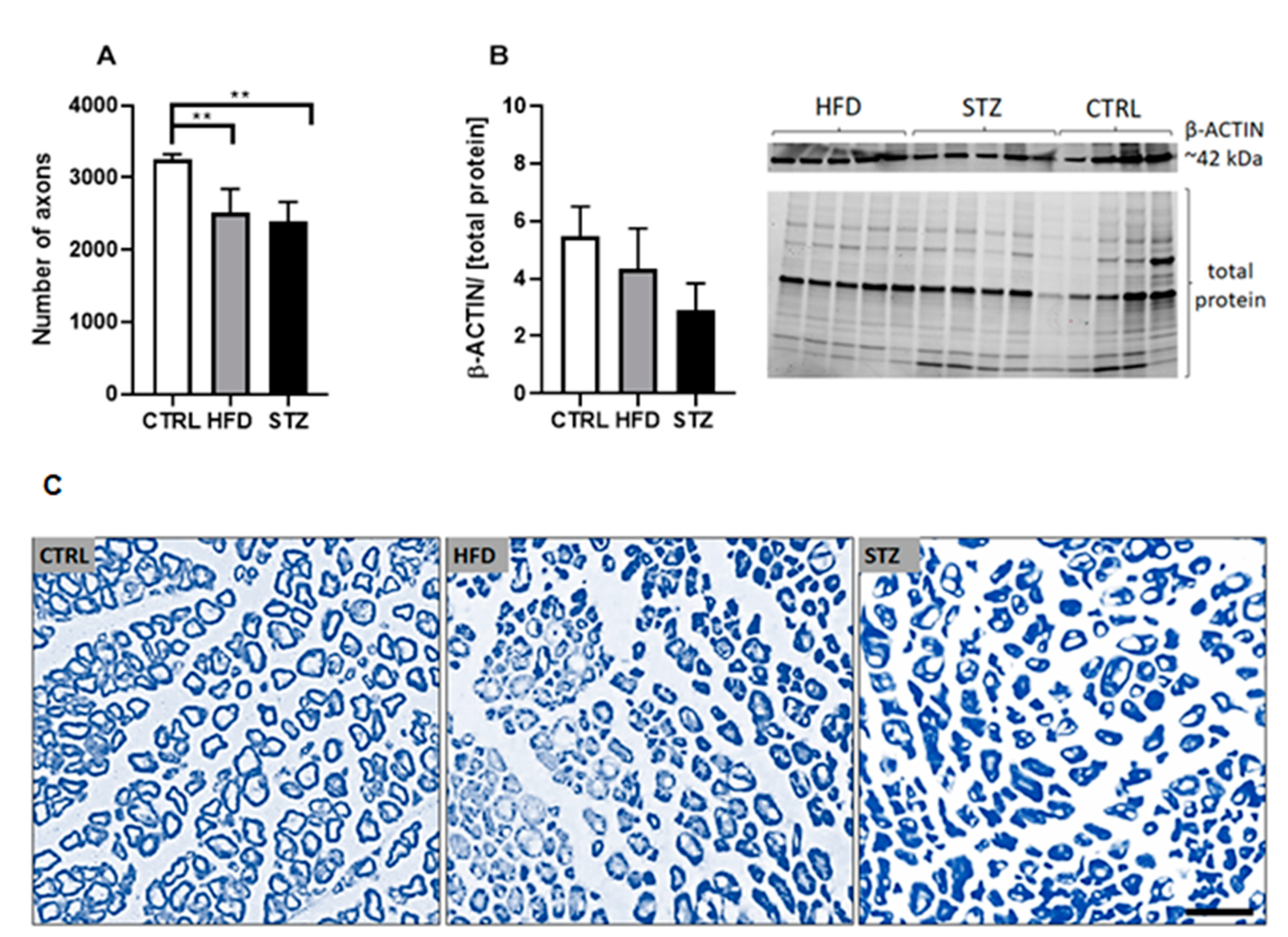
3.3. STZ Injections and HFD-Feeding Lead to Morphometric and Morphological Changes in Mouse Peripheral Nerves
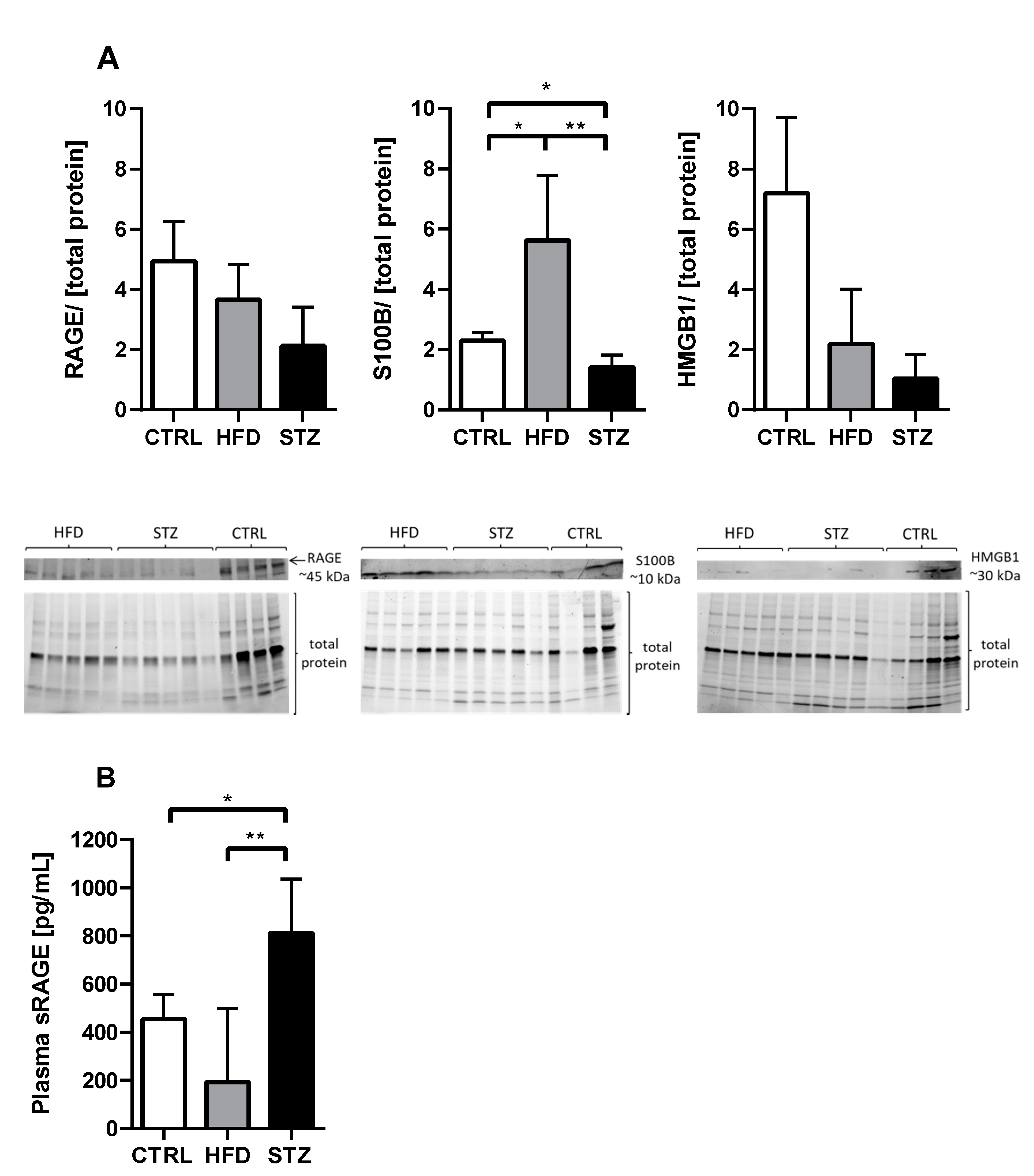
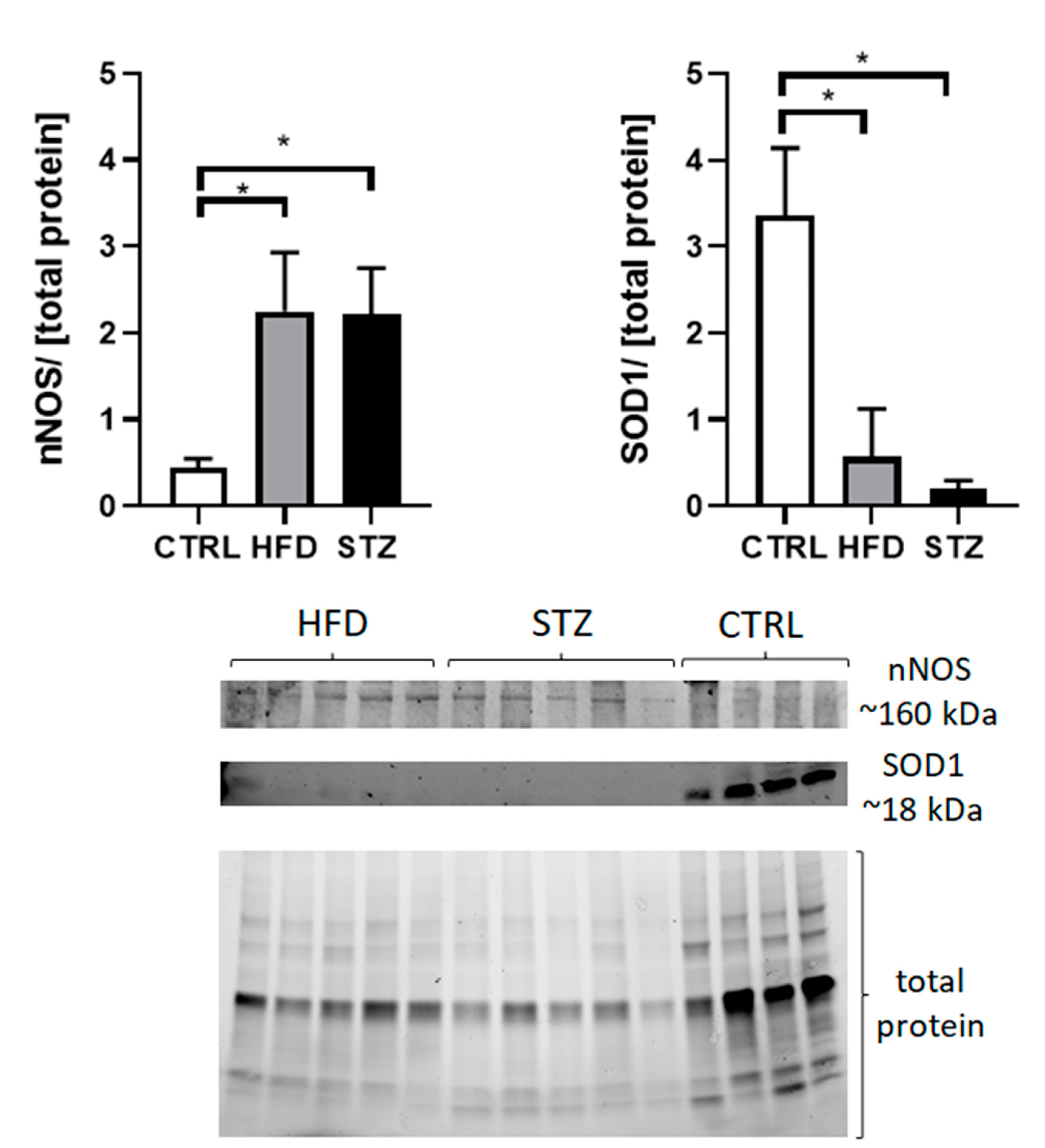
3.4. STZ/HFD Mice Manifest Impaired Oxidative/Antioxidative Balance in Peripheral Nerves
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.-A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juranek, J.; Ray, R.; Banach, M.; Rai, V. Receptor for advanced glycation end-products in neurodegenerative diseases. Rev. Neurosci. 2015, 26, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.A. Diabetic neuropathy: Pathogenetic background, current and future therapies. Expert Rev. Neurother. 2001, 1, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Sadidi, M.; Feldman, E.L. Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Rev. Endocr. Metab. Disord. 2008, 9, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Preguiça, I.; Alves, A.; Nunes, S.; Gomes, P.; Fernandes, R.; Viana, S.D.; Reis, F. Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy. Nutrients 2020, 12, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesfaye, S.; Chaturvedi, N.; Eaton, S.E.; Ward, J.D.; Manes, C.; Ionescu-Tirgoviste, C.; Witte, D.; Fuller, J.H. Vascular Risk Factors and Diabetic Neuropathy. N. Engl. J. Med. 2005, 352, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.M.; Hayes, J.M.; McLean, L.L.; Vivekanandan-Giri, A.; Pennathur, S.; Feldman, E.L. Dyslipidemia-induced neuropathy in mice: The role of oxLDL/LOX-1. Diabetes 2009, 58, 2376–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiggin, T.; Sullivan, K.A.; Pop-Busui, R.; Amato, A.; Sima, A.A.; Feldman, E.L. Elevated Triglycerides Correlate with Progression of Diabetic Neuropathy. Diabetes 2009, 58, 1634–1640. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Walther, C.; Morrison, H.; Bauer, R. In Vivo Electrophysiological Measurements on Mouse Sciatic Nerves. J. Vis. Exp. 2014, e51181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumner, C.; Sheth, S.; Griffin, J.; Cornblath, D.; Polydefkis, M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003, 60, 108–111. [Google Scholar] [CrossRef]
- Pittenger, G.L.; Mehrabyan, A.; Simmons, K.; Amandarice; Dublin, C.; Barlow, P.; Vinik, A.I. Small Fiber Neuropathy Is Associated with the Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2005, 3, 113–121. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Ilnytska, O.; Lyzogubov, V.V.; Pavlov, I.A.; Mashtalir, N.; Nadler, J.L.; Drel, V.R. High-Fat Diet Induced Neuropathy of Pre-Diabetes and Obesity: Effects of “Healthy” Diet and Aldose Reductase Inhibition. Diabetes 2007, 56, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Coppey, L.J.; Gellett, J.S.; Davidson, E.P.; Dunlap, J.A.; Lund, N.D.; Yorek, M.A. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes 2001, 50, 1927–1937. [Google Scholar] [CrossRef] [Green Version]
- Obrosova, I.G.; Mabley, J.G.; Zsengeller, Z.; Charniauskaya, T.; Abatan, O.I.; Groves, J.T.; Szabo, C. Role for nitrosative stress in diabetic neuropathy: Evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005, 19, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.J.; Obrosova, I.; Cao, X.; Van Huysen, C.; Greene, D.A. Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes 2000, 49, 1006–1015. [Google Scholar] [PubMed] [Green Version]
- Sagara, M.; Satoh, J.; Wada, R.; Yagihashi, S.; Takahashi, K.; Fukuzawa, M.; Muto, G.; Muto, Y.; Toyota, T. Inhibition of development of peripheral neuropathy in streptozotocin-induced diabetic rats with N-acetylcysteine. Diabetologia 1996, 39, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.-Y.; Dawson, V.; Dawson, T.M. Nitric oxide in health and disease of the nervous system. Mol. Psychiatry 1997, 2, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Cros, E.T. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demir, I.E.; Heinrich, T.; Carty, D.G.; Saricaoglu Ömer, C.; Klauss, S.; Teller, S.; Kehl, T.; Reyes, C.M.; Tieftrunk, E.; Lazarou, M.; et al. Targeting nNOS ameliorates the severe neuropathic pain due to chronic pancreatitis. EBioMedicine 2019, 46, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.G.; He, S.Q.; Yan, X.T.; Zhang, G.; Wan, L.; Wang, J.; Li, Y.; Tian, X.; Tian, Y.; Luo, A. Variants of neural nitric oxide synthase in the spinal cord of neuropathic rats and their effects on nuclear factor-kappaB (NF-kappaB) activity in PC12 cells. J. Pain 2009, 10, 80–89. [Google Scholar] [CrossRef]
- Kim, H.K.; Park, S.K.; Zhou, J.-L.; Taglialatela, G.; Chung, K.; Coggeshall, R.E.; Chung, J.M. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain 2004, 111, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of Renal Superoxide Dismutase in Progressive Diabetic Nephropathy. J. Am. Soc. Nephrol. 2009, 20, 1303–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niven, J.; Hoare, J.; McGowan, D.; Devarajan, G.; Itohara, S.; Gannage, M.; Teismann, P.; Crane, I. S100B Up-Regulates Macrophage Production of IL1beta and CCL22 and Influences Severity of Retinal Inflammation. PLoS ONE 2015, 10, e0132688. [Google Scholar]
- Toth, C.; Rong, L.L.; Yang, C.; Martinez, J.; Song, F.; Ramji, N.; Brussee, V.; Liu, W.; Durand, J.; Nguyen, M.D.; et al. Receptor for Advanced Glycation End Products (RAGEs) and Experimental Diabetic Neuropathy. Diabetes 2007, 57, 1002–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juranek, J.K.; Geddis, M.S.; Song, F.; Zhang, J.; Garcia, J.; Rosario, R.; Yan, S.F.; Brannagan, T.H.; Schmidt, A.M. RAGE Deficiency Improves Postinjury Sciatic Nerve Regeneration in Type 1 Diabetic Mice. Diabetes 2012, 62, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestall, S.M.; Hulse, R.P.; Blackley, Z.; Swift, M.; Ved, N.; Paton, K.; Beazley-Long, N.; Bates, D.; Donaldson, L.F. Sensory neuronal sensitisation occurs through HMGB-1/RAGE and TRPV1 in high glucose conditions. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.; Hitman, G.; Neil, A.; Livingstone, S.; Charlton-Menys, V.; Bao, W.; DeMicco, D.A.; Preston, G.M.; et al. Total Soluble and Endogenous Secretory Receptor for Advanced Glycation End Products as Predictive Biomarkers of Coronary Heart Disease Risk in Patients With Type 2 Diabetes: An Analysis From the CARDS Trial. Diabetes 2011, 60, 2379–2385. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Yan, S.D.; Brett, J.; Mora, R.; Nowygrod, R.; Stern, D. Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J. Clin. Investig. 1993, 91, 2155–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.M.; Hori, O.; Brett, J.; Yan, S.D.; Wautier, J.L.; Stern, D. Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arter. Thromb. A J. Vasc. Biol. 1994, 14, 1521–1528. [Google Scholar] [CrossRef] [Green Version]
- Wautier, J.L.; Zoukourian, C.; Chappey, O.; Wautier, M.P.; Guillausseau, P.J.; Cao, R.; Hori, O.; Stern, D.; Schmidt, A.M. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J. Clin. Investig. 1996, 97, 238–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challier, M.; Jacqueminet, S.; Benabdesselam, O.; Grimaldi, A.; Beaudeux, J.-L. Increased Serum Concentrations of Soluble Receptor for Advanced Glycation Endproducts in Patients with Type 1 Diabetes. Clin. Chem. 2005, 51, 1749–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrha, J., Jr.; Kalousova, M.; Svarcova, J.; Muravska, A.; Kvasnicka, J.; Landova, L.; Zima, T.; Skrha, J. Relationship of Soluble RAGE and RAGE Ligands HMGB1 and EN-RAGE to Endothelial Dysfunction in Type 1 and Type 2 Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2012, 120, 277–281. [Google Scholar] [PubMed]
- Salonen, K.M.; Ryhanen, S.J.; Forbes, J.M.; Harkonen, T.; Ilonen, J.; Laine, A.P.; Groop, P.H.; Knip, M. Circulating concentrations of soluble receptor for AGE are associated with age and AGER gene polymorphisms in children with newly diagnosed type 1 diabetes. Diabetes Care 2014, 37, 1975–1981. [Google Scholar] [CrossRef] [Green Version]
- Salonen, K.; Ryhänen, S.; Forbes, J.; Härkönen, T.; Ilonen, J.; Simell, O.; Veijola, R.; Groop, P.-H.; Knip, M. A drop in the circulating concentrations of soluble receptor for advanced glycation end products is associated with seroconversion to autoantibody positivity but not with subsequent progression to clinical disease in children en route to type 1 diabetes. Diabetes/Metabolism Res. Rev. 2017, 33, e2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazzana, N.; Guagnano, M.T.; Cuccurullo, C.; Ferrante, E.; Lattanzio, S.; Liani, R.; Romano, M.; Davi, G. Endogenous secretory RAGE in obese women: Association with platelet activation and oxidative stress. J. Clin. Endocrinol. Metab. 2012, 97, E1726–E1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Adamo, E.; Giannini, C.; Chiavaroli, V.; de Giorgis, T.; Verrotti, A.; Chiarelli, F.; Mohn, A. What is the significance of soluble and endogenous secretory receptor for advanced glycation end products in liver steatosis in obese prepubertal children? Antioxid. Redox Signal 2011, 14, 1167–1172. [Google Scholar] [CrossRef]
- Hinder, L.M.; O’Brien, P.D.; Hayes, J.M.; Backus, C.; Solway, A.P.; Robinson, C.; Feldman, E.L. Dietary reversal of neuropathy in a murine model of prediabetes and the metabolic syndrome. Dis. Model. Mech. 2017, 10, 717–725. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaroslawska, J.; Korytko, A.; Zglejc-Waszak, K.; Antonowski, T.; Pomianowski, A.S.; Wasowicz, K.; Wojtkiewicz, J.; Juranek, J.K. Peripheral Neuropathy Presents Similar Symptoms and Pathological Changes in Both High-Fat Diet and Pharmacologically Induced Pre- and Diabetic Mouse Models. Life 2021, 11, 1267. https://doi.org/10.3390/life11111267
Jaroslawska J, Korytko A, Zglejc-Waszak K, Antonowski T, Pomianowski AS, Wasowicz K, Wojtkiewicz J, Juranek JK. Peripheral Neuropathy Presents Similar Symptoms and Pathological Changes in Both High-Fat Diet and Pharmacologically Induced Pre- and Diabetic Mouse Models. Life. 2021; 11(11):1267. https://doi.org/10.3390/life11111267
Chicago/Turabian StyleJaroslawska, Julia, Agnieszka Korytko, Kamila Zglejc-Waszak, Tomasz Antonowski, Andrzej S. Pomianowski, Krzysztof Wasowicz, Joanna Wojtkiewicz, and Judyta K. Juranek. 2021. "Peripheral Neuropathy Presents Similar Symptoms and Pathological Changes in Both High-Fat Diet and Pharmacologically Induced Pre- and Diabetic Mouse Models" Life 11, no. 11: 1267. https://doi.org/10.3390/life11111267