
Structure, Activity, and Function of the Protein Lysine Methyltransferase G9a



Abstract
1. Introduction
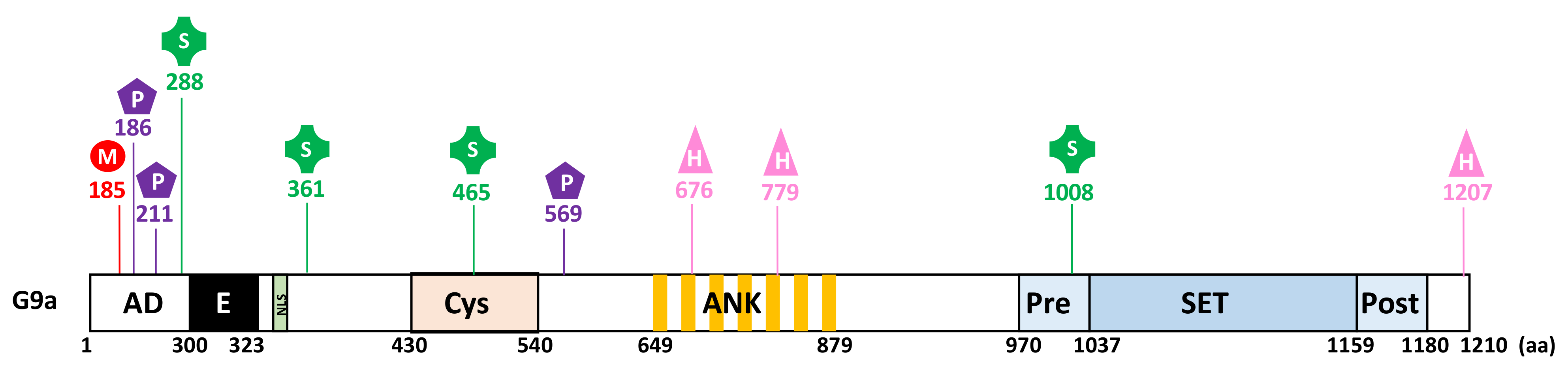
2. Structural Features
2.1. Structure and Domain Architecture
2.2. GLP, a G9a Paralog
3. Biochemical Features
3.1. Sequence Specificity
3.2. Product Specificity
3.3. Regulation
3.3.1. PTMs
3.3.2. Stability
3.4. Substrates
3.4.1. Histone Substrates
3.4.2. Non-Histone Substrates
3.5. Inhibitors
3.5.1. Substrate Competitive Inhibitors
3.5.2. SAM Competitive Inhibitors
3.5.3. Inhibition by Ejection of Structural Zn2+
4. Cellular Features
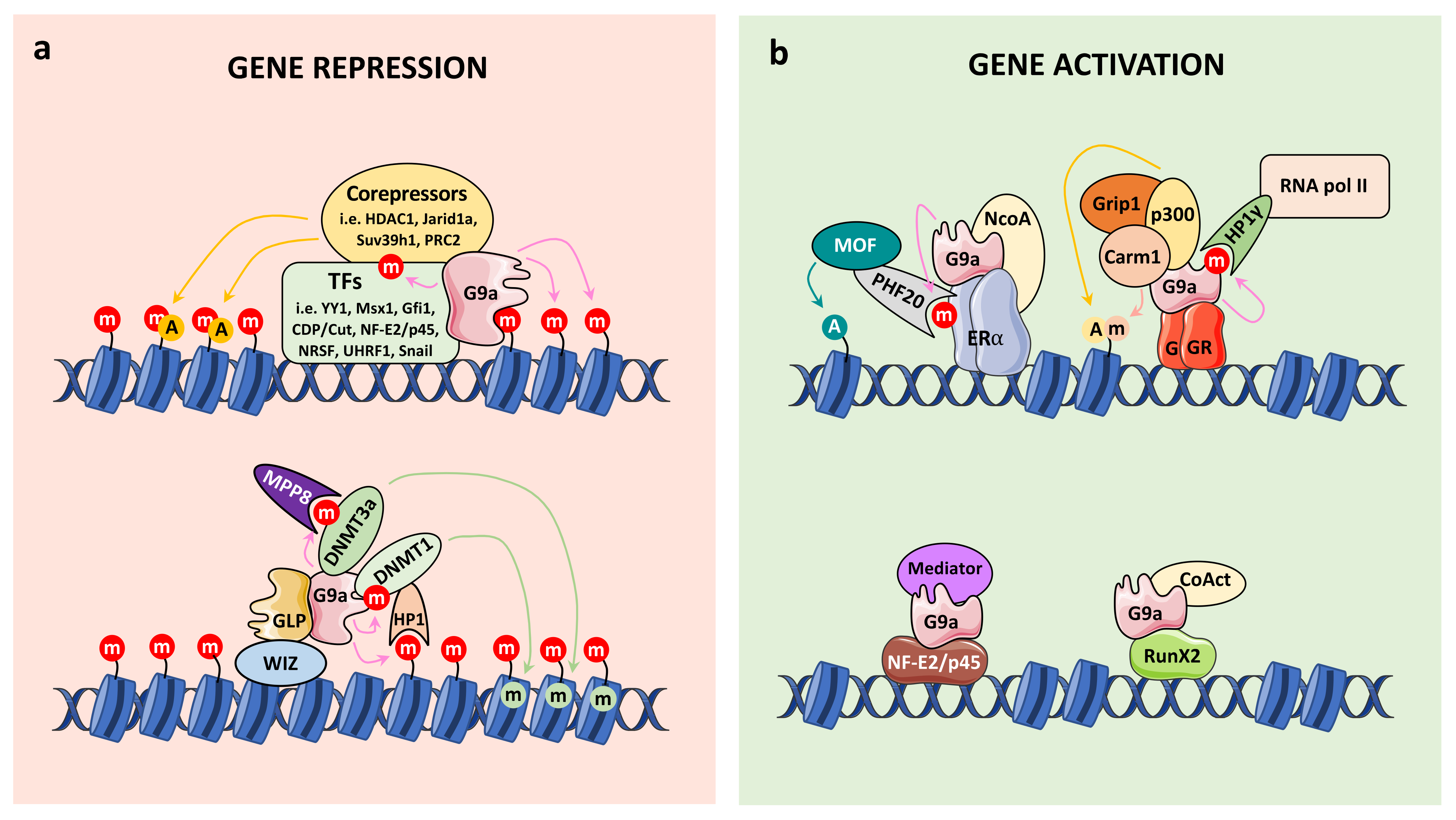
4.1. Connection with Chromatin Regulation
4.1.1. G9a Corepressor Functions
- G9a in Euchromatin
- G9a in heterochromatin
- G9a and DNA methylation
4.1.2. G9a Coactivator Functions
4.2. Cellular Roles and Functions
4.2.1. Embryonic Development
- Germ Cell Development
- Cardiac Development
- Neuronal Development
- Bone Formation
- Other Mechanisms
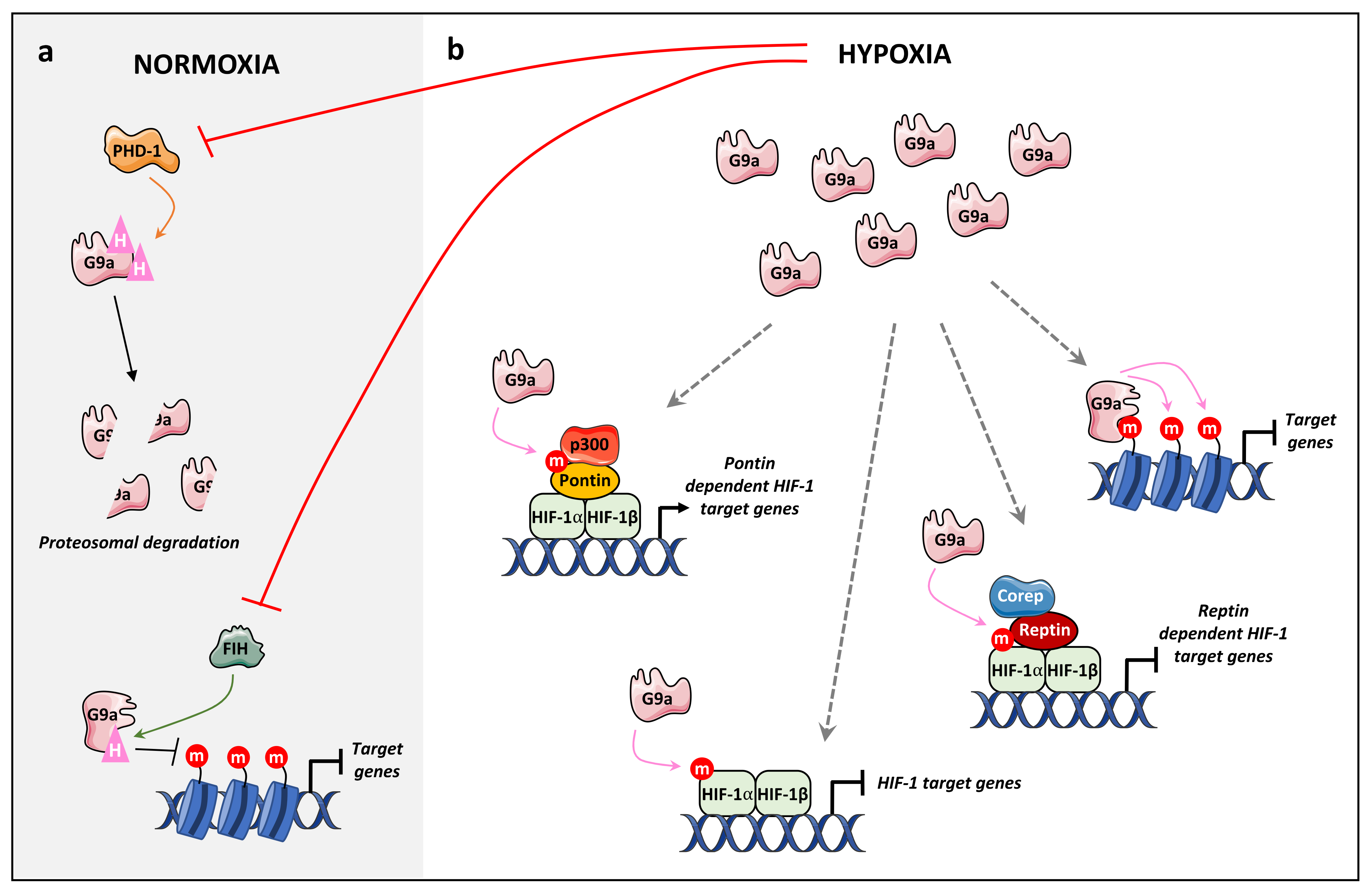
4.2.2. Hypoxia
4.2.3. DNA Damage and DNA Repair
5. G9a in Cancer
5.1. G9a Oncogenic Role
5.1.1. Breast Cancer
5.1.2. Gastric Cancer
5.1.3. Human Reproductive Cancers
5.1.4. Lung Cancer
5.1.5. Colorectal Cancer
5.1.6. Hepatocellular Carcinoma
5.1.7. Urinary Bladder Cancer
5.1.8. Hematological Cancers
5.1.9. Other Cancers
5.2. G9a Tumor Suppressive Role
6. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, Z.; Connolly, J.; Biggar, K.K. Beyond Histones—The Expanding Roles of Protein Lysine Methylation. FEBS J. 2017, 284, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-Domain Protein Superfamily: Protein Lysine Methyltransferases. Genome Biol. 2005, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Falnes, P.; Jakobsson, M.E.; Davydova, E.; Ho, A.; Malecki, J. Protein Lysine Methylation by Seven-β-Strand Methyltransferases. Biochem. J. 2016, 473, 1995–2009. [Google Scholar] [CrossRef] [PubMed]
- Milner, C.M.; Campbell, R.D. The G9a Gene in the Human Major Histocompatibility Complex Encodes a Novel Protein Containing Ankyrin-like Repeats the Class III Region of the Human Major Histocompatibility Complex Spans Approx. Biochem. J. 1993, 290, 811–818. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a Histone Methyltransferase Plays a Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and Is Essential for Early Embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone Methyltransferases G9a and GLP Form Heteromeric Complexes and Are Both Crucial for Methylation of Euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef]
- Tachibana, M.; Matsumura, Y.; Fukuda, M.; Kimura, H.; Shinkai, Y. G9a/GLP Complexes Independently Mediate H3K9 and DNA Methylation to Silence Transcription. EMBO J. 2008, 27, 2681–2690. [Google Scholar] [CrossRef]
- Bittencourt, D.; Wu, D.Y.; Jeong, K.W.; Gerke, D.S.; Herviou, L.; Ianculescu, I.; Chodankar, R.; Siegmund, K.D.; Stallcup, M.R. G9a Functions as a Molecular Scaffold for Assembly of Transcriptional Coactivators on a Subset of Glucocorticoid Receptor Target Genes. Proc. Natl. Acad. Sci. USA 2012, 109, 19673–19678. [Google Scholar] [CrossRef]
- Lee, D.Y.; Northrop, J.P.; Kuo, M.H.; Stallcup, M.R. Histone H3 Lysine 9 Methyltransferase G9a Is a Transcriptional Coactivator for Nuclear Receptors. J. Biol. Chem. 2006, 281, 8476–8485. [Google Scholar] [CrossRef]
- Purcell, D.J.; Jeong, K.W.; Bittencourt, D.; Gerke, D.S.; Stallcup, M.R. A Distinct Mechanism for Coactivator versus Corepressor Function by Histone Methyltransferase G9a in Transcriptional Regulation. J. Biol. Chem. 2011, 286, 41963–41971. [Google Scholar] [CrossRef]
- Purcell, D.J.; Khalid, O.; Ou, C.Y.; Little, G.H.; Frenkel, B.; Baniwal, S.K.; Stallcup, M.R. Recruitment of Coregulator G9a by Runx2 for Selective Enhancement or Suppression of Transcription. J. Cell. Biochem. 2012, 113, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, C.P.; Hosey, A.M.; Palii, C.; Perez-Iratxeta, C.; Nakatani, Y.; Ranish, J.A.; Dilworth, F.J.; Brand, M. Dual Role for the Methyltransferase G9a in the Maintenance of β-Globin Gene Transcription in Adult Erythroid Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18303–18308. [Google Scholar] [CrossRef] [PubMed]
- Mauger, O.; Klinck, R.; Chabot, B.; Muchardt, C.; Allemand, E.; Batschébatsch´batsché, E. Alternative Splicing Regulates the Expression of G9A and SUV39H2 Methyltransferases, and Dramatically Changes SUV39H2 Functions. Nucleic Acids Res. 2015, 43, 1869–1882. [Google Scholar] [CrossRef]
- Fiszbein, A.; Giono, L.E.; Quaglino, A.; Berardino, B.G.; Sigaut, L.; von Bilderling, C.; Schor, I.E.; Steinberg, J.H.E.; Rossi, M.; Pietrasanta, L.I.; et al. Alternative Splicing of G9a Regulates Neuronal Differentiation. Cell Rep. 2016, 14, 2797–2808. [Google Scholar] [CrossRef]
- Shankar, S.R.; Bahirvani, A.G.; Rao, V.K.; Bharathy, N.; Ow, J.R.; Taneja, R. G9a, a Multipotent Regulator of Gene Expression. Epigenetics 2013, 8, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Schotta, G.; Ebert, A.; Krauss, V.; Fischer, A.; Hoffmann, J.; Rea, S.; Jenuwein, T.; Dorn, R.; Reuter, G. Central Role of Drosophila SU(VAR)3–9 in Histone H3-K9 Methylation and Heterochromatic Gene Silencing. EMBO J. 2002, 21, 1121–1131. [Google Scholar] [CrossRef]
- Jenuwein, T.; Laible, G.; Dorn, R.; Reuter, G. SET Domain Proteins Modulate Chromatin Domains in Eu-and Heterochromatin. Cell. Mol. Life Sci 1998, 54, 80–93. [Google Scholar] [CrossRef]
- Taylor, W.R.; Xiao, B.; Gamblin, S.J.; Lin, K. A Knot or Not a Knot? SETting the Record ‘Straight’ on Proteins. Comput. Biol. Chem. 2003, 27, 11–15. [Google Scholar] [CrossRef]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.A.; Houtz, R.L.; Hurley, J.H. Structure and Catalytic Mechanism of a SET Domain Protein Methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef]
- Qian, C.; Zhou, M.M. SET Domain Protein Lysine Methyltransferases: Structure, Specificity and Catalysis. Cell. Mol. Life Sci. 2006, 63, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Min, J.; Lunin, V.V.; Antoshenko, T.; Dombrovski, L.; Zeng, H.; Allali-Hassani, A.; Campagna-Slater, V.; Vedadi, M.; Arrowsmith, C.H.; et al. Structural Biology of Human H3K9 Methyltransferases. PLoS ONE 2010, 5, e8570. [Google Scholar] [CrossRef]
- Lenstra, D.C.; al Temimi, A.H.K.; Mecinović, J. Inhibition of Histone Lysine Methyltransferases G9a and GLP by Ejection of Structural Zn(II). Bioorganic Med. Chem. Lett. 2018, 28, 1234–1238. [Google Scholar] [CrossRef]
- Schapira, M. Structural Chemistry of Human SET Domain Protein Methyltransferases. Curr. Chem. Genom. 2011, 5, 85–94. [Google Scholar] [CrossRef]
- Collins, R.E.; Northrop, J.P.; Horton, J.R.; Lee, D.Y.; Zhang, X.; Stallcup, M.R.; Cheng, X. The Ankyrin Repeats of G9a and GLP Histone Methyltransferases Are Mono- and Dimethyllysine Binding Modules. Nat. Struct. Mol. Biol. 2008, 15, 245–250. [Google Scholar] [CrossRef]
- Estève, P.O.; Patnaik, D.; Chin, H.G.; Benner, J.; Teitell, M.A.; Pradhan, S. Functional Analysis of the N- and C-Terminus of Mammalian G9a Histone H3 Methyltransferase. Nucleic Acids Res. 2005, 33, 3211–3223. [Google Scholar] [CrossRef] [PubMed]
- Corpet, F. Multiple Sequence Alignment with Hierarchical Clustering. Nucleic Acids Res. 1988, 16, 10881. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Zhang, X.; Horton, J.R.; Upadhyay, A.K.; Spannhoff, A.; Liu, J.; Snyder, J.P.; Bedford, M.T.; Cheng, X. Structural Basis for G9a-like Protein Lysine Methyltransferase Inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, Z.; Wu, H.; Jiang, Y.; Meng, L.; Xiong, J.; Zhao, Z.; Zhou, X.; Li, J.; Li, H.; et al. Recognition of H3K9 Methylation by GLP Is Required for Efficient Establishment of H3K9 Methylation, Rapid Target Gene Repression, and Mouse Viability. Genes Dev. 2015, 29, 379–393. [Google Scholar] [CrossRef]
- Shinkai, Y.; Tachibana, M. H3K9 Methyltransferase G9a and the Related Molecule GLP. Genes Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Ueda, J.; Tachibana, M.; Ikura, T.; Shinkai, Y. Zinc Finger Protein Wiz Links G9a/GLP Histone Methyltransferases to the Co-Repressor Molecule CtBP. J. Biol. Chem. 2006, 281, 20120–20128. [Google Scholar] [CrossRef]
- Bian, C.; Chen, Q.; Yu, X. The Zinc Finger Proteins ZNF644 and WIZ Regulate the G9A/GLP Complex for Gene Repression. eLife 2015, 4, e05606. [Google Scholar] [CrossRef] [PubMed]
- Poulard, C.; Bittencourt, D.; Wu, D.; Hu, Y.; Gerke, D.S.; Stallcup, M.R. A Post-translational Modification Switch Controls Coactivator Function of Histone Methyltransferases G9a and GLP. EMBO Rep. 2017, 18, 1442–1459. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.G.; Pradhan, M.; Estève, P.O.; Patnaik, D.; Evans, T.C.; Pradhan, S. Sequence Specificity and Role of Proximal Amino Acids of the Histone H3 Tail on Catalysis of Murine G9a Lysine 9 Histone H3 Methyltransferase. Biochemistry 2005, 44, 12998–13006. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.G.; Estève, P.O.; Pradhan, M.; Benner, J.; Patnaik, D.; Carey, M.F.; Pradhan, S. Automethylation of G9a and Its Implication in Wider Substrate Specificity and HP1 Binding. Nucleic Acids Res. 2007, 35, 7313–7323. [Google Scholar] [CrossRef] [PubMed]
- Sampath, S.C.; Marazzi, I.; Yap, K.L.; Sampath, S.C.; Krutchinsky, A.N.; Mecklenbräuker, I.; Viale, A.; Rudensky, E.; Zhou, M.M.; Chait, B.T.; et al. Methylation of a Histone Mimic within the Histone Methyltransferase G9a Regulates Protein Complex Assembly. Mol. Cell 2007, 27, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Dhayalan, A.; Murakami, M.; Zhang, X.; Tamas, R.; Jurkowska, R.; Komatsu, Y.; Shinkai, Y.; Cheng, X.; Jeltsch, A. Protein Lysine Methyltransferase G9a Acts on Non-Histone Targets. Nat. Chem. Biol. 2008, 4, 344–346. [Google Scholar] [CrossRef]
- Patnaik, D.; Hang, G.C.; Estève, P.O.; Benner, J.; Jacobsen, S.E.; Pradhan, S. Substrate Specificity and Kinetic Mechanism of Mammalian G9a Histone H3 Methyltransferase. J. Biol. Chem. 2004, 279, 53248–53258. [Google Scholar] [CrossRef]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1–Chromatin Binding by Histone H3 Methylation and Phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Lipp, J.J.; Toh, B.-H.; Peters, J.-M. Histone H3 Serine 10 Phosphorylation by Aurora B Causes HP1 Dissociation from Heterochromatin. Nature 2005, 438, 1176–1180. [Google Scholar] [CrossRef]
- Poulard, C.; Baulu, E.; Lee, B.H.; Pufall, M.A.; Stallcup, M.R. Increasing G9a Automethylation Sensitizes B Acute Lymphoblastic Leukemia Cells to Glucocorticoid-Induced Death. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Ginjala, V.; Rodriguez-Colon, L.; Ganguly, B.; Gangidi, P.; Gallina, P.; Al-Hraishawi, H.; Kulkarni, A.; Tang, J.; Gheeya, J.; Simhadri, S.; et al. Protein-Lysine Methyltransferases G9a and GLP1 Promote Responses to DNA Damage. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, Q.; Lu, X.; Du, Y.; Cao, L.; Shen, C.; Hou, T.; Li, M.; Li, Z.; Liu, C.; et al. G9a Coordinates with the RPA Complex to Promote DNA Damage Repair and Cell Survival. Proc. Natl. Acad. Sci. USA 2017, 114, E6054–E6063. [Google Scholar] [CrossRef]
- Srinivasan, S.; Shankar, S.R.; Wang, Y.; Taneja, R. SUMOylation of G9a Regulates Its Function as an Activator of Myoblast Proliferation. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Casciello, F.; Al-Ejeh, F.; Kelly, G.; Brennan, D.J.; Ngiow, S.F.; Young, A.; Stoll, T.; Windloch, K.; Hill, M.M.; Smyth, M.J.; et al. G9a Drives Hypoxia-Mediated Gene Repression for Breast Cancer Cell Survival and Tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 7077–7082. [Google Scholar] [CrossRef]
- Kang, J.; Shin, S.H.; Yoon, H.; Huh, J.; Shin, H.W.; Chun, Y.S.; Park, J.W. FIH Is an Oxygen Sensor in Ovarian Cancer for G9a/GLP-Driven Epigenetic Regulation of Metastasis-Related Genes. Cancer Res. 2018, 78, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. SET Domain-Containing Protein, G9a, Is a Novel Lysine-Preferring Mammalian Histone Methyltransferase with Hyperactivity and Specific Selectivity to Lysines 9 and 27 of Histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef]
- Richards, E.J.; Elgin, S.C.R. Epigenetic Codes for Heterochromatin Formation and Silencing: Rounding up the Usual Suspects. Cell 2002, 108, 489–500. [Google Scholar] [CrossRef]
- Wu, H.; Chen, X.; Xiong, J.; Li, Y.; Li, H.; Ding, X.; Liu, S.; Chen, S.; Gao, S.; Zhu, B. Histone Methyltransferase G9a Contributes to H3K27 Methylation in Vivo. Cell Res. 2011, 21, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Song, C.; Zhang, Q.; DiMaggio, P.A.; Garcia, B.A.; York, A.; Carey, M.F.; Grunstein, M. Histone H3 Lysine 56 Methylation Regulates DNA Replication through Its Interaction with PCNA. Mol. Cell 2012, 46, 7–17. [Google Scholar] [CrossRef]
- Trojer, P.; Zhang, J.; Yonezawa, M.; Schmidt, A.; Zheng, H.; Jenuwein, T.; Reinberg, D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-Containing JMJD2/KDM4 Proteins. J. Biol. Chem. 2009, 284, 8395–8405. [Google Scholar] [CrossRef]
- Weiss, T.; Hergeth, S.; Zeissler, U.; Izzo, A.; Tropberger, P.; Zee, B.M.; Dundr, M.; Garcia, B.A.; Daujat, S.; Schneider, R. Histone H1 Variant-Specific Lysine Methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin 2010, 3, 1–13. [Google Scholar] [CrossRef]
- Cao, H.; Li, L.; Deying, Y.; Liming, Z.; Yewei, X.; Yu, B.; Liao, G.; Chen, J. Recent Progress in Histone Methyltransferase (G9a) Inhibitors as Anticancer Agents. Eur. J. Med. Chem. 2019, 179, 537–546. [Google Scholar] [CrossRef]
- Xiong, Y.; Li, F.; Babault, N.; Wu, H.; Dong, A.; Zeng, H.; Chen, X.; Arrowsmith, C.H.; Brown, P.J.; Liu, J.; et al. Structure-Activity Relationship Studies of G9a-like Protein (GLP) Inhibitors. Bioorganic Med. Chem. 2017, 25, 4414–4423. [Google Scholar] [CrossRef]
- Pless, O.; Kowenz-Leutz, E.; Knoblich, M.; Lausen, J.; Beyermann, M.; Walsh, M.J.; Leutz, A. G9a-Mediated Lysine Methylation Alters the Function of CCAAT/Enhancer-Binding Protein-β. J. Biol. Chem. 2008, 283, 26357–26363. [Google Scholar] [CrossRef]
- Ling, B.M.T.; Bharathy, N.; Chung, T.-K.; Kok, W.K.; Li, S.; Tan, Y.H.; Rao, V.K.; Gopinadhan, S.; Sartorelli, V.; Walsh, M.J.; et al. Lysine Methyltransferase G9a Methylates the Transcription Factor MyoD and Regulates Skeletal Muscle Differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jang, H.; Kim, H.; Lee, J.H.; Kim, S.T.; Cho, E.J.; Youn, H.D. Modulation of Lysine Methylation in Myocyte Enhancer Factor 2 during Skeletal Muscle Cell Differentiation. Nucleic Acids Res. 2014, 42, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dorsey, J.; Chuikov, S.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp Methylate Lysine 373 in the Tumor Suppressor P53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Peng, D.; Xi, Y.; Yuan, C.; Sagum, C.A.; Klein, B.J.; Tanaka, K.; Wen, H.; Kutateladze, T.G.; Li, W.; et al. G9a-Mediated Methylation of ERα Links the PHF20/MOF Histone Acetyltransferase Complex to Hormonal Gene Expression. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Kim, J.Y.; Park, J.W.; Kim, K.B.; Oh, H.; Lee, K.H.; Seo, S.B. FOXO1 Degradation via G9a-Mediated Methylation Promotes Cell Proliferation in Colon Cancer. Nucleic Acids Res. 2019, 47, 1692–1705. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.E.; Carlson, S.M.; Camp, N.D.; Cheung, P.; James, R.G.; Chua, K.F.; Wolf-Yadlin, A.; Gozani, O. A General Molecular Affinity Strategy for Global Detection and Proteomic Analysis of Lysine Methylation. Mol. Cell 2013, 50, 444–456. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, Y.; Bhin, J.; Shin, H.-J.R.; Nam, H.J.; Lee, H.; Yoon, J.-B.; Binda, O.; Hwang, D.; Baek, S.H. Hypoxia-Induced Methylation of a Pontin Chromatin Remodeling Factor. Proc. Natl. Acad. Sci. USA 2011, 108, 13510–13515. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, Y.; Kim, I.S.; Kim, B.; Choi, H.J.; Lee, J.M.; Shin, H.J.R.; Kim, J.H.; Kim, J.Y.; Seo, S.B.; et al. Negative Regulation of Hypoxic Responses via Induced Reptin Methylation. Mol. Cell 2010, 39, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Chen, Y.; Lai, H.T.; Wu, S.Y.; Wang, J.E.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.M.; et al. Methylation of Hypoxia-Inducible Factor (HIF)-1α by G9a/GLP Inhibits HIF-1 Transcriptional Activity and Cell Migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Li, D.Q.; Kumar, R. A Core Chromatin Remodeling Factor Instructs Global Chromatin Signaling through Multivalent Reading of Nucleosome Codes. Mol. Cell 2013, 49, 704–718. [Google Scholar] [CrossRef]
- Tsusaka, T.; Kikuchi, M.; Shimazu, T.; Suzuki, T.; Sohtome, Y.; Akakabe, M.; Sodeoka, M.; Dohmae, N.; Umehara, T.; Shinkai, Y. Tri-Methylation of ATF7IP by G9a/GLP Recruits the Chromodomain Protein MPP8. Epigenetics Chromatin 2018, 11, 1–16. [Google Scholar] [CrossRef]
- Chang, Y.; Sun, L.; Kokura, K.; Horton, J.R.; Fukuda, M.; Espejo, A.; Izumi, V.; Koomen, J.M.; Bedford, M.T.; Zhang, X.; et al. MPP8 Mediates the Interactions between DNA Methyltransferase Dnmt3a and H3K9 Methyltransferase GLP/G9a. Nat. Commun. 2011, 2, 1–10. [Google Scholar] [CrossRef]
- Watanabe, S.; Iimori, M.; Chan, D.V.; Hara, E.; Kitao, H.; Maehara, Y. MDC1 Methylation Mediated by Lysine Methyltransferases EHMT1 and EHMT2 Regulates Active ATM Accumulation Flanking DNA Damage Sites. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.-Y.; Zhao, X.; Duan, H.; Cheng, B.; Liu, Y.; Zhao, M.; Shu, W.; Mei, Y.; Wen, Z.; et al. A Methylation-Phosphorylation Switch Determines Plk1 Kinase Activity and Function in DNA Damage Repair. Sci. Adv. 2019, 5, eaau7566. [Google Scholar] [CrossRef]
- Ferry, L.; Fournier, A.; Tsusaka, T.; Adelmant, G.; Shimazu, T.; Matano, S.; Kirsh, O.; Amouroux, R.; Dohmae, N.; Suzuki, T.; et al. Methylation of DNA Ligase 1 by G9a/GLP Recruits UHRF1 to Replicating DNA and Regulates DNA Methylation. Mol. Cell 2017, 67, 550–565. [Google Scholar] [CrossRef]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a Small-Molecule Inhibitor for the G9a Histone Methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wasney, G.A.; Dong, A.; Barsyte, D.; Kozieradzki, I.; Senisterra, G.; Chau, I.; et al. Discovery of a 2,4-Diamino-7-Aminoalkoxyquinazoline as a Potent and Selective Inhibitor of Histone Lysine Methyltransferase G9a. J. Med. Chem. 2009, 52, 7950–7953. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Barsyte-Lovejoy, D.; Liu, F.; Rival-Gervier, S.; Allali-Hassani, A.; Labrie, V.; Wigle, T.J.; DiMaggio, P.A.; Wasney, G.A.; Siarheyeva, A.; et al. A Chemical Probe Selectively Inhibits G9a and GLP Methyltransferase Activity in Cells. Nat. Chem. Biol. 2011, 7, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Milite, C.; Feoli, A.; Horton, J.R.; Rescigno, D.; Cipriano, A.; Pisapia, V.; Viviano, M.; Pepe, G.; Amendola, G.; Novellino, E.; et al. Discovery of a Novel Chemotype of Histone Lysine Methyltransferase EHMT1/2 (GLP/G9a) Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Co-Crystal Structure. J. Med. Chem. 2019, 62, 2666–2689. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A Small-Molecule Probe of the Histone Methyltransferase G9a Induces Cellular Senescence in Pancreatic Adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Nishio, H.; Walsh, M.J. CCAAT Displacement Protein/Cut Homolog Recruits G9a Histone Lysine Methyltransferase to Repress Transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 11257–11262. [Google Scholar] [CrossRef]
- Duan, Z.; Zarebski, A.; Montoya-Durango, D.; Grimes, H.L.; Horwitz, M. Gfi1 Coordinates Epigenetic Repression of P21Cip/WAF1 by Recruitment of Histone Lysine Methyltransferase G9a and Histone Deacetylase 1. Mol. Cell. Biol. 2005, 25, 10338–10351. [Google Scholar] [CrossRef]
- Győry, I.; Wu, J.; Fejér, G.; Seto, E.; Wright, K.L. PRDI-BF1 Recruits the Histone H3 Methyltransferase G9a in Transcriptional Silencing. Nat. Immunol. 2004, 5, 299–308. [Google Scholar] [CrossRef]
- Roopra, A.; Qazi, R.; Schoenike, B.; Daley, T.J.; Morrison, J.F. Localized Domains of G9a-Mediated Histone Methylation Are Required for Silencing of Neuronal Genes. Mol. Cell 2004, 14, 727–738. [Google Scholar] [CrossRef]
- Kim, J.K.; Estève, P.O.; Jacobsen, S.E.; Pradhan, S. UHRF1 Binds G9a and Participates in P21 Transcriptional Regulation in Mammalian Cells. Nucleic Acids Res. 2009, 37, 493–505. [Google Scholar] [CrossRef]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air Noncoding RNA Epigenetically Silences Transcription by Targeting G9a to Chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The Histone H3 Lysine 9 Methyltransferases G9a and GLP Regulate Polycomb Repressive Complex 2-Mediated Gene Silencing. Mol. Cell 2014, 53, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.; Wallrath, L.; Urrutia, R. The Heterochromatin Protein 1 Family. Genome Biol. 2006, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, L.; Robin, P.; Mathieu, J.R.R.; Souidi, M.; Hinaux, H.; Rougeulle, C.; Harel-Bellan, A.; Ameyar-Zazoua, M.; Ait-Si-Ali, S. A Subset of the Histone H3 Lysine 9 Methyltransferases Suv39h1, G9a, GLP, and SETDB1 Participate in a Multimeric Complex. Mol. Cell 2010, 37, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Epsztejn-Litman, S.; Feldman, N.; Abu-Remaileh, M.; Shufaro, Y.; Gerson, A.; Ueda, J.; Deplus, R.; Fuks, F.; Shinkai, Y.; Cedar, H.; et al. De Novo DNA Methylation Promoted by G9a Prevents Reprogramming of Embryonically Silenced Genes. Nat. Struct. Mol. Biol. 2008, 15, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, D.; Lee, B.H.; Gao, L.; Gerke, D.S.; Stallcup, M.R. Role of Distinct Surfaces of the G9a Ankyrin Repeat Domain in Histone and DNA Methylation during Embryonic Stem Cell Self-Renewal and Differentiation. Epigenetics Chromatin 2014, 7, 1–12. [Google Scholar] [CrossRef]
- Smallwood, A.; Estève, P.O.; Pradhan, S.; Carey, M. Functional Cooperation between HP1 and DNMT1 Mediates Gene Silencing. Genes Dev. 2007, 21, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Kim, K.B.; Chae, Y.C.; Kang, J.Y.; Hahn, Y.; Seo, S.B. H3K9 Histone Methyltransferase G9a-Mediated Transcriptional Activation of P21. FEBS Lett. 2014, 588, 685–691. [Google Scholar] [CrossRef]
- Poulard, C.; Kim, H.N.; Fang, M.; Kruth, K.; Gagnieux, C.; Gerke, D.S.; Bhojwani, D.; Kim, Y.M.; Kampmann, M.; Stallcup, M.R.; et al. Relapse-Associated AURKB Blunts the Glucocorticoid Sensitivity of B Cell Acute Lymphoblastic Leukemia. Proc. Natl. Acad. Sci. USA 2019, 116, 3052–3061. [Google Scholar] [CrossRef]
- Chaturvedi, C.P.; Somasundaram, B.; Singh, K.; Carpenedo, R.L.; Stanford, W.L.; Dilworth, F.J.; Brand, M. Maintenance of Gene Silencing by the Coordinate Action of the H3K9 Methyltransferase G9a/KMT1C and the H3K4 Demethylase Jarid1a/KDM5A. Proc. Natl. Acad. Sci. USA 2012, 109, 18845–18850. [Google Scholar] [CrossRef]
- Tachibana, M.; Nozaki, M.; Takeda, N.; Shinkai, Y. Functional Dynamics of H3K9 Methylation during Meiotic Prophase Progression. EMBO J. 2007, 26, 3346–3359. [Google Scholar] [CrossRef]
- Inagawa, M.; Nakajima, K.; Makino, T.; Ogawa, S.; Kojima, M.; Ito, S.; Ikenishi, A.; Hayashi, T.; Schwartz, R.J.; Nakamura, K.; et al. Histone H3 Lysine 9 Methyltransferases, G9a and GLP Are Essential for Cardiac Morphogenesis. Mech. Dev. 2013, 130, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Sampath, S.C.; Intrator, A.; Min, A.; Gertler, T.S.; Surmeier, D.J.; Tarakhovsky, A.; Greengard, P. Control of Cognition and Adaptive Behavior by the GLP/G9a Epigenetic Suppressor Complex. Neuron 2009, 64, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Nicolay-Kritter, K.; Lassalle, J.; Guillou, J.L.; Mons, N. The Histone H3 Lysine 9 Methyltransferase G9a/GLP Complex Activity Is Required for Long-Term Consolidation of Spatial Memory in Mice. Neurobiol. Learn. Mem. 2021, 179, 107406. [Google Scholar] [CrossRef]
- Maze, I.; Covington, H.E.; Dietz, D.M.; LaPlant, Q.; Renthal, W.; Russo, S.J.; Mechanic, M.; Mouzon, E.; Neve, R.L.; Haggarty, S.J.; et al. Essential Role of the Histone Methyltransferase G9a in Cocaine-Induced Plasticity. Science 2010, 327, 213–216. [Google Scholar] [CrossRef]
- Maze, I.; Chaudhury, D.; Dietz, D.M.; von Schimmelmann, M.; Kennedy, P.J.; Lobo, M.K.; Sillivan, S.E.; Miller, M.L.; Bagot, R.C.; Sun, H.; et al. G9a Influences Neuronal Subtype Specification in Striatum. Nat. Neurosci. 2014, 17, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Kamiunten, T.; Ideno, H.; Shimada, A.; Arai, Y.; Terashima, T.; Tomooka, Y.; Nakamura, Y.; Nakashima, K.; Kimura, H.; Shinkai, Y.; et al. Essential Roles of G9a in Cell Proliferation and Differentiation during Tooth Development. Exp. Cell Res. 2017, 357, 202–210. [Google Scholar] [CrossRef]
- Ideno, H.; Nakashima, K.; Komatsu, K.; Araki, R.; Abe, M.; Arai, Y.; Kimura, H.; Shinkai, Y.; Tachibana, M.; Nifuji, A. G9a Is Involved in the Regulation of Cranial Bone Formation through Activation of Runx2 Function during Development. Bone 2020, 137, 115332. [Google Scholar] [CrossRef] [PubMed]
- Higashihori, N.; Lehnertz, B.; Sampaio, A.; Underhill, T.M.; Rossi, F.; Richman, J.M. Methyltransferase G9A Regulates Osteogenesis via Twist Gene Repression. J. Dent. Res. 2017, 96, 1136–1144. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef]
- Zhang, R.H.; Judson, R.N.; Liu, D.Y.; Kast, J.; Rossi, F.M.V. The Lysine Methyltransferase Ehmt2/G9a Is Dispensable for Skeletal Muscle Development and Regeneration. Skelet. Muscle 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Katoh, K.; Yamazaki, R.; Onishi, A.; Sanuki, R.; Furukawa, T. G9a Histone Methyltransferase Activity in Retinal Progenitors Is Essential for Proper Differentiation and Survival of Mouse Retinal Cells. J. Neurosci. 2012, 32, 17658–17670. [Google Scholar] [CrossRef]
- Olsen, J.B.; Wong, L.; Deimling, S.; Miles, A.; Guo, H.; Li, Y.; Zhang, Z.; Greenblatt, J.F.; Emili, A.; Tropepe, V. G9a and ZNF644 Physically Associate to Suppress Progenitor Gene Expression during Neurogenesis. Stem Cell Rep. 2016, 7, 454–470. [Google Scholar] [CrossRef][Green Version]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic Stress Induces Dimethylated Histone H3 Lysine 9 through Histone Methyltransferase G9a in Mammalian Cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like Kinase-1 Is Activated by Aurora A to Promote Checkpoint Recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef]
- Agarwal, P.; Jackson, S.P. G9a Inhibition Potentiates the Anti-Tumour Activity of DNA Double-Strand Break Inducing Agents by Impairing DNA Repair Independent of P53 Status. Cancer Lett. 2016, 380, 467–475. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Shen, Y.; He, P.; Ding, J.; Chen, Y. G9a Stimulates CRC Growth by Inducing P53 Lys373 Dimethylation-Dependent Activation of Plk1. Theranostics 2018, 8, 2884–2895. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Turner, B.M. Altered Histone Modifications in Cancer. Adv. Exp. Med. Biol. 2013, 754, 81–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Su, Y.; Shen, Y.; Jiang, D.; Hou, Y.; Geng, M.; Ding, J.; Chen, Y. G9a Regulates Breast Cancer Growth by Modulating Iron Homeostasis through the Repression of Ferroxidase Hephaestin. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Qian, Q.; Zhu, Z.; Shen, X.; Yu, S.; Yu, Z.; Sun, R.; Li, Y.; Guo, D.; Fan, H. Increased Expression of EHMT2 Associated with H3K9me2 Level Contributes to the Poor Prognosis of Gastric Cancer. Oncol. Lett. 2020, 20, 1734–1742. [Google Scholar] [CrossRef]
- Hua, K.-T.; Wang, M.-Y.; Chen, M.-W.; Wei, L.-H.; Chen, C.-K.; Ko, C.-H.; Jeng, Y.-M.; Sung, P.-L.; Jan, Y.-H.; Hsiao, M.; et al. The H3K9 Methyltransferase G9a Is a Marker of Aggressive Ovarian Cancer That Promotes Peritoneal Metastasis. Mol. Cancer 2014, 13, 1–13. [Google Scholar] [CrossRef]
- Chen, G.; Yu, X.; Zhang, M.; Zheng, A.; Wang, Z.; Zuo, Y.; Liang, Q.; Jiang, D.; Chen, Y.; Zhao, L.; et al. Inhibition of Euchromatic Histone Lysine Methyltransferase 2 (EHMT2) Suppresses the Proliferation and Invasion of Cervical Cancer Cells. Cytogenet. Genome Res. 2019, 158, 205–212. [Google Scholar] [CrossRef]
- Hsiao, S.M.; Chen, M.W.; Chen, C.A.; Chien, M.H.; Hua, K.T.; Hsiao, M.; Kuo, M.L.; Wei, L.H. The H3K9 Methyltransferase G9a Represses E-Cadherin and Is Associated with Myometrial Invasion in Endometrial Cancer. Ann. Surg. Oncol. 2015, 22, 1556–1565. [Google Scholar] [CrossRef]
- Fan, H.T.; Shi, Y.Y.; Lin, Y.; Yang, X.P. EHMT2 Promotes the Development of Prostate Cancer by Inhibiting PI3K/AKT/MTOR Pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7808–7815. [Google Scholar] [CrossRef]
- Qin, J.; Zeng, Z.; Luo, T.; Li, Q.; Hao, Y.; Chen, L. Clinicopathological Significance of G9A Expression in Colorectal Carcinoma. Oncol. Lett. 2018, 15, 8611–8619. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chiu, D.K.C.; Tsang, F.H.C.; Law, C.T.; Cheng, C.L.H.; Au, S.L.K.; Lee, J.M.F.; Wong, C.C.L.; Ng, I.O.L.; Wong, C.M. Histone Methyltransferase G9a Promotes Liver Cancer Development by Epigenetic Silencing of Tumor Suppressor Gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef]
- Guo, A.S.; Huang, Y.Q.; Ma, X.D.; Lin, R.S. Mechanism of G9a Inhibitor BIX-01294 Acting on U251 Glioma Cells. Mol. Med. Rep. 2016, 14, 4613–4621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Rajagopalan, D.; Chung, T.H.; Hooi, L.; Toh, T.B.; Tian, J.S.; Rashid, M.B.M.A.; Sahib, N.R.B.M.; Gu, M.; Lim, J.J.; et al. Frequent Upregulation of G9a Promotes RelB-Dependent Proliferation and Survival in Multiple Myeloma. Exp. Hematol. Oncol. 2020, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Dang, N.N.; Jiao, J.; Meng, X.; An, Y.; Han, C.; Huang, S. Abnormal Overexpression of G9a in Melanoma Cells Promotes Cancer Progression via Upregulation of the Notch1 Signaling Pathway. Aging 2020, 12, 2393–2407. [Google Scholar] [CrossRef]
- Ma, W.; Han, C.; Zhang, J.; Song, K.; Chen, W.; Kwon, H.; Wu, T. The Histone Methyltransferase G9a Promotes Cholangiocarcinogenesis through Regulation of the Hippo Pathway Kinase LATS2 and YAP Signaling Pathway. FASEB J. 2020, 34, 1283–1297. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, J.; Yang, L.; Yuan, Y.-C.; Tong, T.R.; Wu, J.; Yun, X.; Bonner, M.; Pangeni, R.; Liu, Z.; et al. Targeting Histone Methyltransferase G9a Inhibits Growth and Wnt Signaling Pathway by Epigenetically Regulating HP1α and APC2 Gene Expression in Non-Small Cell Lung Cancer. Mol. Cancer 2018, 17, 1–15. [Google Scholar] [CrossRef]
- Cui, J.; Sun, W.; Hao, X.; Wei, M.; Su, X.; Zhang, Y.; Su, L.; Liu, X. EHMT2 Inhibitor BIX-01294 Induces Apoptosis through PMAIP1-USP9X-MCL1 Axis in Human Bladder Cancer Cells. Cancer Cell Int. 2015, 15, 1–9. [Google Scholar] [CrossRef]
- Casciello, F.; Al-Ejeh, F.; Miranda, M.; Kelly, G.; Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. G9a-Mediated Repression of CDH10 in Hypoxia Enhances Breast Tumour Cell Motility and Associates with Poor Survival Outcome. Theranostics 2020, 10, 4515–4529. [Google Scholar] [CrossRef]
- Chen, M.-W.; Hua, K.-T.; Kao, H.-J.; Chi, C.-C.; Wei, L.-H.; Johansson, G.; Shiah, S.-G.; Chen, P.-S.; Jeng, Y.-M.; Cheng, T.-Y.; et al. H3K9 Histone Methyltransferase G9a Promotes Lung Cancer Invasion and Metastasis by Silencing the Cell Adhesion Molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef]
- Yin, C.; Ke, X.; Zhang, R.; Hou, J.; Dong, Z.; Wang, F.; Zhang, K.; Zhong, X.; Yang, L.; Cui, H. G9a Promotes Cell Proliferation and Suppresses Autophagy in Gastric Cancer by Directly Activating MTOR. FASEB J. 2019, 33, 14036–14050. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, X.; Guan, X.; Zhang, H.; Ma, Y.; Zhang, S.; Wang, E.; Zhang, L.; Han, Y. Overexpression of G9a and MCM7 in Oesophageal Squamous Cell Carcinoma Is Associated with Poor Prognosis. Histopathology 2015, 66, 192–200. [Google Scholar] [CrossRef]
- Ho, J.C.; Abdullah, L.N.; Pang, Q.Y.; Jha, S.; Chow, E.K.H.; Yang, H.; Kato, H.; Poellinger, L.; Ueda, J.; Lee, K.L. Inhibition of the H3K9 Methyltransferase G9A Attenuates Oncogenicity and Activates the Hypoxia Signaling Pathway. PLoS ONE 2017, 12, e0188051. [Google Scholar] [CrossRef]
- Liu, X.R.; Zhou, L.H.; Hu, J.X.; Liu, L.M.; Wan, H.P.; Zhang, X.Q. UNC0638, a G9a Inhibitor, Suppresses Epithelial-Mesenchymal Transition-Mediated Cellular Migration and Invasion in Triple Negative Breast Cancer. Mol. Med. Rep. 2018, 17, 2239–2244. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.B.; Shiah, Y.J.; Lourenco, C.; Mullen, P.J.; Dingar, D.; Redel, C.; Tamachi, A.; Ba-Alawi, W.; Aman, A.; Al-awar, R.; et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer Cell 2018, 34, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.J.; Klimecki, W.T.; Lau, S.S.; Feinstein, Y.; Futscher, B.W. 5-Aza-2′-Deoxycytidine-Mediated Reductions in G9A Histone Methyltransferase and Histone H3 K9 Di-Methylation Levels Are Linked to Tumor Suppressor Gene Reactivation. Oncogene 2007, 26, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a Interacts with Snail and Is Critical for Snail-Mediated E-Cadherin Repression in Human Breast Cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yao, D.; Jiang, Y.; Huang, J.; Yang, S.; Wang, J. Synthesis and Biological Evaluation of Benzimidazole Derivatives as the G9a Histone Methyltransferase Inhibitors That Induce Autophagy and Apoptosis of Breast Cancer Cells. Bioorganic Chem. 2017, 72, 168–181. [Google Scholar] [CrossRef]
- Mabe, N.W.; Garcia, N.M.G.; Wolery, S.E.; Newcomb, R.; Meingasner, R.C.; Vilona, B.A.; Lupo, R.; Lin, C.C.; Chi, J.T.; Alvarez, J.V. G9a Promotes Breast Cancer Recurrence through Repression of a Pro-Inflammatory Program. Cell Rep. 2020, 33, 108341. [Google Scholar] [CrossRef]
- Lin, X.; Huang, Y.; Zou, Y.; Chen, X.; Ma, X. Depletion of G9a Gene Induces Cell Apoptosis in Human Gastric Carcinoma. Oncol. Rep. 2016, 35, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.-G. Kaempferol Induces Autophagic Cell Death via IRE1-JNK-CHOP Pathway and Inhibition of G9a in Gastric Cancer Cells. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Kim, T.W.; Cheon, C.; Ko, S.-G. SH003 Activates Autophagic Cell Death by Activating ATF4 and Inhibiting G9a under Hypoxia in Gastric Cancer Cells. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W. Cinnamaldehyde Induces Autophagy-Mediated Cell Death through ER Stress and Epigenetic Modification in Gastric Cancer Cells. Acta Pharmacol. Sin. 2021, 2021, 1–12. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, J.; Kim, W.-H.; Lee, Y.M. Hypoxic Silencing of Tumor Suppressor RUNX3 by Histone Modification in Gastric Cancer Cells. Oncogene 2009, 28, 184–194. [Google Scholar] [CrossRef]
- Hu, L.; Zang, M.; Wang, H.; Zhang, B.; Wang, Z.; Fan, Z.; Wu, H.; Li, J.; Su, L.; Yan, M.; et al. G9A Promotes Gastric Cancer Metastasis by Upregulating ITGB3 in a SET Domain-Independent Manner. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Watson, Z.L.; Yamamoto, T.M.; McMellen, A.; Kim, H.; Hughes, C.J.; Wheeler, L.J.; Post, M.D.; Behbakht, K.; Bitler, B.G. Histone Methyltransferases EHMT1 and EHMT2 (GLP/G9A) Maintain PARP Inhibitor Resistance in High-Grade Serous Ovarian Carcinoma. Clin. Epigenetics 2019, 11, 1–16. [Google Scholar] [CrossRef]
- Liu, M.; Thomas, S.L.; DeWitt, A.K.; Zhou, W.; Madaj, Z.B.; Ohtani, H.; Baylin, S.B.; Liang, G.; Jones, P.A. Dual Inhibition of DNA and Histone Methyltransferases Increases Viral Mimicry in Ovarian Cancer Cells. Cancer Res. 2018, 78, 5754–5766. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.-J.; Shun, C.-T.; Yen, M.-L.; Chou, C.-H.; Lin, M.-C.; Chen, R.-J.; Shun, C.-T.; Yen, M.-L.; Chou, C.-H.; Lin, M.-C. Methyltransferase G9a Promotes Cervical Cancer Angiogenesis and Decreases Patient Survival. Oncotarget 2017, 8, 62081–62098. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Soejima, K.; Yasuda, H.; Kawada, I.; Nakachi, I.; Yoda, S.; Naoki, K.; Ishizaka, A. Deregulation of Histone Lysine Methyltransferases Contributes to Oncogenic Transformation of Human Bronchoepithelial Cells. Cancer Cell Int. 2008, 8, 1–12. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, P.; Li, W.; Zhao, T.; Zhang, Z.; Chen, S.; Yang, Y.; Feng, Y.; Li, F.; Shirley Liu, X.; et al. G9A Promotes Tumor Cell Growth and Invasion by Silencing CASP1 in Non-Small-Cell Lung Cancer Cells. Cell Death Dis. 2017, 8, e2726. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, Y.; Dai, M.; Wu, J.; Yu, B.; Zhang, H.; Kong, W.; Wu, H.; Yu, X. Snail2 Induced E-Cadherin Suppression and Metastasis in Lung Carcinoma Facilitated by G9a and HDACs. Cell Adhes. Migr. 2019, 13, 285–292. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, K.; Pangeni, R.P.; Wu, J.; Li, W.; Du, Y.; Guo, Y.; Chaurasiya, S.; Arvanitis, L.; Raz, D.J. G9a Promotes Invasion and Metastasis of Non–Small Cell Lung Cancer through Enhancing Focal Adhesion Kinase Activation via NF-ΚB Signaling Pathway. Mol. Cancer Res. 2021, 19, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Pangeni, R.P.; Yang, L.; Zhang, K.; Wang, J.; Li, W.; Guo, C.; Yun, X.; Sun, T.; Wang, J.; Raz, D.J. G9a Regulates Tumorigenicity and Stemness through Genome-Wide DNA Methylation Reprogramming in Non-Small Cell Lung Cancer. Clin. Epigenetics 2020, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bergin, C.J.; Zouggar, A.; Haebe, J.R.; Masibag, A.N.; Desrochers, F.M.; Reilley, S.Y.; Agrawal, G.; Benoit, Y.D. G9a Controls Pluripotent-like Identity and Tumor-Initiating Function in Human Colorectal Cancer. Oncogene 2020, 40, 1191–1202. [Google Scholar] [CrossRef]
- Bárcena-Varela, M.; Caruso, S.; Llerena, S.; Álvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Rebouissou, S.; Alvarez, L.; Jimenez, M.; et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology 2019, 69, 587–603. [Google Scholar] [CrossRef]
- Yokoyama, M.; Chiba, T.; Zen, Y.; Oshima, M.; Kusakabe, Y.; Noguchi, Y.; Yuki, K.; Koide, S.; Tara, S.; Saraya, A.; et al. Histone Lysine Methyltransferase G9a Is a Novel Epigenetic Target for the Treatment of Hepatocellular Carcinoma. Oncotarget 2017, 8, 21315–21326. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, Y.; Dai, M.; Wang, X.; Wu, J.; Yu, B.; Zhang, H.; Cui, Y.; Kong, W.; Wu, H.; et al. G9a and Histone Deacetylases Are Crucial for Snail2-Mediated E-Cadherin Repression and Metastasis in Hepatocellular Carcinoma. Cancer Sci. 2019, 110, 3442–3452. [Google Scholar] [CrossRef]
- Nakatsuka, T.; Tateishi, K.; Kato, H.; Fujiwara, H.; Yamamoto, K.; Kudo, Y.; Nakagawa, H.; Tanaka, Y.; Ijichi, H.; Ikenoue, T.; et al. Inhibition of Histone Methyltransferase G9a Attenuates Liver Cancer Initiation by Sensitizing DNA-Damaged Hepatocytes to P53-Induced Apoptosis. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Segovia, C.; San José-Enériz, E.; Munera-Maravilla, E.; Martínez-Fernández, M.; Garate, L.; Miranda, E.; Vilas-Zornoza, A.; Lodewijk, I.; Rubio, C.; Segrelles, C.; et al. Inhibition of a G9a/DNMT Network Triggers Immune-Mediated Bladder Cancer Regression. Nat. Med. 2019, 25, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Alves-Silva, J.C.; de Carvalho, J.L.; Rabello, D.A.; Serejo, T.R.T.; Rego, E.M.; Neves, F.A.R.; Lucena-Araujo, A.R.; Pittella-Silva, F.; Saldanha-Araujo, F. GLP Overexpression Is Associated with Poor Prognosis in Chronic Lymphocytic Leukemia and Its Inhibition Induces Leukemic Cell Death. Investig. New Drugs 2018, 36, 955–960. [Google Scholar] [CrossRef]
- Huang, Y.; Zou, Y.; Lin, L.; Ma, X.; Huang, X. Effect of BIX-01294 on Proliferation, Apoptosis and Histone Methylation of Acute T Lymphoblastic Leukemia Cells. Leuk. Res. 2017, 62, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kondengaden, S.M.; Luo, L.F.; Huang, K.; Zhu, M.; Zang, L.; Bataba, E.; Wang, R.; Luo, C.; Wang, B.; Li, K.K.; et al. Discovery of Novel Small Molecule Inhibitors of Lysine Methyltransferase G9a and Their Mechanism in Leukemia Cell Lines. Eur. J. Med. Chem. 2016, 122, 382–393. [Google Scholar] [CrossRef]
- Lehnertz, B.; Pabst, C.; Su, L.; Miller, M.; Liu, F.; Yi, L.; Zhang, R.; Krosl, J.; Yung, E.; Kirschner, J.; et al. The Methyltransferase G9a Regulates HoxA9-Dependent Transcription in AML. Genes Dev. 2014, 28, 317–327. [Google Scholar] [CrossRef]
- San José-Enériz, E.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; Estella-Hermoso de Mendoza, A.; et al. Discovery of First-in-Class Reversible Dual Small Molecule Inhibitors against G9a and DNMTs in Hematological Malignancies. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Madrazo, E.; Ruano, D.; Abad, L.; Alonso-Gómez, E.; Sánchez-Valdepeñas, C.; González-Murillo, Á.; Ramírez, M.; Redondo-Muñoz, J. G9a Correlates with VLA-4 Integrin and Influences the Migration of Childhood Acute Lymphoblastic Leukemia Cells. Cancers 2018, 10, 325. [Google Scholar] [CrossRef]
- Nagaraja, S.G.S.; Subramanian, U.; Nagarajan, D. Radiation-Induced H3K9 Methylation on E-Cadherin Promoter Mediated by ROS/Snail Axis: Role of G9a Signaling during Lung Epithelial-Mesenchymal Transition. Toxicol. Vitr. 2021, 70, 105037. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Z.; Cao, K.; Zhang, L.; Ma, Y.; Yu, S.; Jin, H.; Liu, X.; Li, W. G9a Regulates Cell Sensitivity to Radiotherapy via Histone H3 Lysine 9 Trimethylation and Ccdc8 in Lung Cancer. OncoTargets Ther. 2021, 14, 3721–3728. [Google Scholar] [CrossRef]
- Luo, C.W.; Wang, J.Y.; Hung, W.C.; Peng, G.; Tsai, Y.L.; Chang, T.M.; Chai, C.Y.; Lin, C.H.; Pan, M.R. G9a Governs Colon Cancer Stem Cell Phenotype and Chemoradioresistance through PP2A-RPA Axis-Mediated DNA Damage Response. Radiother. Oncol. 2017, 124, 395–402. [Google Scholar] [CrossRef]
- Dobson, T.H.W.; Hatcher, R.J.; Swaminathan, J.; Das, C.M.; Shaik, S.; Tao, R.-H.; Milite, C.; Castellano, S.; Taylor, P.H.; Sbardella, G.; et al. Regulation of USP37 Expression by REST-Associated G9a-Dependent Histone Methylation. Mol. Cancer Res. 2017, 15, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Przanowski, P.; Jackl, J.; Wojtas, B.; Kaminska, B. BIX01294, an Inhibitor of Histone Methyltransferase, Induces Autophagy-Dependent Differentiation of Glioma Stem-like Cells. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Kato, S.; Weng, Q.Y.; Insco, M.L.; Chen, K.Y.; Muralidhar, S.; Pozniak, J.; Diaz, J.M.S.; Drier, Y.; Nguyen, N.; Lo, J.A.; et al. Gain-of-Function Genetic Alterations of G9a Drive Oncogenesis. Cancer Discov. 2020, 10, 980–997. [Google Scholar] [CrossRef]
- Liu, S.; Ye, D.; Guo, W.; Yu, W.; He, Y.; Hu, J.; Wang, Y.; Zhang, L.; Liao, Y.; Song, H.; et al. G9a Is Essential for EMT-Mediated Metastasis and Maintenance of Cancer Stem Cell-like Characters in Head and Neck Squamous Cell Carcinoma. Oncotarget 2015, 6, 6887–6901. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, S.P.; Li, F.; Dost, A.F.M.; Louie, S.M.; Marsh, B.P.; Pessina, P.; Anbarasu, C.R.; Brainson, C.F.; Tuminello, S.J.; Lieberman, A.; et al. H3K9 Methyltransferases and Demethylases Control Lung Tumor-Propagating Cells and Lung Cancer Progression. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Marciniak, M.P.; Jackl, J.; Kaminska, B. Pre-Treatment or Post-Treatment of Human Glioma Cells with BIX01294, the Inhibitor of Histone Methyltransferase G9a, Sensitizes Cells to Temozolomide. Front. Pharmacol. 2018, 9, 1271. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histone Types | Sites | Biological Outcome | References |
|---|---|---|---|
| Histone H3 | H3K9me1 | Transcriptional repression Heterochromatin formation | [5,25,37] |
| H3K9me2 | |||
| H3K9me3 | |||
| Histone H3 | H3K27me1 | Transcriptional repression Heterochromatin formation | [46,48] |
| Histone H3 | H3K56me1 | DNA replication | [49] |
| Histone H1.2 | H1.2K187me | nd | [51] |
| Histone H1.4 | H1.4K26me1 | Transcriptional repression Chromatin structure | [50] |
| H1.4K26me2 |
| Functions | Substrates | Site | Biological Outcome | References |
|---|---|---|---|---|
| Transcription Factors | C/EBPb | K39 | Inhibits transcriptional activity by repressing C/EBPb transactivation | [54] |
| MyoD | K104me1/2 | Inhibits MyoD transcriptional activity | [55] | |
| MEF2D | K267me1/2 | Inhibits MEF2D transcriptional activity by preventing its recruitment on chromatin | [56] | |
| p53 | K373me2 | Inhibits transcriptional activity and p53-dependent apoptosis | [57] | |
| ERα | K235me2 | Induces transcriptional activity by recruiting the PHF20/MOF HAT complex | [58] | |
| Foxo1 | K273me1/2 | Induces Foxo1 degradation | [59] | |
| KLF12 | K313 | nd | [36] | |
| Chromatin remodeling factors and coregulators | G9a | K185me2/3 | Induces specific glucocorticoid receptor transcriptional activity by recruiting HP1γ | [32,34,35] |
| GLP | K205me2 | Induces specific glucocorticoid receptor transcriptional activity by recruiting HP1γ | [32] | |
| Sirt1 | K662 | nd | [60] | |
| Pontin | K265, K267, K268, K274, K281, K285 | Induces HIF-1 transcriptional activity by enhancing p300 recruitment | [61] | |
| Reptin | K67me1 | Inhibits HIF transcriptional activity by recruiting corepressors | [62] | |
| HDAC1 | K432 | nd | [36] | |
| HIFα | K674me1/2 | Inhibits HIF-1 transcriptional activity | [63] | |
| CSB | K170, K297, K448, K1054 | nd | [36] | |
| MTA1 | K532me1 | Inhibits transcription by recruiting the assembly of the NuRD repressive complex | [64] | |
| ATF7IP (hAM) | K16me3 | Induces transgene silencing by recruiting MPP8 | [65] | |
| Chromatin binding protein | CDYL1 | K135me3 | Decreases its interaction with H3K9me3 | [36] |
| WIZ | K305me3 | nd | [36] | |
| DNA methyltransferases | DNMT1 | K70me2 | nd | [36] |
| DNMT3 | K47me2 | Inhibits transcription by recruiting MPP8/DNMT3/G9a/GLP repressive complex | [66] | |
| Others | Acinus | K654me2 | nd | [36] |
| MDC1 | K45me2 | Induces ATM accumulation on damage sites | [67] | |
| Plk1 | K209me1 | Antagonizes T210 phosphorylation to inhibit Plk1 activity on DNA replication | [68] | |
| Lig1 | K126me2/3 | Maintenance in DNA methylation by promoting UHRF1 recruitment to replication foci | [69] |
| G9a Roles | Cancer Types | G9a Biological Roles | References |
|---|---|---|---|
| Oncogenic | Breast Cancer | Suppresses tumor suppressor genes Enhances EMT Disrupts iron homeostasis Inhibits autophagy | [44,130] [123,131] [109] [132] |
| Gastric Cancer | Suppresses tumor suppressor genes Inhibits apoptosis and autophagy Promotes metastasis | [138] [125,135,136,137] [139] | |
| Ovarian Cancer | Promotes metastasis Suppresses tumor suppressor genes Maintains PARP-inhibitor resistance | [111] [45,111] [140] | |
| Cervical Cancer | Induces angiogenesis Enhances tissue invasion | [142] [112] | |
| Endometrial Cancer | Enhances tissue invasion | [113] | |
| Prostate Cancer | Stimulates proliferation | [114] | |
| Lung Cancer | Enhances EMT Activates WNT signaling pathway Maintains lung cancer stemness Supports resistance to radiotherapy | [124,144,145,160] [121] [147] [161] | |
| Colorectal Cancer | Stimulates proliferation Enhances self-renewal and stemness Promotes resistance to chemotherapy | [59,106] [148] [162] | |
| Liver Cancer | Suppresses tumor suppressor genes Enhances EMT Inhibits cell apoptosis | [116] [151] [152] | |
| Bladder Cancer | Inhibits cell apoptosis and autophagy | [122,153] | |
| Brain Cancer | Stimulates proliferation Inhibits autophagy | [117,163] [164] | |
| Hematological malignancies | Enhances self-renewal and stemness Promotes migration Inhibits apoptosis and stimulates proliferation | [157] [159] [118,155] | |
| Skin Cancer | Promotes progression | [119,165] | |
| Head and Neck Cancer | Enhances EMT | [166] | |
| Bile duct Cancer | Suppresses tumor suppressor genes | [120] | |
| Anti- oncogenic | Lung Cancer | Inhibits cancer progression | [167] |
| Brain Cancer | Inhibits HIF-induced migration Inhibits cancer stemness | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poulard, C.; Noureddine, L.M.; Pruvost, L.; Le Romancer, M. Structure, Activity, and Function of the Protein Lysine Methyltransferase G9a. Life 2021, 11, 1082. https://doi.org/10.3390/life11101082
Poulard C, Noureddine LM, Pruvost L, Le Romancer M. Structure, Activity, and Function of the Protein Lysine Methyltransferase G9a. Life. 2021; 11(10):1082. https://doi.org/10.3390/life11101082
Chicago/Turabian StylePoulard, Coralie, Lara M. Noureddine, Ludivine Pruvost, and Muriel Le Romancer. 2021. "Structure, Activity, and Function of the Protein Lysine Methyltransferase G9a" Life 11, no. 10: 1082. https://doi.org/10.3390/life11101082
APA StylePoulard, C., Noureddine, L. M., Pruvost, L., & Le Romancer, M. (2021). Structure, Activity, and Function of the Protein Lysine Methyltransferase G9a. Life, 11(10), 1082. https://doi.org/10.3390/life11101082