Implication of Contactins in Demyelinating Pathologies



Abstract
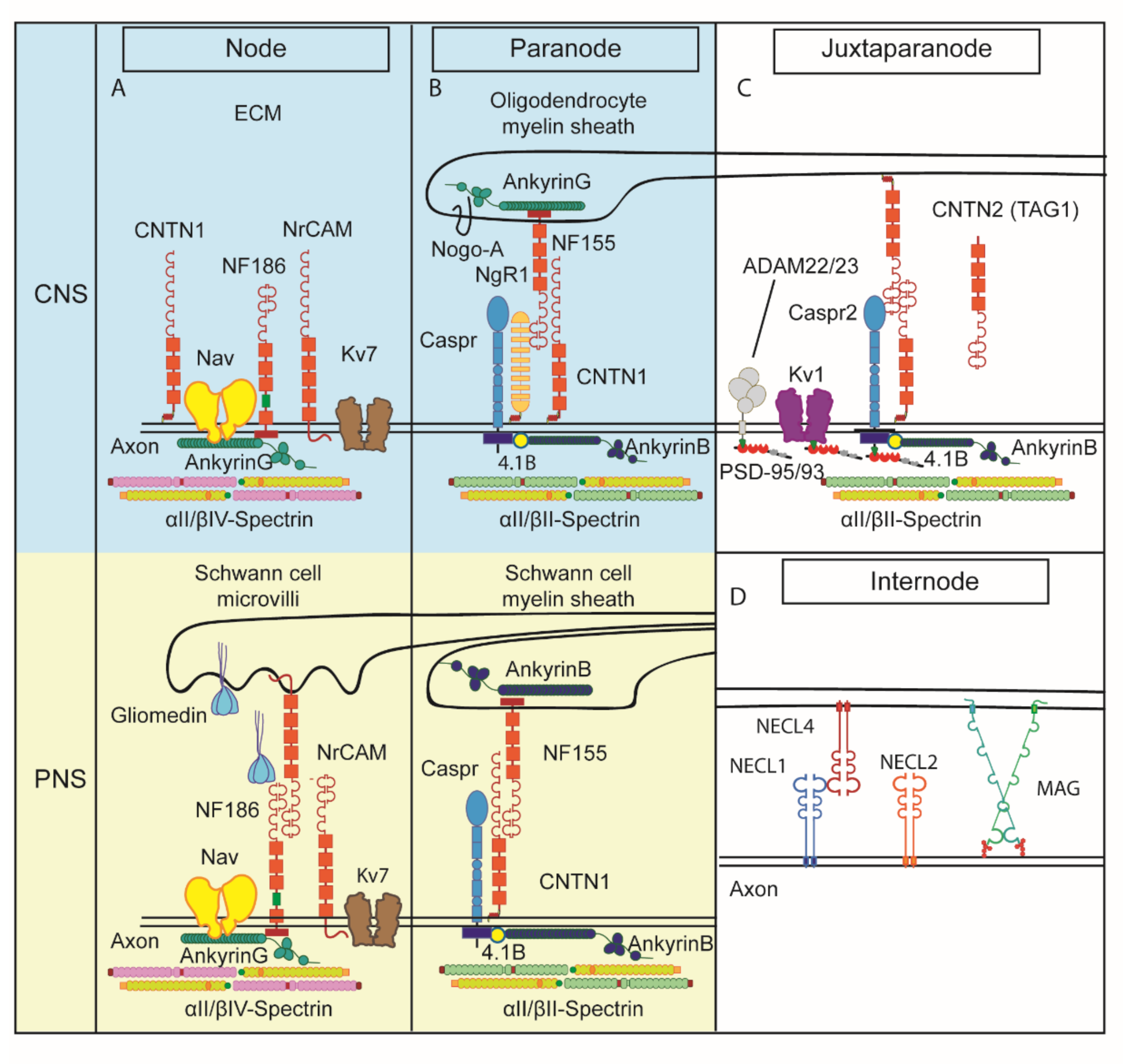
1. The Role of Contactins in the Organization of Myelinated Axons
1.1. Node of Ranvier
1.2. Paranode
1.3. Juxtaparanode
1.4. Internode
2. Implication of Perinodal Contactins and Their Interactors in Demyelination
2.1. Contactin-1 and CIDP
2.2. Contactin-2 and MS
2.3. Contactins as Biomarkers?
3. Perinodal Contactins and Their Interactors in Animal Models of Demyelination
3.1. Contactins and EAE
3.2. Contactins and the Cuprizone Model
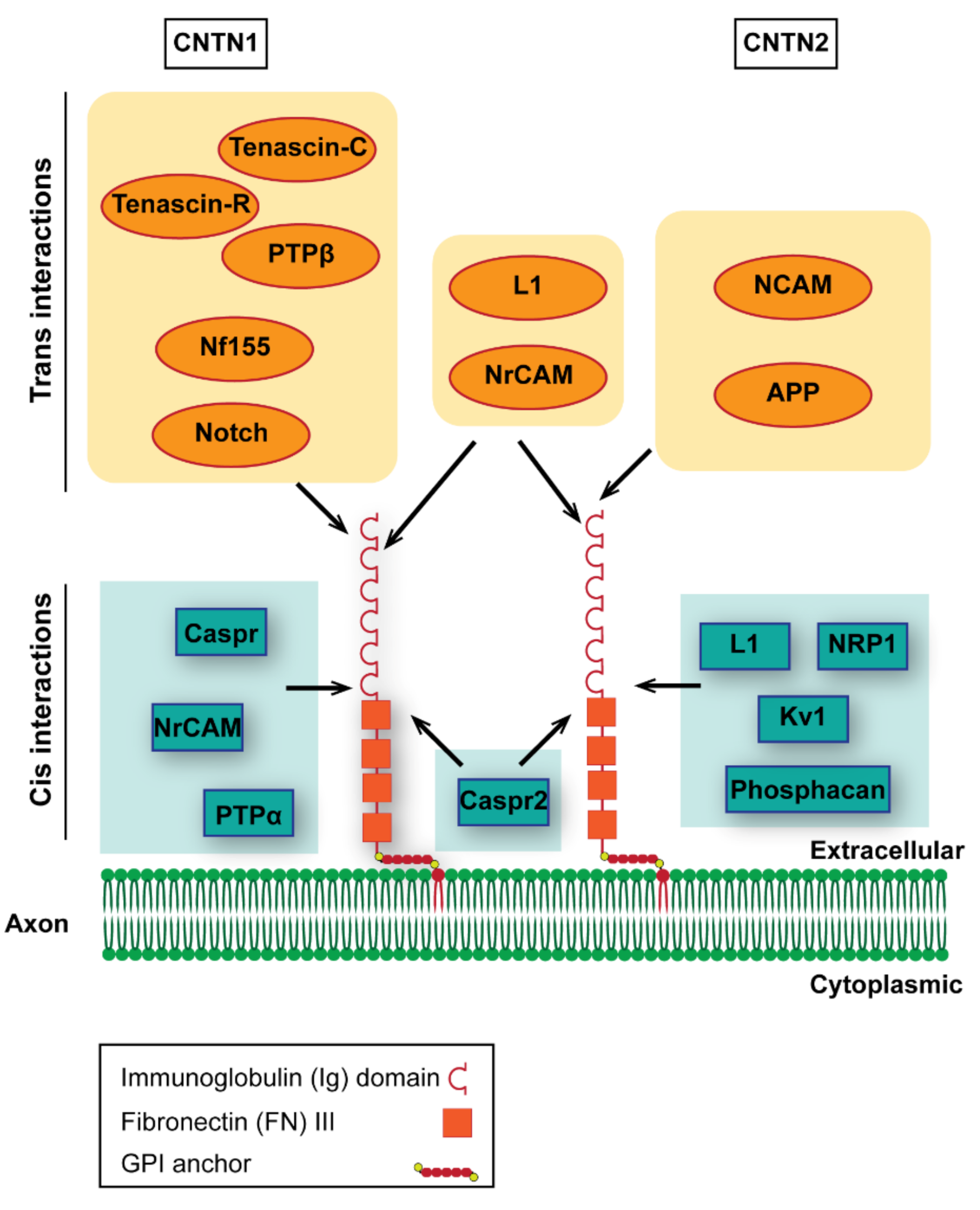
4. Other Functions of Perinodal CNTN1 and 2
4.1. Contactin-1
4.2. Contactin-2
5. Concluding Paragraph
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arroyo, E.J.; Scherer, S.S. On the molecular architecture of myelinated fibers. Histochem. Cell Biol. 2000, 113, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Peles, E.; Salzer, J.L. Molecular domains of myelinated axons. Curr. Opin. Neurobiol. 2000, 10, 558–565. [Google Scholar] [CrossRef]
- Girault, J.-A.; Peles, E. Development of nodes of Ranvier. Curr. Opin. Neurobiol. 2002, 12, 476–485. [Google Scholar] [CrossRef]
- Poliak, S.; Peles, E. The local differentiation of myelinated axons at nodes of Ranvier. Nat. Rev. Neurosci. 2003, 4, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Salzer, J.L.; Brophy, P.J.; Peles, E. Molecular domains of myelinated axons in the peripheral nervous system. Glia 2008, 56, 1532–1540. [Google Scholar] [CrossRef]
- Faivre-Sarrailh, C.; Devaux, J. Neuro-glial interactions at the nodes of Ranvier: Implication in health and diseases. Front. Cell. Neurosci. 2013, 7, 196. [Google Scholar] [CrossRef]
- Rasband, M.N.; Peles, E. The Nodes of Ranvier: Molecular Assembly and Maintenance. Cold Spring Harb. Perspect. Biol. 2015, 8, a020495. [Google Scholar] [CrossRef]
- Lubetzki, C.; Sol-Foulon, N.; Desmazières, A. Nodes of Ranvier During Development and Re-pair in the Cns. Nat. Rev. Neurol. 2020, 16, 426–439. [Google Scholar] [CrossRef]
- Zoupi, L.; Savvaki, M.; Karagogeos, D. Axons and myelinating glia: An intimate contact. IUBMB Life 2011, 63, 730–735. [Google Scholar] [CrossRef]
- Moll, C.; Mourre, C.; Lazdunski, M.; Ulrich, J. Increase of sodium channels in demyelinated lesions of multiple sclerosis. Brain Res. 1991, 556, 311–316. [Google Scholar] [CrossRef]
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173. [Google Scholar] [CrossRef] [PubMed]
- Coman, I.; Aigrot, M.S.; Seilhean, D.; Reynolds, R.; Girault, J.A.; Zalc, B.; Lubetzki, C. Nodal, Paranodal and Juxtaparanodal Axonal Proteins During Demyelination and Remyelination in Multiple Sclerosis. Brain 2006, 129 Pt 12, 3186–3195. [Google Scholar] [CrossRef]
- Kastriti, M.E.; Sargiannidou, I.; Kleopa, K.A.; Karagogeos, D. Differential modulation of the juxtaparanodal complex in Multiple Sclerosis. Mol. Cell. Neurosci. 2015, 67, 93–103. [Google Scholar] [CrossRef]
- Karagogeos, D. Neural GPI anchored cell adhesion molecules. Front. Biosci. 2003, 8, s1304–s1320. [Google Scholar] [CrossRef] [PubMed]
- Katidou, M.; Vidaki, M.; Strigini, M.; Karagogeos, D. The immunoglobulin superfamily of neuronal cell adhesion molecules: Lessons from animal models and correlation with human disease. Biotechnol. J. 2008, 3, 1564–1580. [Google Scholar] [CrossRef] [PubMed]
- Gennarini, G.; Bizzoca, A.; Picocci, S.; Puzzo, D.; Corsi, P.; Furley, A.J. The role of Gpi-anchored axonal glycoproteins in neural development and neurological disorders. Mol. Cell. Neurosci. 2017, 81, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Ranvier, L.J. Contributions à L’histologie Et à La Physiologie Des Nerfs Périphériques. Comptes Rendus l’Académie Sci. 1871, 73, 1168–1171. [Google Scholar]
- Jenkins, S.M.; Bennett, V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 2001, 155, 739–746. [Google Scholar] [CrossRef]
- Bennett, V.; Lambert, S. Physiological roles of axonal ankyrins in survival of premyelinated axons and localization of voltage-gated sodium channels. J. Neurocytol. 2000, 28, 303–318. [Google Scholar] [CrossRef]
- Salzer, J.L. Clustering Sodium Channels at the Node of Ranvier: Close Encounters of the Axon–Glia Kind. Neuron 1997, 18, 843–846. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Schaller, K.L.; Lasher, R.S.; Peles, E.; Levinson, S.R. Sodium channel Nav1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 5616–5620. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kazen-Gillespie, K.; Hortsch, M.; Isom, L.L. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J. Biol. Chem. 2000, 275, 11383–11388. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, K.; Eshed-Eisenbach, Y.; Frechter, S.; Amor, V.; Salomon, D.; Sabanay, H.; DuPree, J.L.; Grumet, M.; Brophy, P.J.; Shrager, P.; et al. A Glial Signal Consisting of Gliomedin and NrCAM Clusters Axonal Na+ Channels during the Formation of Nodes of Ranvier. Neuron 2010, 65, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Collinson, J.M.; Marshall, D.; Gillespie, C.S.; Brophy, P.J. Transient expression of neurofascin by oligodendrocytes at the onset of myelinogenesis: Implications for mechanisms of axon-glial interaction. Glia 1998, 23, 11–23. [Google Scholar] [CrossRef]
- Ratcliffe, C.F.; Westenbroek, R.E.; Curtis, R.; Catterall, W.A. Sodium Channel Beta1 and Beta3 Subunits Associate with Neurofascin through Their Extracellular Immunoglobulin-Like Domain. J. Cell Biol. 2001, 154, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.L.; Tait, S.; Melrose, S.; Johnson, R.; Zonta, B.; Court, F.A.; Macklin, W.B.; Meek, S.; Smith, A.J.; Cottrell, D.F.; et al. Neurofascins Are Required to Establish Axonal Domains for Saltatory Conduction. Neuron 2005, 48, 737–742. [Google Scholar] [CrossRef]
- Freeman, S.A.; Desmazières, A.; Fricker, D.; Lubetzki, C.; Sol-Foulon, N. Mechanisms of So-dium Channel Clustering and Its Influence on Axonal Impulse Conduction. Cell Mol. Life Sci. 2016, 73, 723–735. [Google Scholar]
- Freeman, S.A.; Desmazières, A.; Simonnet, J.; Gatta, M.; Pfeiffer, F.; Aigrot, M.S.; Rappeneau, Q.; Guerreiro, S.; Michel, P.P.; Yanagawa, Y.; et al. Acceleration of Conduction Velocity Linked to Clustering of Nodal Components Pre-cedes Myelination. Proc. Natl. Acad. Sci. USA 2015, 112, E321–E328. [Google Scholar] [CrossRef]
- Dubessy, A.L.; Mazuir, E.; Rappeneau, Q.; Ou, S.; Abi Ghanem, C.; Piquand, K.; Aigrot, M.S.; Thétiot, M.; Desmazières, A.; Chan, E.; et al. Role of a Contactin Multi-Molecular Complex Secreted by Oli-godendrocytes in Nodal Protein Clustering in the Cns. Glia 2019, 67, 2248–2263. [Google Scholar] [CrossRef]
- Bennett, V.; Lorenzo, D.N. Spectrin- and Ankyrin-Based Membrane Domains and the Evolution of Vertebrates. Curr. Top Membr. 2013, 72, 1–37. [Google Scholar]
- Zhang, A.; Desmazières, A.; Zonta, B.; Melrose, S.; Campbell, G.R.; Mahad, N.; Li, Q.; Sherman, D.L.; Reynolds, R.; Brophy, P.J. Neurofascin 140 is an embryonic neuronal neurofascin isoform that promotes the assembly of the node of Ranvier. J. Neurosci. 2015, 35, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- Custer, A.W.; Kazarinova-Noyes, K.; Sakurai, T.; Xu, X.; Simon, W.; Grumet, M.; Shrager, P. The Role of the Ankyrin-Binding Protein NrCAM in Node of Ranvier Formation. J. Neurosci. 2003, 23, 10032–10039. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.Q.; Lambert, S.; Bennett, V. Molecular Composition of the Node of Ranvier: Identi-fication of Ankyrin-Binding Cell Adhesion Molecules Neurofascin (Mucin+/Third Fniii Domain-) and Nrcam at Nodal Axon Segments. J. Cell Biol. 1996, 135, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Kazarinova-Noyes, K.; Malhotra, J.D.; McEwen, D.P.; Mattei, L.N.; Berglund, E.O.; Ranscht, B.; Levinson, S.R.; Schachner, M.; Shrager, P.; Isom, L.L.; et al. Contactin Associates with Na+ Channels and Increases Their Functional Expression. J. Neurosci. 2001, 21, 7517–7525. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, J. Multiple functions of the paranodal junction of myelinated nerve fibers. J. Neurosci. Res. 2009, 87, 3250–3258. [Google Scholar] [CrossRef]
- Boyle, M.E.; Berglund, E.O.; Murai, K.K.; Weber, L.; Peles, E.; Ranscht, B. Contactin Orches-trates Assembly of the Septate-Like Junctions at the Paranode in Myelinated Peripheral Nerve. Neuron 2001, 30, 385–397. [Google Scholar] [CrossRef]
- Susuki, K.; Chang, K.-J.; Zollinger, D.R.; Liu, Y.; Ogawa, Y.; Eshed-Eisenbach, Y.; Dours-Zimmermann, M.T.; Oses-Prieto, J.A.; Burlingame, A.L.; Seidenbecher, C.I.; et al. Three Mechanisms Assemble Central Nervous System Nodes of Ranvier. Neuron 2013, 78, 469–482. [Google Scholar] [CrossRef]
- Peles, E.; Joho, K.; Plowman, G.D.; Schlessinger, J. Close Similarity between Drosophila Neu-rexin Iv and Mammalian Caspr Protein Suggests a Conserved Mechanism for Cellular Interactions. Cell 1997, 88, 745–746. [Google Scholar] [CrossRef]
- Menegoz, M.; Gaspar, P.; Le Bert, M.; Galvez, T.; Burgaya, F.; Palfrey, C.; Ezan, P.; Arnos, F.; Girault, J.-A. Paranodin, a glycoprotein of neuronal paranodal membranes. Neuron 1997, 19, 319–331. [Google Scholar] [CrossRef]
- Einheber, S.; Zanazzi, G.; Ching, W.; Scherer, S.; Milner, T.A.; Peles, E.; Salzer, J.L. "The Axonal Membrane Protein Caspr, a Homologue of Neurexin Iv, is a Component of the Septate-Like Par-anodal Junctions That Assemble During Myelination. J. Cell Biol. 1997, 139, 1495–1506. [Google Scholar] [CrossRef]
- Bhat, M.A.; Rios, J.C.; Lu, Y.; Garcia-Fresco, G.P.; Ching, W.; St Martin, M.; Li, J.; Einheber, S.; Chesler, M.; Rosenbluth, J.; et al. Axon-Glia Interactions and the Domain Organization of Myelinated Axons Requires Neurexin Iv/Caspr/Paranodin. Neuron 2001, 30, 369–383. [Google Scholar] [CrossRef]
- Tait, S.; Gunn-Moore, F.; Collinson, J.M.; Huang, J.; Lubetzki, C.; Pedraza, L.; Sherman, D.L.; Colman, D.R.; Brophy, P.J. An Oligodendrocyte Cell Adhesion Molecule at the Site of Assembly of the Paranodal Axo-Glial Junction. J. Cell Biol. 2000, 150, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.; Tait, S.; Faivre-Sarrailh, C.; Barbin, G.; Gunn-Moore, F.; Denisenko-Nehrbass, N.; Guennoc, A.M.; Girault, J.A.; Brophy, P.J.; Lubetzki, C. Neurofascin Is a Glial Receptor for the Par-anodin/Caspr-Contactin Axonal Complex at the Axoglial Junction. Curr. Biol. 2002, 12, 217–220. [Google Scholar] [CrossRef]
- Yermakov, L.M.; Hong, L.A.; Drouet, D.E.; Griggs, R.B.; Susuki, K. Functional Domains in Myelinated Axons. Adv. Exp. Med. Biol. 2019, 1190, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.M.; Thaxton, C.; Pribisko, A.L.; Cheng, J.G.; Dupree, J.L.; Bhat, M.A. Spatiotemporal Ablation of Myelinating Glia-Specific Neurofascin (Nfasc Nf155) in Mice Reveals Gradual Loss of Paranodal Axoglial Junctions and Concomitant Disorganization of Axonal Domains. J. Neurosci. Res. 2009, 87, 1773–1793. [Google Scholar]
- Çolakoğlu, G.; Bergstrom-Tyrberg, U.; Berglund, E.O.; Ranscht, B. Contactin-1 Regulates My-elination and Nodal/Paranodal Domain Organization in the Central Nervous System. Proc. Natl. Acad. Sci. USA 2014, 111, E394–E403. [Google Scholar] [CrossRef]
- Ogawa, Y.; Schafer, D.P.; Horresh, I.; Bar, V.; Hales, K.; Yang, Y.; Susuki, K.; Peles, E.; Stankewich, M.C.; Rasband, M.N. Spectrins and Ankyrinb Constitute a Specialized Paranodal Cy-toskeleton. J. Neurosci. 2006, 26, 5230–5239. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuen, S.; Peles, E.; Salzer, J.L. Accumulation of Neurofascin at Nodes of Ranvier Is Regulated by a Paranodal Switch. J. Neurosci. 2020, 40, 5709–5723. [Google Scholar] [CrossRef]
- Brivio, V.; Faivre-Sarrailh, C.; Peles, E.; Sherman, D.L.; Brophy, P.J. Assembly of CNS Nodes of Ranvier in Myelinated Nerves Is Promoted by the Axon Cytoskeleton. Curr. Biol. 2017, 27, 1068–1073. [Google Scholar] [CrossRef]
- Wang, H.; Kunkel, D.D.; Martin, T.M.; Schwartzkroin, P.A.; Tempel, B.L. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nat. Cell Biol. 1993, 365, 75–79. [Google Scholar] [CrossRef]
- Rasband, M.N.; Trimmer, J.S.; Schwarz, T.L.; Levinson, S.R.; Ellisman, M.H.; Schachner, M.; Shrager, P. Potassium Channel Distribution, Clustering, and Function in Remyelinating Rat Axons. J. Neurosci. 1998, 18, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Vabnick, I.; Trimmer, J.S.; Schwarz, T.L.; Levinson, S.R.; Risal, D.; Shrager, P. Dynamic Po-tassium Channel Distributions During Axonal Development Prevent Aberrant Firing Patterns. J. Neurosci. 1999, 19, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Messing, A.; Chiu, S.Y. Determinants of Excitability at Transition Zones in Kv1.1-Deficient Myelinated Nerves. J. Neurosci. 1999, 19, 5768–5781. [Google Scholar] [CrossRef] [PubMed]
- Poliak, S.; Salomon, D.; Elhanany, H.; Sabanay, H.; Kiernan, B.; Pevny, L.; Stewart, C.L.; Xu, X.; Chiu, S.-Y.; Shrager, P.; et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003, 162, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Dupree, J.L.; Popko, B.; Karagogeos, D. The Neuronal Adhesion Protein Tag-1 Is Expressed by Schwann Cells and Oligodendrocytes and Is Localized to the Juxtaparanodal Region of Myelinated Fibers. J. Neurosci. 2002, 22, 3016–3024. [Google Scholar] [CrossRef]
- Traka, M.; Goutebroze, L.; Denisenko, N.; Bessa, M.; Nifli, A.; Havaki, S.; Iwakura, Y.; Fukamauchi, F.; Watanabe, K.; Soliven, B.; et al. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J. Cell Biol. 2003, 162, 1161–1172. [Google Scholar] [CrossRef]
- Savvaki, M.; Panagiotaropoulos, T.; Stamatakis, A.; Sargiannidou, I.; Karatzioula, P.; Watanabe, K.; Stylianopoulou, F.; Karagogeos, D.; Kleopa, K.A. Impairment of learning and memory in TAG-1 deficient mice associated with shorter CNS internodes and disrupted juxtaparanodes. Mol. Cell. Neurosci. 2008, 39, 478–490. [Google Scholar] [CrossRef]
- Savvaki, M.; Theodorakis, K.; Zoupi, L.; Stamatakis, A.; Tivodar, S.; Kyriacou, K.; Stylianopoulou, F.; Karagogeos, D. The Expression of TAG-1 in Glial Cells Is Sufficient for the Formation of the Juxtaparanodal Complex and the Phenotypic Rescue of Tag-1 Homozygous Mutants in the CNS. J. Neurosci. 2010, 30, 13943–13954. [Google Scholar] [CrossRef]
- Denisenko-Nehrbass, N.; Oguievetskaia, K.; Goutebroze, L.; Galvez, T.; Yamakawa, H.; Ohara, O.; Carnaud, M.; Girault, J.A. Protein 4.1b Associates with Both Caspr/Paranodin and Caspr2 at Par-anodes and Juxtaparanodes of Myelinated Fibres. Eur. J. Neurosci. 2003, 17, 411–416. [Google Scholar]
- Horresh, I.; Bar, V.; Kissil, J.L.; Peles, E. Organization of Myelinated Axons by Caspr and Caspr2 Requires the Cytoskeletal Adapter Protein 4.1B. J. Neurosci. 2010, 30, 2480–2489. [Google Scholar] [CrossRef]
- Cifuentes-Diaz, C.; Chareyre, F.; García, M.; Devaux, J.; Carnaud, M.; Levasseur, G.; Niwa-Kawakita, M.; Harroch, S.; Girault, J.-A.; Giovannini, M.; et al. Protein 4.1B Contributes to the Organization of Peripheral Myelinated Axons. PLoS ONE 2011, 6, e25043. [Google Scholar] [CrossRef]
- Einheber, S.; Meng, X.; Rubin, M.; Lam, I.; Mohandas, N.; An, X.; Shrager, P.; Kissil, J.; Maurel, P.; Salzer, J.L. The 4.1B cytoskeletal protein regulates the domain organization and sheath thickness of myelinated axons. Glia 2012, 61, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Hivert, B.; Pinatel, D.; Labasque, M.; Tricaud, N.; Goutebroze, L.; Faivre-Sarrailh, C. Assembly of juxtaparanodes in myelinating DRG culture: Differential clustering of the Kv1/Caspr2 complex and scaffolding protein 4.1B. Glia 2016, 64, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Oses-Prieto, J.A.; Kim, M.Y.; Horresh, I.; Peles, E.; Burlingame, A.L.; Trimmer, J.S.; Meijer, D.; Rasband, M.N. ADAM22, A Kv1 Channel-Interacting Protein, Recruits Membrane-Associated Guanylate Kinases to Juxtaparanodes of Myelinated Axons. J. Neurosci. 2010, 30, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Ozkaynak, E.; Abello, G.; Jaegle, M.; van Berge, L.; Hamer, D.; Kegel, L.; Driegen, S.; Sagane, K.; Bermingham, J.R., Jr.; Meijer, D. Adam22 Is a Major Neuronal Receptor for Lgi4-Mediated Schwann Cell Signaling. J. Neurosci. 2010, 30, 3857–3864. [Google Scholar] [CrossRef]
- Kegel, L.; Jaegle, M.; Driegen, M.; Aunin, E.; Leslie, K.; Fukata, Y.; Watanabe, M.; Fukata, M.; Meijer, D. Functional phylogenetic analysis of LGI proteins identifies an interaction motif crucial for myelination. Development 2014, 141, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Zoupi, L.; Savvaki, M.; Kalemaki, K.; Kalafatakis, I.; Sidiropoulou, K.; Karagogeos, D. The Function of Contac-tin-2/Tag-1 in Oligodendrocytes in Health and Demyelinating Pathology. Glia 2018, 66, 576–591. [Google Scholar] [CrossRef]
- Hivert, B.; Marien, L.; Agbam, K.N.; Faivre-Sarrailh, C. ADAM22 and ADAM23 modulate the targeting of the Kv1 channel-associated protein LGI1 to the axon initial segment. J. Cell Sci. 2019, 132, jcs219774. [Google Scholar] [CrossRef]
- Maurel, P.; Einheber, S.; Galinska, J.; Thaker, P.; Lam, I.; Rubin, M.B.; Scherer, S.S.; Murakami, Y.; Gutmann, D.H.; Salzer, J.L. Nectin-Like Proteins Mediate Axon Schwann Cell Interactions Along the Internode and Are Essential for Myelination. J. Cell Biol. 2007, 178, 861–874. [Google Scholar] [CrossRef]
- Spiegel, I.; Adamsky, K.; Eshed, Y.; Milo, R.; Sabanay, H.; Sarig-Nadir, O.; Horresh, I.; Scherer, S.S.; Rasband, M.N.; Peles, E. A central role for Necl4 (SynCAM4) in Schwann cell–axon interaction and myelination. Nat. Neurosci. 2007, 10, 861–869. [Google Scholar] [CrossRef]
- Djannatian, M.; Timmler, S.; Arends, M.; Luckner, M.; Weil, M.-T.; Alexopoulos, I.; Snaidero, N.; Schmid, B.; Misgeld, T.; Möbius, W.; et al. Two adhesive systems cooperatively regulate axon ensheathment and myelin growth in the CNS. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, Y.; Kawasaki, M.; Tamada, A.; Nagata, S.; Kagamiyama, H.; Mori, K. Overlapping and differential expression of BIG-2, BIG-1, TAG-1, and F3: Four members of an axon-associated cell adhesion molecule subgroup of the immunoglobulin superfamily. J. Neurobiol. 1995, 28, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Kaneko, H.; Masuda, T.; Nagata, S.; Hosoya, H.; Watanabe, K. Novel neural adhesion molecules in the Contactin/F3 subgroup of the immunoglobulin superfamily: Isolation and characterization of cDNAs from rat brain. Neurosci. Lett. 1996, 218, 173–176. [Google Scholar] [CrossRef]
- Oguro-Ando, A.; Zuko, A.; Kleijer, K.T.E.; Burbach, J.P.H. A Current View on Contactin-4, -5, and -6: Implica-tions in Neurodevelopmental Disorders. Mol. Cell Neurosci. 2017, 81, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Querol, L.; Devaux, J.; Rojas-Garcia, R.; Illa, I. Autoantibodies in chronic inflammatory neuropathies: Diagnostic and therapeutic implications. Nat. Rev. Neurol. 2017, 13, 533–547. [Google Scholar] [CrossRef]
- Lefter, S.; Hardiman, O.; Ryan, A.M. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology 2017, 88, 304–313. [Google Scholar] [CrossRef]
- Mahdi-Rogers, M.; Hughes, R.A.C. Epidemiology of chronic inflammatory neuropathies in southeast England. Eur. J. Neurol. 2014, 21, 28–33. [Google Scholar] [CrossRef]
- Dyck, P.J.B.; Tracy, J.A. History, Diagnosis, and Management of Chronic Inflammatory Demyelinating Polyradiculoneuropathy. Mayo Clin. Proc. 2018, 93, 777–7793. [Google Scholar] [CrossRef]
- Lewis, R.A. Chronic inflammatory demyelinating polyneuropathy. Curr. Opin. Neurol. 2017, 30, 508–512. [Google Scholar] [CrossRef]
- Querol, L.; Illa, I. Paranodal and other autoantibodies in chronic inflammatory neuropathies. Curr. Opin. Neurol. 2015, 28, 474–479. [Google Scholar] [CrossRef]
- Miura, Y.; Devaux, J.J.; Fukami, Y.; Manso, C.; Belghazi, M.; Wong, A.H.Y.; Yuki, N. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain 2015, 138, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Devaux, J.; Odaka, M.; Yuki, N. Nodal proteins are target antigens in Guillain-Barré syndrome. J. Peripher. Nerv. Syst. 2012, 17, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Querol, L.; Siles, A.M.; Alba-Rovira, R.; Jáuregui, A.; Devaux, J.; Faivre-Sarrailh, C.; Araque, J.; Rojas-Garcia, R.; Di-az-Manera, J.; Cortés-Vicente, E.; et al. Antibodies against Pe-ripheral Nerve Antigens in Chronic Inflammatory Demyelinating Polyradiculoneuropathy. Sci. Rep. 2017, 7, 14411. [Google Scholar] [CrossRef] [PubMed]
- Doppler, K.; Appeltshauser, L.; Villmann, C.; Martin, C.; Peles, E.; Krämer, H.H.; Haarmann, A.; Buttmann, M.; Sommer, C. Auto-Antibodies to Contactin-Associated Protein 1 (Caspr) in Two Patients with Painful Inflammatory Neuropathy. Brain 2016, 139 Pt 10, 2617–2630. [Google Scholar] [CrossRef]
- Labasque, M.; Hivert, B.; Nogales-Gadea, G.; Querol, L.; Illa, I.; Faivre-Sarrailh, C. Specific ContactinN-Glycans Are Implicated in Neurofascin Binding and Autoimmune Targeting in Peripheral Neuropathies. J. Biol. Chem. 2014, 289, 7907–7918. [Google Scholar] [CrossRef]
- Doppler, K.; Appeltshauser, L.; Wilhelmi, K.; Villmann, C.; Dib-Hajj, S.D.; Waxman, S.G.; Mäurer, M.; Weishaupt, A.; Sommer, C. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J. Neurol. Neurosurg. Psychiatry 2015, 86, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, M.G.; Querol, L.; Niks, E.H.; Plomp, J.J.; Van Der Maarel, S.M.; Graus, F.; Dalmau, J.; Illa, I.; Verschuuren, J.J.G.M. The expanding field of IgG4-mediated neurological autoimmune disorders. Eur. J. Neurol. 2015, 22, 1151–1161. [Google Scholar] [CrossRef]
- Manso, C.; Querol, L.; Mekaouche, M.; Illa, I.; Devaux, J.J. Contactin-1 Igg4 Antibodies Cause Paranode Dismantling and Conduction Defects. Brain 2016, 139 Pt 6, 1700–1712. [Google Scholar] [CrossRef]
- Koike, H.; Kadoya, M.; Kaida, K.-I.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Kato, D.; Ogata, H.; Yamasaki, R.; Matsukawa, N.; et al. Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 465–473. [Google Scholar] [CrossRef]
- Querol, L.; Nogales-Gadea, G.; Rojas-Garcia, R.; Martinez-Hernandez, E.; Diaz-Manera, J.; Suárez-Calvet, X.; Navas, M.; Araque, J.; Gallardo, E.; Illa, I. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann. Neurol. 2013, 73, 370–380. [Google Scholar] [CrossRef]
- Ogata, H.; Yamasaki, R.; Hiwatashi, A.; Oka, N.; Kawamura, N.; Matsuse, D.; Kuwahara, M.; Suzuki, H.; Kusunoki, S.; Fujimoto, Y.; et al. Characterization of Igg4 Anti-Neurofascin 155 Antibody-Positive Polyneuropathy. Ann. Clin. Transl. Neurol. 2015, 2, 960–971. [Google Scholar]
- Vallat, J.-M.; Yuki, N.; Sekiguchi, K.; Kokubun, N.; Oka, N.; Mathis, S.; Magy, L.; Sherman, D.L.; Brophy, P.J.; Devaux, J.J. Paranodal lesions in chronic inflammatory demyelinating polyneuropathy associated with anti-Neurofascin 155 antibodies. Neuromuscul. Disord. 2017, 27, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Vallat, J.M.; Nizon, M.; Magee, A.; Isidor, B.; Magy, L.; Péréon, Y.; Richard, L.; Ouvrier, R.; Cogné, B.; Devaux, J.; et al. Contactin-Associated Protein 1 (Cntnap1) Mutations Induce Characteristic Lesions of the Paranodal Region. J. Neuropathol. Exp. Neurol. 2016, 75, 1155–1159. [Google Scholar] [CrossRef]
- Iijima, M.; Koike, H.; Katsuno, M.; Sobue, G. Polymorphism of Transient Axonal Glycoprotein-1 in Chronic In-flammatory Demyelinating Polyneuropathy. J. Peripher. Nerv. Syst. 2011, 16 (Suppl. S1), 52–55. [Google Scholar]
- Pang, S.Y.Y.; Chan, K.; Mak, W.W.W.; Kung, M.H.W.; Lee, C.-N.; Tsoi, T.-H.; Yip, E.K.K.; Ho, S.-L. Single-nucleotide polymorphism of transient axonal glycoprotein-1 and its correlation with clinical features and prognosis in chronic inflammatory demyelinating polyneuropathy. J. Peripher. Nerv. Syst. 2012, 17, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Rosch, R.; Bamford, A.; Hacohen, Y.; Wraige, E.; Vincent, A.; Mewasingh, L.; Lim, M. Guillain-Barré syndrome associated with CASPR2 antibodies: Two paediatric cases. J. Peripher. Nerv. Syst. 2014, 19, 246–249. [Google Scholar] [CrossRef]
- Pugliatti, M.; Sotgiu, S.; Rosati, G. The worldwide prevalence of multiple sclerosis. Clin. Neurol. Neurosurg. 2002, 104, 182–191. [Google Scholar] [CrossRef]
- Zoupi, L.; Markoullis, K.; Kleopa, K.A.; Karagogeos, D. Alterations of juxtaparanodal domains in two rodent models of CNS demyelination. Glia 2013, 61, 1236–1249. [Google Scholar] [CrossRef]
- Howell, O.W.; Palser, A.; Polito, A.; Melrose, S.; Zonta, B.; Scheiermann, C.; Vora, A.J.; Brophy, P.J.; Reynolds, R. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain 2006, 129, 3173–3185. [Google Scholar] [CrossRef]
- Howell, O.W.; Rundle, J.L.; Garg, A.; Komada, M.; Brophy, P.J.; Reynolds, R. Activated Microglia Mediate Axoglial Disruption That Contributes to Axonal Injury in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 1017–1033. [Google Scholar] [CrossRef]
- Derfuss, T.; Parikh, K.; Velhin, S.; Braun, M.; Mathey, E.; Krumbholz, M.; Kümpfel, T.; Moldenhauer, A.; Rader, C.; Sonderegger, P.; et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc. Natl. Acad. Sci. USA 2009, 106, 8302–8307. [Google Scholar] [CrossRef]
- Boronat, A.; Sepulveda, M.; Llufriu, S.; Sabater, L.; Blanco, Y.; Gabilondo, I.; Solà-Valls, N.; Villoslada, P.; Dalmau, J.; Graus, F.; et al. Analysis of antibodies to surface epitopes of contactin-2 in multiple sclerosis. J. Neuroimmunol. 2012, 244, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.A.; Trapp, B.D. Gray-Matter Injury in Multiple Sclerosis. N. Engl. J. Med. 2009, 361, 1505–1506. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Koel-Simmelink, M.J.; Verberk, I.M.W.; Killestein, J.; Vrenken, H.; Enzinger, C.; Ropele, S.; Fazekas, F.; Khalil, M.; Teunissen, C.E. Contactin-1 and contactin-2 in cerebrospinal fluid as potential biomarkers for axonal domain dysfunction in multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2018, 4, 104. [Google Scholar] [CrossRef] [PubMed]
- Schutzer, S.E.; Angel, T.E.; Liu, T.; Schepmoes, A.A.; Xie, F.; Bergquist, J.; Vécsei, L.; Zadori, D.; Camp, D.G., II; Holland, B.K.; et al. Gray Matter Is Targeted in First-Attack Multiple Sclerosis. PLoS ONE 2013, 8, e66117. [Google Scholar]
- Singh, V.; Van Pelt, E.D.; Stoop, M.P.; Stingl, C.; Ketelslegers, I.A.; Neuteboom, R.F.; Catsman-Berrevoets, C.E.; Luider, T.M.; Hintzen, R.Q. Gray matter–related proteins are associated with childhood-onset multiple sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2015, 2, e155. [Google Scholar] [CrossRef]
- Merrill, J. In Vitro and In Vivo Pharmacological Models to Assess Demyelination and Remyelination. Neuropsychopharmacology 2008, 34, 55–73. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Flores-Díaz, M.; Alape-Girón, A. Biological models in multiple sclerosis. J. Neurosci. Res. 2020, 98, 491–508. [Google Scholar] [CrossRef]
- Traka, M.; Arasi, K.; Avila, R.L.; Podojil, J.R.; Christakos, A.; Miller, S.D.; Soliven, B.; Popko, B. A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain 2010, 133, 3017–3029. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S.S. How to Successfully Apply Animal Studies in Experimental Allergic Encepha-lomyelitis to Research on Multiple Sclerosis. Ann. Neurol. 2006, 60, 12–21. [Google Scholar]
- Markoullis, K.; Sargiannidou, I.; Gardner, C.; Hadjisavvas, A.; Reynolds, R.; Kleopa, K.A. Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 2012, 60, 1053–1066. [Google Scholar] [CrossRef]
- Lindner, M.; Ng, J.K.M.; Hochmeister, S.; Meinl, E.; Linington, C. Neurofascin 186 specific autoantibodies induce axonal injury and exacerbate disease severity in experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 247, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Redler, Y.; Levy, M. Rodent Models of Optic Neuritis. Front. Neurol. 2020, 11, 580951. [Google Scholar] [CrossRef] [PubMed]
- Stojic, A.; Bojcevski, J.; Williams, S.K.; Diem, R.; Fairless, R. Early Nodal and Paranodal Disruption in Autoimmune Optic Neuritis. J. Neuropathol. Exp. Neurol. 2018, 77, 361–373. [Google Scholar] [CrossRef]
- Dembla, M.; Kesharwani, A.; Natarajan, S.; Fecher-Trost, C.; Fairless, R.; Williams, S.K.; Flockerzi, V.; Diem, R.; Schwarz, K.; Schmitz, F. Early auto-immune targeting of photoreceptor ribbon synapses in mouse models of multiple sclerosis. EMBO Mol. Med. 2018, 10, e8926. [Google Scholar] [CrossRef]
- Marella, M.; Patki, G.; Matsuno-Yagi, A.; Yagi, T. Complex I Inhibition in the Visual Pathway Induces Disorganization of the Node of Ranvier. Neurobiol. Dis. 2013, 58, 281–288. [Google Scholar] [CrossRef][Green Version]
- Kipp, M.; Clarner, T.; Dang, J.; Copray, S.; Beyer, C. The cuprizone animal model: New insights into an old story. Acta Neuropathol. 2009, 118, 723–736. [Google Scholar] [CrossRef]
- Matsushima, G.K.; Morell, P. The Neurotoxicant, Cuprizone, as a Model to Study Demyelination and Remyelination in the Central Nervous System. Brain Pathol. 2006, 11, 107–116. [Google Scholar] [CrossRef]
- Aparicio, E.; Mathieu, P.; Luppi, M.P.; Gubiani, M.F.A.; Adamo, A.M. The notch signaling pathway: Its role in focal CNS demyelination and apotransferrin-induced remyelination. J. Neurochem. 2013, 127, 819–836. [Google Scholar] [CrossRef]
- Ranscht, B. Sequence of contactin, a 130-kD glycoprotein concentrated in areas of interneuronal contact, defines a new member of the immunoglobulin supergene family in the nervous system. J. Cell Biol. 1988, 107, 1561–1573. [Google Scholar] [CrossRef]
- Massaro, A.; Bizzoca, A.; Corsi, P.; Pinto, M.F.; Carratù, M.R.; Gennarini, G. Significance of F3/Contactin gene expression in cerebral cortex and nigrostriatal development. Mol. Cell. Neurosci. 2012, 50, 221–237. [Google Scholar] [CrossRef]
- Koch, T.; Brugger, T.; Bach, A.; Gennarini, G.; Trotter, J. Expression of the immunoglobulin superfamily cell adhesion molecule F3 by oligodendrocyte-lineage cells. Glia 1997, 19, 199–212. [Google Scholar] [CrossRef]
- Puzzo, D.; Bizzoca, A.; Privitera, L.; Furnari, D.; Giunta, S.; Girolamo, F.; Pinto, M.; Gennarini, G.; Palmeri, A. F3/Contactin promotes hippocampal neurogenesis, synaptic plasticity, and memory in adult mice. Hippocampus 2013, 23, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, K.; Hosoya, H.; Takeda, Y.; Kobayashi, S.; Watanabe, K. Age-related decline of F3/Contactin in rat hippocampus. Neurosci. Lett. 1998, 245, 117–120. [Google Scholar] [CrossRef]
- Puzzo, D.; Bizzoca, A.; Loreto, C.; Guida, C.A.; Gulisano, W.; Frasca, G.; Bellomo, M.; Castorina, S.; Gennarini, G.; Palmeri, A. Role of F3/contactin expression profile in synaptic plasticity and memory in aged mice. Neurobiol. Aging 2015, 36, 1702–1715. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Shimojo, H.; Sakamoto, M.; Ohtsuka, T.; Kageyama, R. Genetic visualization of notch signaling in mammalian neurogenesis. Cell. Mol. Life Sci. 2012, 70, 2045–2057. [Google Scholar] [CrossRef]
- Tiberi, L.; Ameele, J.V.D.; Dimidschstein, J.; Piccirilli, J.; Gall, D.; Herpoel, A.; Bilheu, A.; Bonnefont, J.; Iacovino, M.; Kyba, M.; et al. BCL6 controls neurogenesis through Sirt1-dependent epigenetic repression of selective Notch targets. Nat. Neurosci. 2012, 15, 1627–1635. [Google Scholar] [CrossRef]
- Pierfelice, T.; Auber, L.A.; Gaiano, N. Notch in the Vertebrate Nervous System: An Old Dog with New Tricks. Neuron 2011, 69, 840–855. [Google Scholar] [CrossRef]
- Cau, E.; Blader, P. Notch activity in the nervous system: To switch or not switch? Neural Dev. 2009, 4, 36. [Google Scholar] [CrossRef]
- Cui, X.-Y.; Hu, Q.-D.; Tekaya, M.; Shimoda, Y.; Ang, B.-T.; Nie, D.-Y.; Sun, L.; Hu, W.-P.; Karsak, M.; Duka, T.; et al. NB-3/Notch1 Pathway via Deltex1 Promotes Neural Progenitor Cell Differentiation into Oligodendrocytes. J. Biol. Chem. 2004, 279, 25858–25865. [Google Scholar] [CrossRef]
- Taylor, M.K.; Yeager, K.; Morrison, S.J. Physiological Notch Signaling Promotes Gliogenesis in the Developing Peripheral and Central Nervous Systems. Development 2007, 134, 2435–2447. [Google Scholar] [CrossRef]
- Bizzoca, A.; Corsi, P.; Polizzi, A.; Pinto, M.F.; Xenaki, D.; Furley, A.J.; Gennarini, G. F3/Contactin acts as a modulator of neurogenesis during cerebral cortex development. Dev. Biol. 2012, 365, 133–151. [Google Scholar] [CrossRef][Green Version]
- Hu, Q.-D.; Ang, B.-T.; Karsak, M.; Hu, W.-P.; Cui, X.-Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.-K.; et al. F3/Contactin Acts as a Functional Ligand for Notch during Oligodendrocyte Maturation. Cell 2003, 115, 163–175. [Google Scholar] [CrossRef]
- Canoll, P.D.; Petanceska, S.; Schlessinger, J.; Musacchio, J.M. Three Forms of Rptp-Beta Are Differentially Expressed During Gliogenesis in the Developing Rat Brain and During Glial Cell Differentiation in Culture. J. Neurosci. Res. 1996, 44, 199–215. [Google Scholar] [CrossRef]
- Harroch, S.; Palmeri, M.; Rosenbluth, J.; Custer, A.; Okigaki, M.; Shrager, P.; Blum, M.; Buxbaum, J.D.; Schlessinger, J. No Obvious Abnormality in Mice Deficient in Receptor Protein Tyrosine Phosphatase Beta. Mol. Cell Biol. 2000, 20, 7706–7715. [Google Scholar] [CrossRef] [PubMed]
- Faissner, A.; Heck, N.; Dobbertin, A.; Garwood, J. DSD-1-Proteoglycan/Phosphacan and Receptor Protein Tyrosine Phosphatase-Beta Isoforms during Development and Regeneration of Neural Tissues. Neurotransm. Interact. Cogn. Funct. 2007, 557, 25–53. [Google Scholar] [CrossRef]
- Lamprianou, S.; Chatzopoulou, E.; Thomas, J.-L.; Bouyain, S.; Harroch, S. A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17498–17503. [Google Scholar] [CrossRef]
- Haenisch, C.; Diekmann, H.; Klinger, M.; Gennarini, G.; Kuwada, J.Y.; Stuermer, C.A. The Neuronal Growth and Regeneration Associated Cntn1 (F3/F11/Contactin) Gene Is Duplicated in Fish: Expression During Development and Retinal Axon Regeneration. Mol. Cell Neurosci. 2005, 28, 361–374. [Google Scholar] [CrossRef]
- Zeng, L.; D’Alessandri, L.; Kalousek, M.B.; Vaughan, L.; Pallen, C.J. Protein Tyrosine Phosphatase Alpha (Ptpalpha) and Contactin Form a Novel Neuronal Receptor Complex Linked to the Intracellular Tyrosine Kinase Fyn. J. Cell Biol. 1999, 147, 707–714. [Google Scholar] [CrossRef]
- Miyata, S. Cytoskeletal Signal-Regulated Oligodendrocyte Myelination and Remyelination. In Taurine 6; Oja, S.S., Saransaari, P., Eds.; Springer: New York, NY, USA, 2019; Volume 1190, pp. 33–42. [Google Scholar]
- Dodd, J.; Morton, S.B.; Karagogeos, D.; Yamamoto, M.; Jessell, T.M. Spatial Regulation of Axonal Glyco-protein Expression on Subsets of Embryonic Spinal Neurons. Neuron 1988, 1, 105–116. [Google Scholar] [CrossRef]
- Karagogeos, D.; Morton, S.B.; Casano, F.; Dodd, J.; Jessell, T.M. Developmental expression of the axonal glycoprotein TAG-1: Differential regulation by central and peripheral neurons in vitro. Development 1991, 112, 51–67. [Google Scholar]
- Wolfer, D.P.; Henehan-Beatty, A.; Stoeckli, E.T.; Sonderegger, P.; Lipp, H.-P. Distribution of TAG-1/Axonin-1 in fibre tracts and migratory streams of the developing mouse nervous system. J. Comp. Neurol. 1994, 345, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Prinetti, A.; Prioni, S.; Chigorno, V.; Karagogeos, D.; Tettamanti, G.; Sonnino, S. Immunoseparation of sphingolipid-enriched membrane domains enriched in Src family protein tyrosine kinases and in the neuronal adhesion molecule TAG-1 by anti-GD3 ganglioside monoclonal antibody. J. Neurochem. 2001, 78, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Loberto, N.; Prioni, S.; Prinetti, A.; Ottico, E.; Chigorno, V.; Karagogeos, D.; Sonnino, S. The adhesion protein TAG-1 has a ganglioside environment in the sphingolipid-enriched membrane domains of neuronal cells in culture. J. Neurochem. 2003, 85, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Furley, A.J.; Morton, S.B.; Manalo, D.; Karagogeos, D.; Dodd, J.; Jessell, T.M. The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell 1990, 61, 157–170. [Google Scholar] [CrossRef]
- Stoeckli, E.T.; Kuhn, T.B.; Duc, C.O.; Ruegg, M.A.; Sonderegger, P. The axonally secreted protein axonin-1 is a potent substratum for neurite growth. J. Cell Biol. 1991, 112, 449–455. [Google Scholar] [CrossRef]
- Stoeckli, E.T.; Landmesser, L.T. Axonin-1, Nr-CAM, and Ng-CAM play different roles in the in vivo guidance of chick commissural neurons. Neuron 1995, 14, 1165–1179. [Google Scholar] [CrossRef]
- Stoeckli, E.T.; Sonderegger, P.; Pollerberg, G.; Landmesser, L.T. Interference with Axonin-1 and NrCAM Interactions Unmasks a Floor-Plate Activity Inhibitory for Commissural Axons. Neuron 1997, 18, 209–221. [Google Scholar] [CrossRef]
- Fitzli, D.; Stoeckli, E.T.; Kunz, S.; Siribour, K.; Rader, C.; Kunz, B.; Kozlov, S.V.; Buchstaller, A.; Lane, R.P.; Suter, D.M.; et al. A Direct Interaction of Axonin-1 with Ngcam-Related Cell Adhesion Molecule (Nrcam) Results in Guidance, but Not Growth of Commissural Axons. J. Cell Biol. 2000, 149, 951–968. [Google Scholar] [CrossRef]
- Denaxa, M.; Chan, C.H.; Schachner, M.; Parnavelas, J.G.; Karagogeos, D. The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development 2001, 128, 4635–4644. [Google Scholar]
- Perrin, F.E.; Rathjen, F.G.; Stoeckli, E.T. Distinct Subpopulations of Sensory Afferents Require F11 or Axonin-1 for Growth to Their Target Layers within the Spinal Cord of the Chick. Neuron 2001, 30, 707–723. [Google Scholar] [CrossRef]
- Kyriakopoulou, K.; de Diego, I.; Wassef, M.; Karagogeos, D. A Combination of Chain and Neurophilic Mi-gration Involving the Adhesion Molecule Tag-1 in the Caudal Medulla. Development 2002, 129, 287–296. [Google Scholar] [PubMed]
- Wolman, M.; Sittaramane, V.K.; Essner, J.J.; Yost, H.J.; Chandrasekhar, A.; Halloran, M.C. Transient axonal glycoprotein-1 (TAG-1) and laminin-alpha1 regulate dynamic growth cone behaviors and initial axon direction in vivo. Neural Dev. 2008, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Tsiotra, P.C.; Theodorakis, K.; Papamatheakis, J.; Karagogeos, D. The Fibronectin Domains of the Neural Adhesion Molecule TAX-1 Are Necessary and Sufficient for Homophilic Binding. J. Biol. Chem. 1996, 271, 29216–29222. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dahl, S.P.; Kimberling, W.J.; Gorin, M.B.; Weston, M.D.; Furman, J.M.; Pikus, A.; Moller, C. Genetic heterogeneity of Usher syndrome type II. J. Med Genet. 1993, 30, 843–848. [Google Scholar] [CrossRef]
- Schròck, E.; Thiel, G.; Lozanova, T.; Du Manoir, S.; Meffert, M.-C.; Jauch, A.; Speicher, M.R.; Nürnberg, P.; Vogel, S.; Janisch, W.; et al. Comparative Genomic Hybridization of Human Malignant Gliomas Reveals Multiple Amplification Sites and Nonrandom Chromosomal Gains and Losses. Am. J. Pathol. 1994, 144, 1203–1218. [Google Scholar]
- Sander, A.; Schmelzle, R.; Murray, J. Evidence for a Microdeletion in 1q32-41 Involving the Gene Responsible for Van Der Woude Syndrome. Hum. Mol. Genet. 1994, 3, 575–578. [Google Scholar]
- Ma, Q.H.; Futagawa, T.; Yang, W.L.; Jiang, X.D.; Zeng, L.; Takeda, Y.; Xu, R.X.; Bagnard, D.; Schachner, M.; Furley, A.J.; et al. A Tag1-App Signalling Pathway through Fe65 Nega-tively Modulates Neurogenesis. Nat. Cell Biol. 2008, 10, 283–294. [Google Scholar]
- Dang, P.; Smythe, E.; Furley, A.J. Tag1 Regulates the Endocytic Trafficking and Signaling of the Semaphorin3a Receptor Complex. J. Neurosci. 2012, 32, 10370–10382. [Google Scholar]



{kind=link}
{kind=link}
| Contactin and Contactin Interactors-Related Demyelination Antibodies | Associated Demyelinating Diseases |
|---|---|
| Anti-CNTN1 | CIDP |
| Anti-NF155 | CIDP |
| Anti-CASPR1 | GBS, CIDP |
| Anti-CNTN2 | MS |
| Anti-CASPR2 | GBS, MS |
| Anti-NFASC | MS |
| Anti-NrCAM | MS |
| Anti-ADAM22 | MS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalafatakis, I.; Savvaki, M.; Velona, T.; Karagogeos, D. Implication of Contactins in Demyelinating Pathologies. Life 2021, 11, 51. https://doi.org/10.3390/life11010051
Kalafatakis I, Savvaki M, Velona T, Karagogeos D. Implication of Contactins in Demyelinating Pathologies. Life. 2021; 11(1):51. https://doi.org/10.3390/life11010051
Chicago/Turabian StyleKalafatakis, Ilias, Maria Savvaki, Theodora Velona, and Domna Karagogeos. 2021. "Implication of Contactins in Demyelinating Pathologies" Life 11, no. 1: 51. https://doi.org/10.3390/life11010051
APA StyleKalafatakis, I., Savvaki, M., Velona, T., & Karagogeos, D. (2021). Implication of Contactins in Demyelinating Pathologies. Life, 11(1), 51. https://doi.org/10.3390/life11010051