Mitonuclear Interactions in the Maintenance of Mitochondrial Integrity



Abstract
1. Eukariotic Cell: A Chimeric Union with Two Genomes
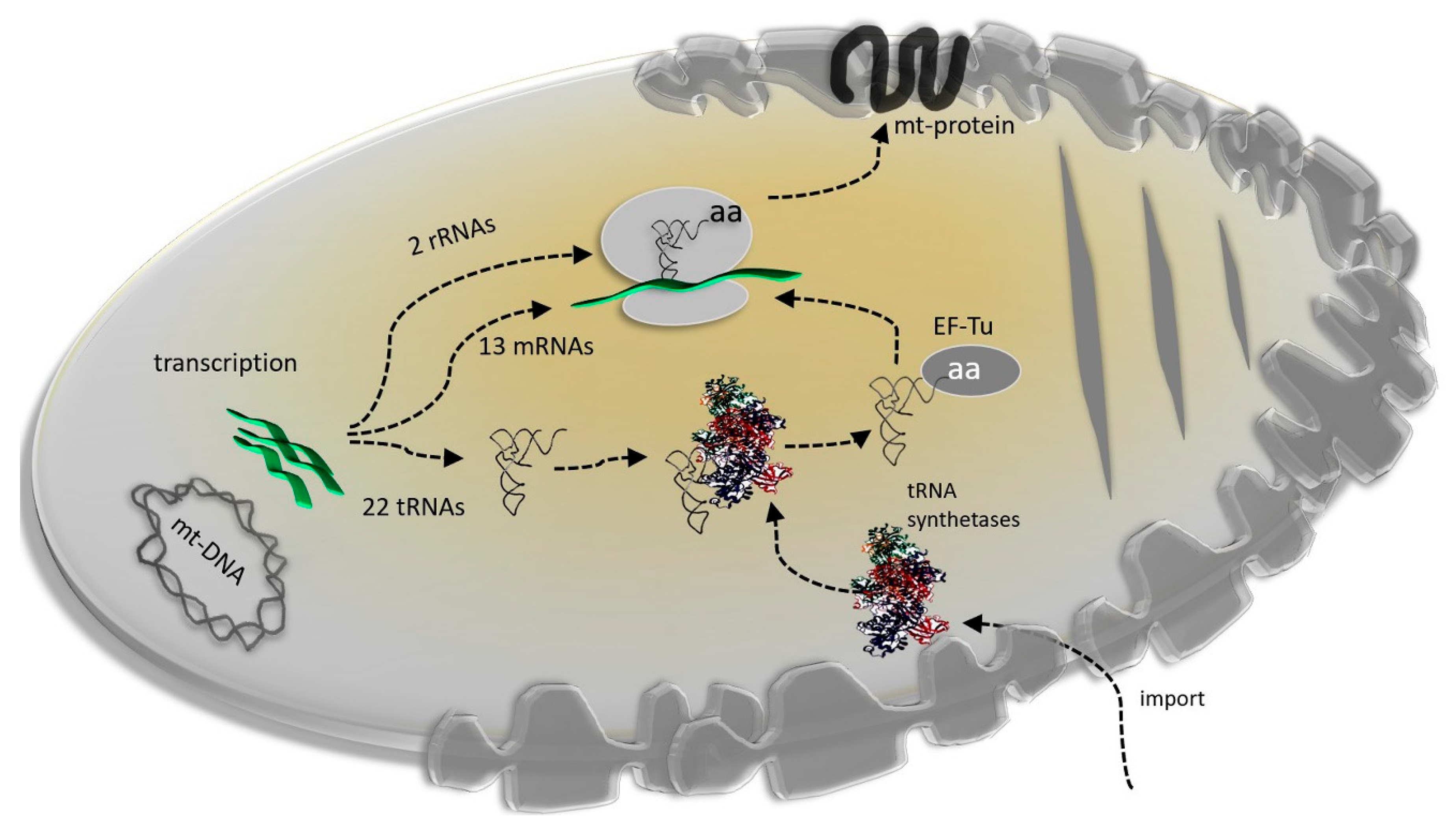
2. Mitochondrial Genome: Evolving While Keeping Essentials
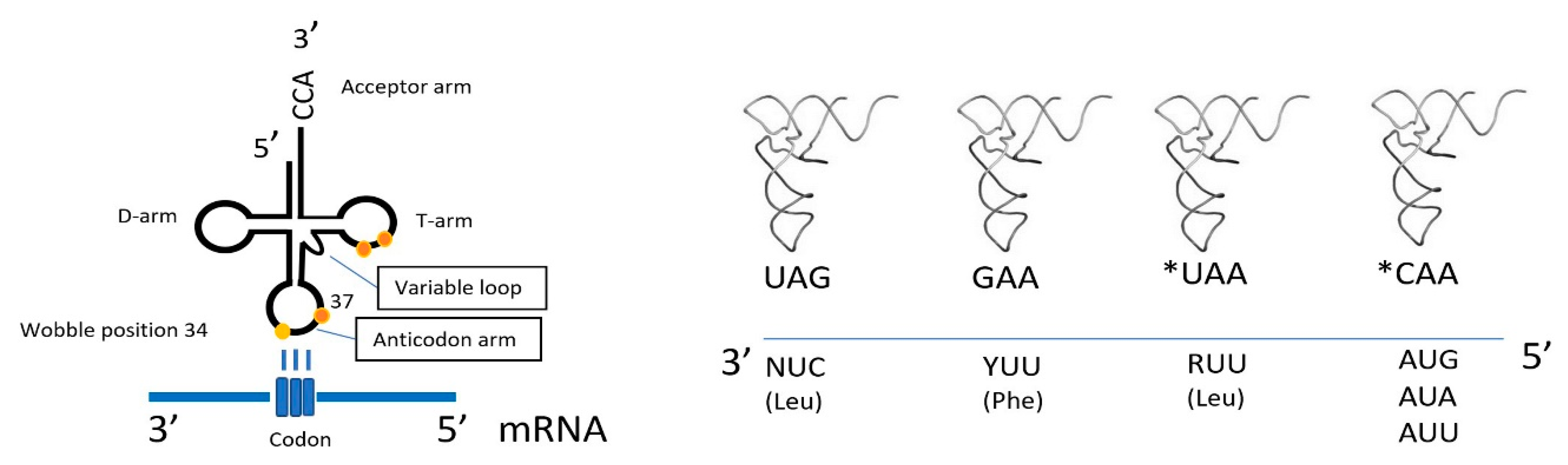
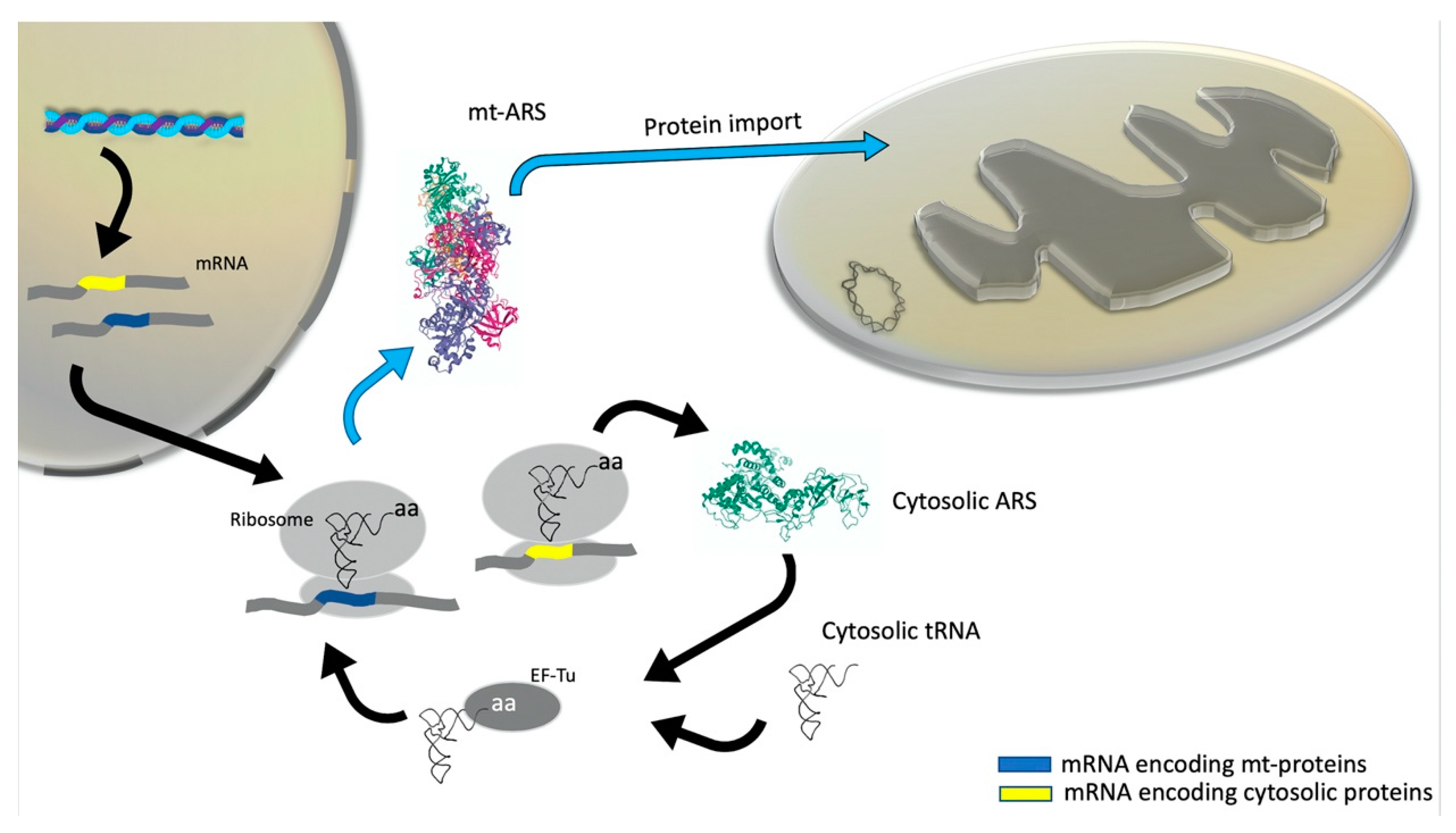
3. Mitonuclear Coordination of Mitochondrial Translation
4. Mitonuclear Coevolution
5. Mitonuclear Communication
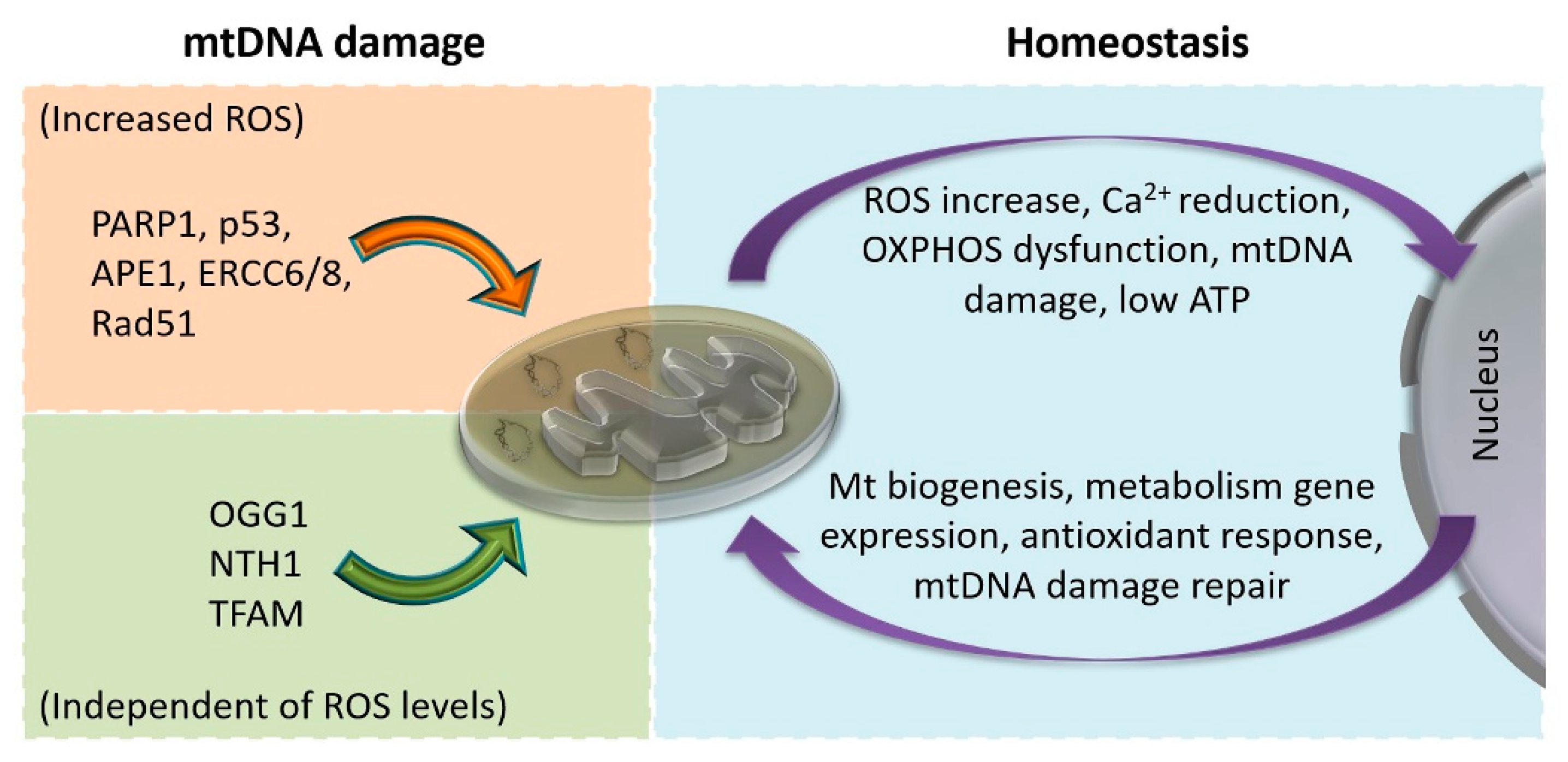
5.1. Mitonuclear Communication in Homeostasis and Stress
5.2. Mitonuclear Communication in mtDNA Damage and Repair
6. Defects in Mitochondria in Aging and Disease
6.1. Human Mitochondrial Diseases
6.2. Aging
6.3. Cancer
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Oyaizu, Y.; Oyaizu, H.; Olsen, G.J.; Woese, C.R. Mitochondrial origins. Proc. Natl. Acad. Sci. USA 1985, 82, 4443–4447. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. The origin and early evolution of eukaryotes in the light of phylogenomics. Genome Biol. 2010, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Eukaryotes with no mitochondria. Nature 1987, 326, 332–333. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. Archaeal ancestors of eukaryotes: Not so elusive any more. BMC Biol. 2015, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Speijer, D.; Lukes, J.; Elias, M. Sex is a ubiquitous, ancient, and inherent attribute of eukaryotic life. Proc. Natl. Acad. Sci. USA 2015, 112, 8827–8834. [Google Scholar] [CrossRef] [PubMed]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Backstrom, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U.; et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Saw, J.H.; Jorgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Speijer, D. Alternating terminal electron-acceptors at the basis of symbiogenesis: How oxygen ignited eukaryotic evolution. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Gabaldon, T.; Huynen, M.A. Reconstruction of the proto-mitochondrial metabolism. Science 2003, 301, 609. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Mosaic nature of the mitochondrial proteome: Implications for the origin and evolution of mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 10133–10138. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Mentel, M.; van Hellemond, J.J.; Henze, K.; Woehle, C.; Gould, S.B.; Yu, R.Y.; van der Giezen, M.; Tielens, A.G.; Martin, W.F. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol Mol. Biol Rev. 2012, 76, 444–495. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Meisinger, C.; Sickmann, A.; Pfanner, N. The mitochondrial proteome: From inventory to function. Cell 2008, 134, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M.; Pfanner, N.; van der Laan, M. A dynamic machinery for import of mitochondrial precursor proteins. FEBS Lett. 2007, 581, 2802–2810. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Stoebe, B.; Goremykin, V.; Hapsmann, S.; Hasegawa, M.; Kowallik, K.V. Gene transfer to the nucleus and the evolution of chloroplasts. Nature 1998, 393, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Berg, O.G.; Kurland, C.G. Why mitochondrial genes are most often found in nuclei. Mol. Biol. Evol. 2000, 17, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.L.; Lynch, M. Organellar genes: Why do they end up in the nucleus? Trends Genet. 2000, 16, 315–320. [Google Scholar] [CrossRef]
- Allen, J.F.; Raven, J.A. Free-radical-induced mutation vs. redox regulation: Costs and benefits of genes in organelles. J. Mol. Evol. 1996, 42, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Aanen, D.K.; Spelbrink, J.N.; Beekman, M. What cost mitochondria? The maintenance of functional mitochondrial DNA within and across generations. Philos. Trans. R Soc. Lond B Biol. Sci. 2014, 369, 20130438. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F. You are what you eat: A gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 1998, 14, 307–311. [Google Scholar] [CrossRef]
- Jacobs, H.T. Structural similarities between a mitochondrially encoded polypeptide and a family of prokaryotic respiratory toxins involved in plasmid maintenance suggest a novel mechanism for the evolutionary maintenance of mitochondrial DNA. J. Mol. Evol. 1991, 32, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Popot, J.L.; de Vitry, C. On the microassembly of integral membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 1990, 19, 369–403. [Google Scholar] [CrossRef] [PubMed]
- Von Heijne, G. Why mitochondria need a genome. FEBS Lett. 1986, 198, 1–4. [Google Scholar] [CrossRef]
- Claros, M.G.; Perea, J.; Shu, Y.; Samatey, F.A.; Popot, J.L.; Jacq, C. Limitations to in vivo import of hydrophobic proteins into yeast mitochondria. The case of a cytoplasmically synthesized apocytochrome b. Eur. J. Biochem. 1995, 228, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Bjorkholm, P.; Ernst, A.M.; Hagstrom, E.; Andersson, S.G. Why mitochondria need a genome revisited. FEBS Lett. 2017, 591, 65–75. [Google Scholar] [CrossRef] [PubMed]
- De Grey, A.D. Forces maintaining organellar genomes: Is any as strong as genetic code disparity or hydrophobicity? Bioessays 2005, 27, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Schnarrenberger, C. The evolution of the Calvin cycle from prokaryotic to eukaryotic chromosomes: A case study of functional redundancy in ancient pathways through endosymbiosis. Curr. Genet. 1997, 32, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F. Control of gene expression by redox potential and the requirement for chloroplast and mitochondrial genomes. J. Theor Biol 1993, 165, 609–631. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F. The function of genomes in bioenergetic organelles. Philos. Trans. R Soc. Lond B Biol. Sci. 2003, 358, 19–37; discussion 37–38. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, I.M.; Walker, J.E. Two overlapping genes in bovine mitochondrial DNA encode membrane components of ATP synthase. EMBO J. 1986, 5, 2003–2008. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 2008, 9, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Klucnika, A.; Ma, H. A battle for transmission: The cooperative and selfish animal mitochondrial genomes. Open Biol. 2019, 9, 180267. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Sobanski, J.; Bock, R. Why are most organelle genomes transmitted maternally? Bioessays 2015, 37, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta 2013, 1833, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Mai, N.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N. The process of mammalian mitochondrial protein synthesis. Cell Tissue Res. 2017, 367, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.W.; Ramakrishnan, V. Ribosome. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Marechal-Drouard, L. Mitochondrial tRNA import: Are there distinct mechanisms? Trends Cell Biol. 2000, 10, 509–513. [Google Scholar] [CrossRef]
- Salinas-Giege, T.; Giege, R.; Giege, P. tRNA biology in mitochondria. Int. J. Mol. Sci. 2015, 16, 4518–4559. [Google Scholar] [CrossRef] [PubMed]
- Barrell, B.G.; Bankier, A.T.; Drouin, J. A different genetic code in human mitochondria. Nature 1979, 282, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, M.; Karcher, D.; Bock, R. Superwobbling facilitates translation with reduced tRNA sets. Nat. Struct. Mol. Biol. 2008, 15, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Hajieva, P.; Moosmann, B. Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 16496–16501. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.P.; Schneller, J.M.; Stahl, A.J.; Dirheimer, G. Import of nuclear deoxyribonucleic acid coded lysine-accepting transfer ribonucleic acid (anticodon C-U-U) into yeast mitochondria. Biochemistry 1979, 18, 4600–4605. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.A.; Rinehart, J.J.; Krett, B.; Duvezin-Caubet, S.; Reichert, A.S.; Soll, D.; Alfonzo, J.D. Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc. Natl. Acad. Sci. USA 2008, 105, 9186–9191. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Krett, B.; Rubio, M.A.; Alfonzo, J.D.; Soll, D. Saccharomyces cerevisiae imports the cytosolic pathway for Gln-tRNA synthesis into the mitochondrion. Genes Dev. 2005, 19, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Brule, H.; Friede, D.; Giege, R.; Putz, D.; Florentz, C. Search for characteristic structural features of mammalian mitochondrial tRNAs. RNA 2000, 6, 1356–1379. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Lavrov, D.; Beck, N.; SV, S. Mitochondrial tRNA Structure, Identity, and Evolution of the Genetic Code. In Organelle Genetics; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Dubin, D.T.; HsuChen, C.C.; Cleaves, G.R.; Timko, K.D. Sequence and structure of a serine transfer RNA with GCU anticodon from mosquito mitochondria. J. Mol. Biol. 1984, 176, 251–260. [Google Scholar] [CrossRef]
- Steinberg, S.; Gautheret, D.; Cedergren, R. Fitting the structurally diverse animal mitochondrial tRNAs(Ser) to common three-dimensional constraints. J. Mol. Biol. 1994, 236, 982–989. [Google Scholar] [CrossRef]
- Watanabe, Y.; Kawai, G.; Yokogawa, T.; Hayashi, N.; Kumazawa, Y.; Ueda, T.; Nishikawa, K.; Hirao, I.; Miura, K.; Watanabe, K. Higher-order structure of bovine mitochondrial tRNA(SerUGA): Chemical modification and computer modeling. Nucleic Acids Res. 1994, 22, 5378–5384. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.; Cedergren, R. Structural compensation in atypical mitochondrial tRNAs. Nat. Struct. Biol. 1994, 1, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M.; Horn, C.; Brown, M.; Ioudovitch, A.; Steinberg, S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998, 26, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Fender, A.; Gaudry, A.; Juhling, F.; Sissler, M.; Florentz, C. Adaptation of aminoacylation identity rules to mammalian mitochondria. Biochimie 2012, 94, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, I.; Kawai, G.; Watanabe, K. Higher-order structure and thermal instability of bovine mitochondrial tRNASerUGA investigated by proton NMR spectroscopy. J. Mol. Biol. 1998, 284, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Suzuki, T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2014, 42, 7346–7357. [Google Scholar] [CrossRef] [PubMed]
- Ogle, J.M.; Brodersen, D.E.; Clemons, W.M., Jr.; Tarry, M.J.; Carter, A.P.; Ramakrishnan, V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 2001, 292, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Agris, P.F.; Vendeix, F.A.; Graham, W.D. tRNA’s wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007, 366, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. Codon--anticodon pairing: The wobble hypothesis. J. Mol. Biol. 1966, 19, 548–555. [Google Scholar] [CrossRef]
- Moriya, J.; Yokogawa, T.; Wakita, K.; Ueda, T.; Nishikawa, K.; Crain, P.F.; Hashizume, T.; Pomerantz, S.C.; McCloskey, J.A.; Kawai, G.; et al. A novel modified nucleoside found at the first position of the anticodon of methionine tRNA from bovine liver mitochondria. Biochemistry 1994, 33, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Rebelo-Guiomar, P.; Powell, C.A.; Van Haute, L.; Minczuk, M. The mammalian mitochondrial epitranscriptome. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 429–446. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.M.; Schimmel, P. Evidence that a major determinant for the identity of a transfer RNA is conserved in evolution. Biochemistry 1989, 28, 6800–6804. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, M.; Sekine, S.; Chong, Y.E.; Guo, M.; Yang, X.L.; Gamper, H.; Hou, Y.M.; Schimmel, P.; Yokoyama, S. The selective tRNA aminoacylation mechanism based on a single G*U pair. Nature 2014, 510, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P.; Schmidt, E. Making connections: RNA-dependent amino acid recognition. Trends Biochem. Sci. 1995, 20, 1–2. [Google Scholar] [CrossRef]
- Bonnefond, L.; Fender, A.; Rudinger-Thirion, J.; Giege, R.; Florentz, C.; Sissler, M. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: Characterization of AspRS and TyrRS. Biochemistry 2005, 44, 4805–4816. [Google Scholar] [CrossRef] [PubMed]
- Mudge, S.J.; Williams, J.H.; Eyre, H.J.; Sutherland, G.R.; Cowan, P.J.; Power, D.A. Complex organisation of the 5’-end of the human glycine tRNA synthetase gene. Gene 1998, 209, 45–50. [Google Scholar] [CrossRef]
- Tolkunova, E.; Park, H.; Xia, J.; King, M.P.; Davidson, E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual alternative splicing of the primary transcript. J. Biol. Chem. 2000, 275, 35063–35069. [Google Scholar] [CrossRef] [PubMed]
- Fender, A.; Sauter, C.; Messmer, M.; Putz, J.; Giege, R.; Florentz, C.; Sissler, M. Loss of a primordial identity element for a mammalian mitochondrial aminoacylation system. J. Biol. Chem. 2006, 281, 15980–15986. [Google Scholar] [CrossRef] [PubMed]
- Maier, U.G.; Zauner, S.; Woehle, C.; Bolte, K.; Hempel, F.; Allen, J.F.; Martin, W.F. Massively convergent evolution for ribosomal protein gene content in plastid and mitochondrial genomes. Genome Biol. Evol. 2013, 5, 2318–2329. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, E.O.; Bauerschmitt, H.; Becker, T.; Mielke, T.; Frauenfeld, J.; Berninghausen, O.; Neupert, W.; Herrmann, J.M.; Beckmann, R. Parallel Structural Evolution of Mitochondrial Ribosomes and OXPHOS Complexes. Genome Biol. Evol. 2015, 7, 1235–1251. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.R.; Koc, E.C.; Datta, P.P.; Booth, T.M.; Spremulli, L.L.; Agrawal, R.K. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 2003, 115, 97–108. [Google Scholar] [CrossRef]
- Koc, E.C.; Burkhart, W.; Blackburn, K.; Moyer, M.B.; Schlatzer, D.M.; Moseley, A.; Spremulli, L.L. The large subunit of the mammalian mitochondrial ribosome. Analysis of the complement of ribosomal proteins present. J. Biol. Chem. 2001, 276, 43958–43969. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.W. Evolution of a protein-rich mitochondrial ribosome: Implications for human genetic disease. Gene 2002, 286, 73–79. [Google Scholar] [CrossRef]
- Greber, B.J.; Boehringer, D.; Leibundgut, M.; Bieri, P.; Leitner, A.; Schmitz, N.; Aebersold, R.; Ban, N. The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 2014, 515, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Amunts, A.; Brown, A.; Bai, X.C.; Llacer, J.L.; Hussain, T.; Emsley, P.; Long, F.; Murshudov, G.; Scheres, S.H.W.; Ramakrishnan, V. Structure of the yeast mitochondrial large ribosomal subunit. Science 2014, 343, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Amunts, A.; Brown, A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu. Rev. Biochem. 2016, 85, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Graack, H.R.; Wittmann-Liebold, B. Mitochondrial ribosomal proteins (MRPs) of yeast. Biochem. J. 1998, 329 Pt 3, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Puigbo, P.; Koonin, E.V.; Wolf, Y.I. Phylogenomics of prokaryotic ribosomal proteins. PLoS ONE 2012, 7, e36972. [Google Scholar] [CrossRef] [PubMed]
- Desmond, E.; Brochier-Armanet, C.; Forterre, P.; Gribaldo, S. On the last common ancestor and early evolution of eukaryotes: Reconstructing the history of mitochondrial ribosomes. Res. Microbiol. 2011, 162, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, P.S.; Sharma, M.R.; Booth, T.M.; Haque, E.M.; Tung, C.S.; Sanbonmatsu, K.Y.; Spremulli, L.L.; Agrawal, R.K. Cryo-EM structure of the small subunit of the mammalian mitochondrial ribosome. Proc. Natl. Acad. Sci. USA 2014, 111, 7284–7289. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.W. Properties of human mitochondrial ribosomes. IUBMB Life 2003, 55, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Yamano, K. Transport of proteins across or into the mitochondrial outer membrane. Biochim. Biophys. Acta 2010, 1803, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Stojanovski, D.; Bohnert, M.; Pfanner, N.; van der Laan, M. Mechanisms of protein sorting in mitochondria. Cold Spring Harb. Perspect Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Schendzielorz, A.; Rehling, P. Unlocking the presequence import pathway. Trends Cell Biol. 2015, 25, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Mani, J.; Meisinger, C.; Schneider, A. Peeping at TOMs-Diverse Entry Gates to Mitochondria Provide Insights into the Evolution of Eukaryotes. Mol. Biol. Evol. 2016, 33, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M.; Haney, R.A.; Fry, A.J. Cytonuclear coevolution: The genomics of cooperation. Trends Ecol. Evol 2004, 19, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.R.; Wildman, D.E.; Uddin, M.; Opazo, J.C.; Goodman, M.; Grossman, L.I. Rapid electrostatic evolution at the binding site for cytochrome c on cytochrome c oxidase in anthropoid primates. Proc. Natl. Acad. Sci. USA 2005, 102, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Mishmar, D.; Ruiz-Pesini, E.; Mondragon-Palomino, M.; Procaccio, V.; Gaut, B.; Wallace, D.C. Adaptive selection of mitochondrial complex I subunits during primate radiation. Gene 2006, 378, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Meiklejohn, C.D.; Montooth, K.L.; Rand, D.M. Positive and negative selection on the mitochondrial genome. Trends Genet. 2007, 23, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Gershoni, M.; Fuchs, A.; Shani, N.; Fridman, Y.; Corral-Debrinski, M.; Aharoni, A.; Frishman, D.; Mishmar, D. Coevolution predicts direct interactions between mtDNA-encoded and nDNA-encoded subunits of oxidative phosphorylation complex i. J. Mol. Biol. 2010, 404, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Gershoni, M.; Levin, L.; Ovadia, O.; Toiw, Y.; Shani, N.; Dadon, S.; Barzilai, N.; Bergman, A.; Atzmon, G.; Wainstein, J.; et al. Disrupting mitochondrial-nuclear coevolution affects OXPHOS complex I integrity and impacts human health. Genome Biol. Evol. 2014, 6, 2665–2680. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.S.; Burton, R.S. Evidence for compensatory evolution of ribosomal proteins in response to rapid divergence of mitochondrial rRNA. Mol. Biol Evol. 2013, 30, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Garbian, Y.; Ovadia, O.; Dadon, S.; Mishmar, D. Gene expression patterns of oxidative phosphorylation complex I subunits are organized in clusters. PLoS ONE 2010, 5, e9985. [Google Scholar] [CrossRef] [PubMed]
- Van Waveren, C.; Moraes, C.T. Transcriptional co-expression and co-regulation of genes coding for components of the oxidative phosphorylation system. BMC Genom. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Richter-Dennerlein, R.; Oeljeklaus, S.; Lorenzi, I.; Ronsor, C.; Bareth, B.; Schendzielorz, A.B.; Wang, C.; Warscheid, B.; Rehling, P.; Dennerlein, S. Mitochondrial Protein Synthesis Adapts to Influx of Nuclear-Encoded Protein. Cell 2016, 167, 471–483.e410. [Google Scholar] [CrossRef] [PubMed]
- Mick, D.U.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef] [PubMed]
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Churchman, L.S. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Strub, J.M.; Carte, N.; Luche, S.; Van Dorsselaer, A.; Lunardi, J.; Giege, R.; Florentz, C. Comparative proteomics as a new tool for exploring human mitochondrial tRNA disorders. Biochemistry 2002, 41, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.; Ahn, B.Y.; Byun, K.; Cho, Y.M.; Yu, M.H.; Lee, B.; Hwang, D.; Park, K.S. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci Signal. 2013, 6, rs4. [Google Scholar] [CrossRef] [PubMed]
- Ponsuksili, S.; Du, Y.; Hadlich, F.; Siengdee, P.; Murani, E.; Schwerin, M.; Wimmers, K. Correlated mRNAs and miRNAs from co-expression and regulatory networks affect porcine muscle and finally meat properties. BMC Genom. 2013, 14, 533. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, P.; Wang, G. Regulation of mitochondrion-associated cytosolic ribosomes by mammalian mitochondrial ribonuclease T2 (RNASET2). J. Biol. Chem. 2018, 293, 19633–19644. [Google Scholar] [CrossRef] [PubMed]
- Pastukh, V.; Shokolenko, I.; Wang, B.; Wilson, G.; Alexeyev, M. Human mitochondrial transcription factor A possesses multiple subcellular targeting signals. FEBS J. 2007, 274, 6488–6499. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, J.E.; Rogozin, I.B.; Koonin, E.V.; Chumakov, P.M. Transcription of mammalian messenger RNAs by a nuclear RNA polymerase of mitochondrial origin. Nature 2005, 436, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, A.; Sri Sailaja, B.; Kundaje, A.; Levin, L.; Dadon, S.; Shmorak, S.; Shaulian, E.; Meshorer, E.; Mishmar, D. Transcription factors bind negatively selected sites within human mtDNA genes. Genome Biol. Evol. 2014, 6, 2634–2646. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.M.; Sekeris, C.E. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: Role of the mitochondrial glucocorticoid receptor. Biochim. Biophys. Acta 2011, 1813, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Yang, Q.; Shepherd, K.; Smith, Y.; Miller, G.; Testa, C.; Mao, Z. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Investig. 2011, 121, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, K.; Lesnefsky, E.J.; Larner, A.C. Multi-tasking: Nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 2012, 22, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, M.; Falkenberg, M.; Larsson, N.G.; Gustafsson, C.M. The mitochondrial RNA polymerase contributes critically to promoter specificity in mammalian cells. EMBO J. 2004, 23, 4606–4614. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, L.; Moraes, C.T. Expanding the functional human mitochondrial DNA database by the establishment of primate xenomitochondrial cybrids. Proc. Natl. Acad. Sci. USA 1997, 94, 9131–9135. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Lechuga-Vieco, A.V.; Johnston, I.G.; Hamalainen, R.H.; Pellico, J.; Justo-Mendez, R.; Fernandez-Toro, J.M.; Claveria, C.; Guaras, A.; Sierra, R.; et al. Regulation of Mother-to-Offspring Transmission of mtDNA Heteroplasmy. Cell Metab. 2019, 30, 1120–1130.e1125. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.; Blumberg, A.; Barshad, G.; Mishmar, D. Mito-nuclear co-evolution: The positive and negative sides of functional ancient mutations. Front. Genet. 2014, 5, 448. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Ongwijitwat, S.; Wong-Riley, M.T. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J. Biol. Chem. 2008, 283, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.R.; Wilson, B.J.; Huss, J.M.; Kelly, D.P.; Alaynick, W.A.; Downes, M.; Evans, R.M.; Blanchette, M.; Giguere, V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007, 5, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Alaynick, W.A.; Kondo, R.P.; Xie, W.; He, W.; Dufour, C.R.; Downes, M.; Jonker, J.W.; Giles, W.; Naviaux, R.K.; Giguere, V.; et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007, 6, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Arnould, T.; Vankoningsloo, S.; Renard, P.; Houbion, A.; Ninane, N.; Demazy, C.; Remacle, J.; Raes, M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002, 21, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Bond, J.D.; Ingram, V.M. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc. Natl. Acad. Sci. USA 1997, 94, 9705–9710. [Google Scholar] [CrossRef] [PubMed]
- Biswas, G.; Anandatheerthavarada, H.K.; Zaidi, M.; Avadhani, N.G. Mitochondria to nucleus stress signaling: A distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J. Cell Biol. 2003, 161, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Biswas, G.; Adebanjo, O.A.; Freedman, B.D.; Anandatheerthavarada, H.K.; Vijayasarathy, C.; Zaidi, M.; Kotlikoff, M.; Avadhani, N.G. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: A novel mode of inter-organelle crosstalk. EMBO J. 1999, 18, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Hwang, A.B.; Kenyon, C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 2010, 20, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Hwang, A.B.; Ryu, E.A.; Artan, M.; Chang, H.W.; Kabir, M.H.; Nam, H.J.; Lee, D.; Yang, J.S.; Kim, S.; Mair, W.B.; et al. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2014, 111, E4458–E4467. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, E.; Yavari, A.; Mandal, S.; Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 2008, 40, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Freije, W.A.; Guptan, P.; Banerjee, U. Metabolic control of G1-S transition: Cyclin E degradation by p53-induced activation of the ubiquitin-proteasome system. J. Cell Biol. 2010, 188, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Formentini, L.; Sanchez-Arago, M.; Sanchez-Cenizo, L.; Cuezva, J.M. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol. Cell. 2012, 45, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. Apoptosis. Death cycle and Swiss army knives. Nature 1998, 391, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Mitonuclear match: Optimizing fitness and fertility over generations drives ageing within generations. Bioessays 2011, 33, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Niveditha, S.; Deepashree, S.; Ramesh, S.R.; Shivanandappa, T. Sex differences in oxidative stress resistance in relation to longevity in Drosophila melanogaster. J. Comp. Physiol. B 2017, 187, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Borras, C.; Sastre, J.; Garcia-Sala, D.; Lloret, A.; Pallardo, F.V.; Vina, J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic. Biol. Med. 2003, 34, 546–552. [Google Scholar] [CrossRef]
- Li, N.; Arief, N.; Edmands, S. Effects of oxidative stress on sex-specific gene expression in the copepod Tigriopus californicus revealed by single individual RNA-seq. Comp. Biochem. Physiol. Part D Genom. Proteomics 2019, 31, 100608. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, R.; Lepage, V.; Charron, D.; Schachter, F. Mitochondrial genotype associated with French Caucasian centenarians. Gerontology 1998, 44, 349. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Gong, J.S.; Zhang, J.; Yoneda, M.; Yagi, K. Mitochondrial genotype associated with longevity. Lancet 1998, 351, 185–186. [Google Scholar] [CrossRef]
- De Benedictis, G.; Carrieri, G.; Varcasia, O.; Bonafe, M.; Franceschi, C. Inherited variability of the mitochondrial genome and successful aging in humans. Ann. N. Y. Acad. Sci. 2000, 908, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Camus, M.F.; Wolf, J.B.; Morrow, E.H.; Dowling, D.K. Single Nucleotides in the mtDNA Sequence Modify Mitochondrial Molecular Function and Are Associated with Sex-Specific Effects on Fertility and Aging. Curr. Biol. 2015, 25, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M.; Fry, A.; Sheldahl, L. Nuclear-mitochondrial epistasis and drosophila aging: Introgression of Drosophila simulans mtDNA modifies longevity in D. melanogaster nuclear backgrounds. Genetics 2006, 172, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.; Passarino, G.; Carrieri, G.; Altomare, K.; Greco, V.; Bertolini, S.; Bonafe, M.; Franceschi, C.; De Benedictis, G. Paradoxes in longevity: Sequence analysis of mtDNA haplogroup J in centenarians. Eur. J. Hum. Genet. 2001, 9, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Lesko, S.A.; Lorentzen, R.J.; Ts’o, P.O. Role of superoxide in deoxyribonucleic acid strand scission. Biochemistry 1980, 19, 3023–3028. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Aruoma, O.I. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991, 281, 9–19. [Google Scholar] [CrossRef]
- Blakely, W.F.; Fuciarelli, A.F.; Wegher, B.J.; Dizdaroglu, M. Hydrogen peroxide-induced base damage in deoxyribonucleic acid. Radiat. Res. 1990, 121, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Hogue, B.A.; Mildaziene, V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 1997, 29, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 1973, 248, 4793–4796. [Google Scholar] [PubMed]
- Kukat, C.; Wurm, C.A.; Spahr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Davies, K.M.; Wurm, C.A.; Spahr, H.; Bonekamp, N.A.; Kuhl, I.; Joos, F.; Polosa, P.L.; Park, C.B.; Posse, V.; et al. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc. Natl. Acad. Sci. USA 2015, 112, 11288–11293. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, J.H.K.; Bonekamp, N.A.; Mourier, A.; Isokallio, M.A.; Just, A.; Kauppila, T.E.S.; Stewart, J.B.; Larsson, N.G. Base-excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Res. 2018, 46, 6642–6669. [Google Scholar] [CrossRef] [PubMed]
- Brieba, L.G.; Eichman, B.F.; Kokoska, R.J.; Doublie, S.; Kunkel, T.A.; Ellenberger, T. Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J. 2004, 23, 3452–3461. [Google Scholar] [CrossRef] [PubMed]
- Graziewicz, M.A.; Bienstock, R.J.; Copeland, W.C. The DNA polymerase gamma Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-2’-deoxyguanosine. Hum. Mol. Genet. 2007, 16, 2729–2739. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, G.; Makarova, A.V.; Wanrooij, P.H.; Forslund, J.; Burgers, P.M.; Wanrooij, S. Oxidative DNA damage stalls the human mitochondrial replisome. Sci. Rep. 2016, 6, 28942. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Zinovkina, L.A. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublie, S. Base Excision Repair in the Mitochondria. J. Cell Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta 2013, 1833, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Mordas, A.; Tokatlidis, K. The MIA pathway: A key regulator of mitochondrial oxidative protein folding and biogenesis. Acc. Chem. Res. 2015, 48, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y. Regulation of intracellular localization of human MTH1, OGG1, and MYH proteins for repair of oxidative DNA damage. Prog Nucleic Acid Res. Mol. Biol. 2001, 68, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Plotz, G.; Casper, M.; Raedle, J.; Hinrichsen, I.; Heckel, V.; Brieger, A.; Trojan, J.; Zeuzem, S. MUTYH gene expression and alternative splicing in controls and polyposis patients. Hum. Mutat. 2012, 33, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Izumi, T.; Boldogh, I.; Bhakat, K.K.; Chattopadhyay, R.; Szczesny, B. Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein. DNA Repair 2007, 6, 461–469. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar] [PubMed]
- Takao, M.; Kanno, S.; Shiromoto, T.; Hasegawa, R.; Ide, H.; Ikeda, S.; Sarker, A.H.; Seki, S.; Xing, J.Z.; Le, X.C.; et al. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. 2002, 21, 3486–3493. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, M.R.; Yu, W.; Gmyrek, A.M.; White, M.A.; Molineux, I.J.; Lee, J.C.; Yin, Y.W. A domain in human EXOG converts apoptotic endonuclease to DNA-repair exonuclease. Nat. Commun. 2017, 8, 14959. [Google Scholar] [CrossRef] [PubMed]
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 2017, 107, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Frossi, B.; Tell, G.; Spessotto, P.; Colombatti, A.; Vitale, G.; Pucillo, C. H(2)O(2) induces translocation of APE/Ref-1 to mitochondria in the Raji B-cell line. J. Cell Physiol. 2002, 193, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dolle, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Stevnsner, T.; Muftuoglu, M.; Aamann, M.D.; Bohr, V.A. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008, 129, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Sage, J.M.; Knight, K.L. Human Rad51 promotes mitochondrial DNA synthesis under conditions of increased replication stress. Mitochondrion 2013, 13, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Yokota, J. The OGG1 gene encodes a repair enzyme for oxidatively damaged DNA and is involved in human carcinogenesis. Antioxid. Redox Signal. 2001, 3, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kohmoto, T.; Tabata, R.; Seki, Y. Differential intracellular localization of the human and mouse endonuclease III homologs and analysis of the sorting signals. DNA Repair 2002, 1, 847–854. [Google Scholar] [CrossRef]
- Swartzlander, D.B.; Griffiths, L.M.; Lee, J.; Degtyareva, N.P.; Doetsch, P.W.; Corbett, A.H. Regulation of base excision repair: Ntg1 nuclear and mitochondrial dynamic localization in response to genotoxic stress. Nucleic Acids Res. 2010, 38, 3963–3974. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Brunyanszki, A.; Olah, G.; Mitra, S.; Szabo, C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: Implications for the regulation of mitochondrial function. Nucleic Acids Res. 2014, 42, 13161–13173. [Google Scholar] [CrossRef] [PubMed]
- Burkle, A.; Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef] [PubMed]
- Brunyanszki, A.; Szczesny, B.; Virag, L.; Szabo, C. Mitochondrial poly(ADP-ribose) polymerase: The Wizard of Oz at work. Free Radic. Biol. Med. 2016, 100, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Izumi, H.; Torigoe, T.; Ishiguchi, H.; Itoh, H.; Kang, D.; Kohno, K. P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res. 2003, 63, 3729–3734. [Google Scholar] [PubMed]
- Park, J.H.; Zhuang, J.; Li, J.; Hwang, P.M. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016, 590, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Hamasaki, N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: Overview of its multiple roles. Ann. N. Y. Acad. Sci. 2005, 1042, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Wang, P.Y.; Matsumoto, T.; Sung, H.J.; Ma, W.; Choi, J.W.; Anderson, S.A.; Leary, S.C.; Balaban, R.S.; Kang, J.G.; et al. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res. 2009, 105, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Ma, W.; Park, J.Y.; Celi, F.S.; Arena, R.; Choi, J.W.; Ali, Q.A.; Tripodi, D.J.; Zhuang, J.; Lago, C.U.; et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N. Engl. J. Med. 2013, 368, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Croteau, D.L.; Bohr, V.A.; Wilson, D.M., 3rd. Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 7437–7442. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Sykora, P.; Bohr, V.A. Slow mitochondrial repair of 5’-AMP renders mtDNA susceptible to damage in APTX deficient cells. Sci. Rep. 2015, 5, 12876. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Dexheimer, T.S.; Maddali, K.; Pommier, Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 19790–19795. [Google Scholar] [CrossRef] [PubMed]
- Bokov, A.; Chaudhuri, A.; Richardson, A. The role of oxidative damage and stress in aging. Mech. Ageing Dev. 2004, 125, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Alaei-Mahabadi, B.; Sabarinathan, R.; Samuelsson, T.; Gorodkin, J.; Gustafsson, C.M.; Larsson, E. Simultaneous DNA and RNA Mapping of Somatic Mitochondrial Mutations across Diverse Human Cancers. PLoS Genet. 2015, 11, e1005333. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Ozawa, T. Strand asymmetry in human mitochondrial DNA mutations. Genomics 1994, 22, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Gissi, C.; Pesole, G.; Saccone, C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 1998, 15, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb. Perspect Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Tuna, S.; Keogh, M.J.; Smith, K.R.; Aitman, T.J.; Beales, P.L.; Bennett, D.L.; Gale, D.P.; Bitner-Glindzicz, M.A.K.; Black, G.C.; et al. Germline selection shapes human mitochondrial DNA diversity. Science 2019, 364. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA variation in human radiation and disease. Cell 2015, 163, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Hutchin, T.; Cortopassi, G. A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1995, 92, 6892–6895. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Balbi, V.; Balducci, E.; Pirazzini, C.; Rosini, F.; Tavano, F.; Achilli, A.; Siviero, P.; Minicuci, N.; Bellavista, E.; et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE 2010, 5, e12037. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, A.; Derbeneva, O.; Younes, D.; Keator, D.; Bakken, T.; Lvova, M.; Brandon, M.; Guffanti, G.; Reglodi, D.; Saykin, A.; et al. Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol. Aging 2010, 31, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, E.; Gilyazova, I.; Ruiz-Pesini, E.; Derbeneva, O.; Khusainova, R.; Khidiyatova, I.; Magzhanov, R.; Wallace, D.C. A mitochondrial etiology of neurodegenerative diseases: Evidence from Parkinson’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Van der Walt, J.M.; Dementieva, Y.A.; Martin, E.R.; Scott, W.K.; Nicodemus, K.K.; Kroner, C.C.; Welsh-Bohmer, K.A.; Saunders, A.M.; Roses, A.D.; Small, G.W.; et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci. Lett. 2004, 365, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Van der Walt, J.M.; Nicodemus, K.K.; Martin, E.R.; Scott, W.K.; Nance, M.A.; Watts, R.L.; Hubble, J.P.; Haines, J.L.; Koller, W.C.; Lyons, K.; et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet. 2003, 72, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Elson, J.L.; Nooteboom, M.; Grady, J.P.; Taylor, G.A.; Taylor, R.W.; Mathers, J.C.; Kirkwood, T.B.; Turnbull, D.M. Comparison of mitochondrial mutation spectra in ageing human colonic epithelium and disease: Absence of evidence for purifying selection in somatic mitochondrial DNA point mutations. PLoS Genet. 2012, 8, e1003082. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Wilson, I.J.; Yu-Wai-Man, P.; Coxhead, J.; Deehan, D.; Horvath, R.; Taylor, R.W.; Samuels, D.C.; Santibanez-Koref, M.; Chinnery, P.F. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 2013, 22, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo-Jaramillo, B.; Su, M.S.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, D.R. Mitochondrial disorders: Prevalence, myths and advances. J. Inherit. Metab. Dis. 2004, 27, 349–362. [Google Scholar] [CrossRef] [PubMed]
- De Silva, D.; Tu, Y.T.; Amunts, A.; Fontanesi, F.; Barrientos, A. Mitochondrial ribosome assembly in health and disease. Cell Cycle 2015, 14, 2226–2250. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, V.; Pitceathly, R.D.; Turnbull, D.M.; Taylor, R.W.; Sweeney, M.G.; Mudanohwo, E.E.; Rahman, S.; Hanna, M.G.; McFarland, R. The UK MRC Mitochondrial Disease Patient Cohort Study: Clinical phenotypes associated with the m.3243A>G mutation--implications for diagnosis and management. J. Neurol. Neurosurg. Psychiatry 2013, 84, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F.; Towbin, J.A.; Craigen, W.J.; Belmont, J.W.; Smith, E.O.; Neish, S.R.; Ware, S.M.; Hunter, J.V.; Fernbach, S.D.; Vladutiu, G.D.; et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004, 114, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Nguyen, K.V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004, 55, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Sofou, K.; Kollberg, G.; Holmstrom, M.; Davila, M.; Darin, N.; Gustafsson, C.M.; Holme, E.; Oldfors, A.; Tulinius, M.; Asin-Cayuela, J. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol. Genet. Genom. Med. 2015, 3, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Salviati, L.; Sacconi, S.; Otaegui, D.; Camano, P.; Marina, A.; Bacman, S.; Moraes, C.T.; Carlo, J.R.; Garcia, M.; et al. Mitochondrial DNA depletion: Mutations in thymidine kinase gene with myopathy and SMA. Neurology 2002, 59, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Vaz, F.M.; Gardeitchik, T.; Vissers, L.E.; Renkema, G.H.; Schuurs-Hoeijmakers, J.H.; Kulik, W.; Lammens, M.; Christin, C.; Kluijtmans, L.A.; et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 2012, 44, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited mitochondrial optic neuropathies. J. Med. Genet. 2009, 46, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Carelli, V.; Spruijt, L.; Gerards, M.; Mowbray, C.; Achilli, A.; Pyle, A.; Elson, J.; Howell, N.; La Morgia, C.; et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am. J. Hum. Genet. 2007, 81, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.A.; Yu-Wai-Man, P.; Korsten, A.; Leonhardt, M.; Dimitriadis, K.; De Coo, I.F.; Klopstock, T.; Chinnery, P.F. Gene-environment interactions in Leber hereditary optic neuropathy. Brain 2009, 132, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Kearns, T.P.; Sayre, G.P. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: Unusual syndrome with histologic study in one of two cases. AMA Arch. Ophthalmol. 1958, 60, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.G.; Newman, N.J.; Shoffner, J.M.; Kaufman, A.E.; Koontz, D.A.; Wallace, D.C. Variable retinal and neurologic manifestations in patients harboring the mitochondrial DNA 8993 mutation. Arch. Ophthalmol. 1993, 111, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Sommerville, E.W.; Chinnery, P.F.; Gorman, G.S.; Taylor, R.W. Adult-onset Mendelian PEO Associated with Mitochondrial Disease. J. Neuromuscul. Dis. 2014, 1, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.O.; Steinberg, S.V.; Schimmel, P. Functional defects of pathogenic human mitochondrial tRNAs related to structural fragility. Nat. Struct. Biol. 2000, 7, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.D.; Ericson, N.G.; Burton, J.N.; Prolla, T.A.; Silber, J.R.; Shendure, J.; Bielas, J.H. Targeted enrichment and high-resolution digital profiling of mitochondrial DNA deletions in human brain. Aging Cell 2014, 13, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Barreau, E.; Brossas, J.Y.; Courtois, Y.; Treton, J.A. Accumulation of mitochondrial DNA deletions in human retina during aging. Investig. Ophthalmol. Vis. Sci. 1996, 37, 384–391. [Google Scholar]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.H.; Chao, H.T.; Wei, Y.H. Multiple deletions of mitochondrial DNA are associated with the decline of motility and fertility of human spermatozoa. Mol. Hum. Reprod. 1998, 4, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, T.; Suganuma, N.; Nawa, A.; Kikkawa, F.; Tanaka, M.; Ozawa, T.; Tomoda, Y. Rapid accumulation of deleted mitochondrial deoxyribonucleic acid in postmenopausal ovaries. Biol. Reprod. 1993, 49, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Edgar, D.; Shabalina, I.; Camara, Y.; Wredenberg, A.; Calvaruso, M.A.; Nijtmans, L.; Nedergaard, J.; Cannon, B.; Larsson, N.G.; Trifunovic, A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009, 10, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Bielas, J.H.; Kujoth, G.C.; Ladiges, W.C.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007, 39, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Baris, O.R.; Ederer, S.; Neuhaus, J.F.; von Kleist-Retzow, J.C.; Wunderlich, C.M.; Pal, M.; Wunderlich, F.T.; Peeva, V.; Zsurka, G.; Kunz, W.S.; et al. Mosaic Deficiency in Mitochondrial Oxidative Metabolism Promotes Cardiac Arrhythmia during Aging. Cell Metab. 2015, 21, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wanagat, J.; McKiernan, S.H.; Aiken, J.M. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: Analysis by laser-capture microdissection. Nucleic Acids Res. 2001, 29, 4502–4508. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001, 15, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Popadin, K.; Safdar, A.; Kraytsberg, Y.; Khrapko, K. When man got his mtDNA deletions? Aging Cell 2014, 13, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Salas, A.; Yao, Y.G.; Macaulay, V.; Vega, A.; Carracedo, A.; Bandelt, H.J. A critical reassessment of the role of mitochondria in tumorigenesis. PLoS Med. 2005, 2, e296. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Tolkunov, D.; Aviv, H.; Hakimi, A.A.; Yao, M.; Hsieh, J.J.; Ganesan, S.; Chan, C.S.; White, E. The Genomic Landscape of Renal Oncocytoma Identifies a Metabolic Barrier to Tumorigenesis. Cell Rep. 2015, 13, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhou, S.; Chang, S.S.; McFate, T.; Verma, A.; Califano, J.A. Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, W.; Li, H.; Yu, Y.; Tao, J.; Huang, S.; Zeng, Z. Nonsense and missense mutation of mitochondrial ND6 gene promotes cell migration and invasion in human lung adenocarcinoma. BMC Cancer 2015, 15, 346. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; de Coo, I.; Diaz, F.; Smeets, H.; Moraes, C.T. An out-of-frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann. Neurol. 2000, 48, 774–781. [Google Scholar] [CrossRef]
- Geromel, V.; Kadhom, N.; Cebalos-Picot, I.; Ouari, O.; Polidori, A.; Munnich, A.; Rotig, A.; Rustin, P. Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the neurogenic ataxia retinitis pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA. Hum. Mol. Genet. 2001, 10, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Degoul, F.; Diry, M.; Rodriguez, D.; Robain, O.; Francois, D.; Ponsot, G.; Marsac, C.; Desguerre, I. Clinical, biochemical, and molecular analysis of a maternally inherited case of Leigh syndrome (MILS) associated with the mtDNA T8993G point mutation. J. Inherit. Metab. Dis. 1995, 18, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.K.; Calvo, S.E.; Shih, A.R.; Chaves, F.L.; McGuone, D.; Mick, E.; Pierce, K.A.; Li, Y.; Garofalo, A.; Van Allen, E.M.; et al. Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6283–E6290. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
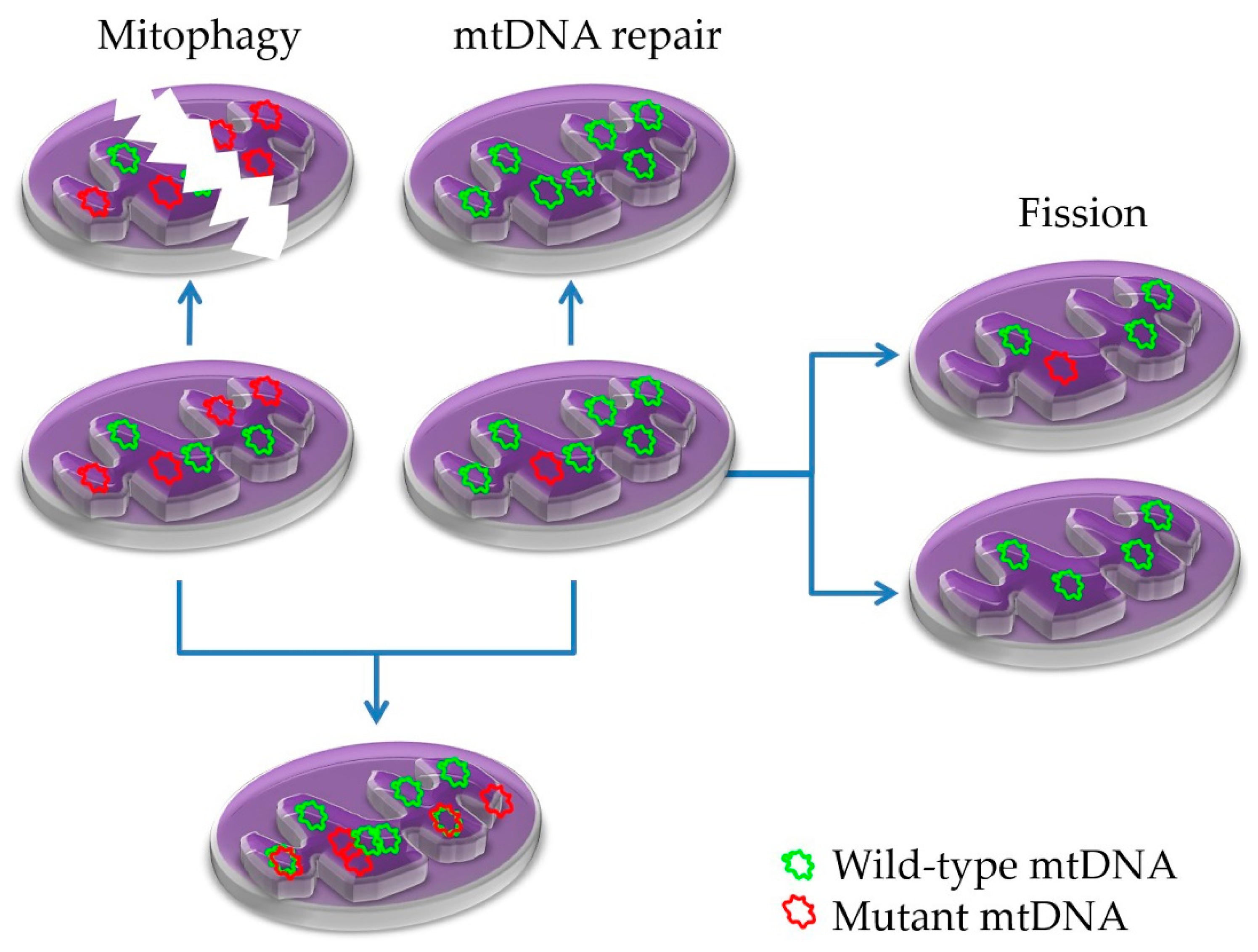{kind=link}
| Repair Pathway | Type of Damage | Key Enzymes |
|---|---|---|
| BER | Deamination | UNG |
| Oxidation | MYH, OGG1, NTHL1, NEIL1, and NEIL2 | |
| Alkylation | MPG | |
| SSBR | Single-strand breaks | Utilizes BER components |
| MMR | Mismatch | MSH2, YB-1, NEIL2 and DNA ligase 3 |
| DSBR | Double-strand breaks | SSB, MGME1, Rad51, Twinkle, and TFAM |
| NER | Bulky adducts | CSA, CSB |
| Syndrome | Mitochondrial Mutations or Mutated Nuclear Genes | Number of Cases per 100,000 Individuals (Adults) |
|---|---|---|
| Leigh syndrome | >75 genes | 3.7 |
| Alpers–Huttenlocher syndrome | POLγ | 0.3 |
| Pearson syndrome | Large mtDNA deletion or rearrangements | 1.5 |
| LHON syndrome | MT-ND4 m.11778G>A, MT-ND6 m.14484T>C and MT-ND1 m.3460G>A | |
| MELAS | MT-TL1 m.3243A>G | 3.5 |
| Kearns–Sayre syndrome | Large mtDNA deletion | 1.5 |
| NARP | MT-ATP6 m.8993T>G or m.8993T>C | |
| CPEO | TYMP, POLG1, POLG2, MPV17, SLC25A4, TK2, DGUOK, OPA1, AFG3L2, DNM2, RNASEH1, DNA2 or C20orf7 | 0.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakaidos, P.; Rampias, T. Mitonuclear Interactions in the Maintenance of Mitochondrial Integrity. Life 2020, 10, 173. https://doi.org/10.3390/life10090173
Karakaidos P, Rampias T. Mitonuclear Interactions in the Maintenance of Mitochondrial Integrity. Life. 2020; 10(9):173. https://doi.org/10.3390/life10090173
Chicago/Turabian StyleKarakaidos, Panagiotis, and Theodoros Rampias. 2020. "Mitonuclear Interactions in the Maintenance of Mitochondrial Integrity" Life 10, no. 9: 173. https://doi.org/10.3390/life10090173
APA StyleKarakaidos, P., & Rampias, T. (2020). Mitonuclear Interactions in the Maintenance of Mitochondrial Integrity. Life, 10(9), 173. https://doi.org/10.3390/life10090173