Targeting the C-Terminal Domain Small Phosphatase 1




Abstract
1. Introduction
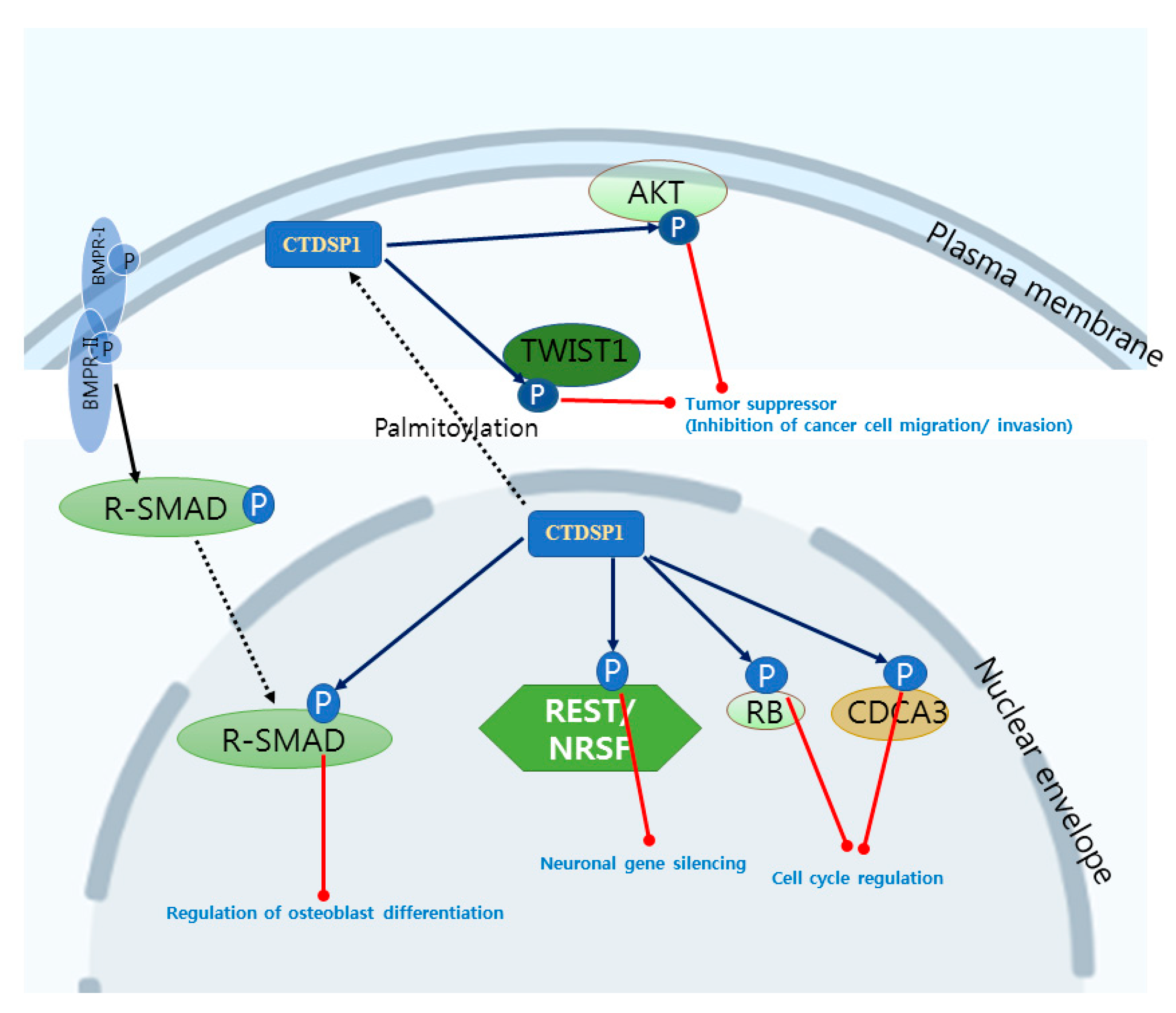
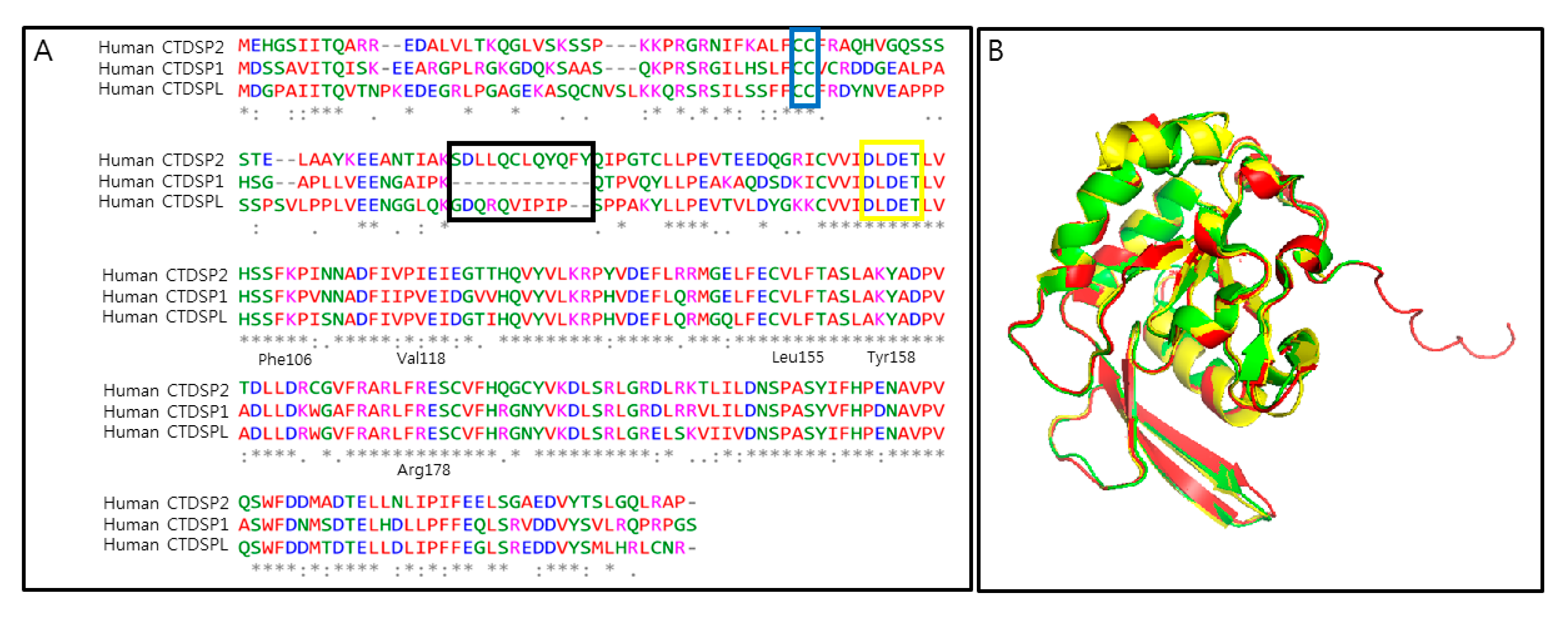
2. CTDSP1 as an Emerging Target
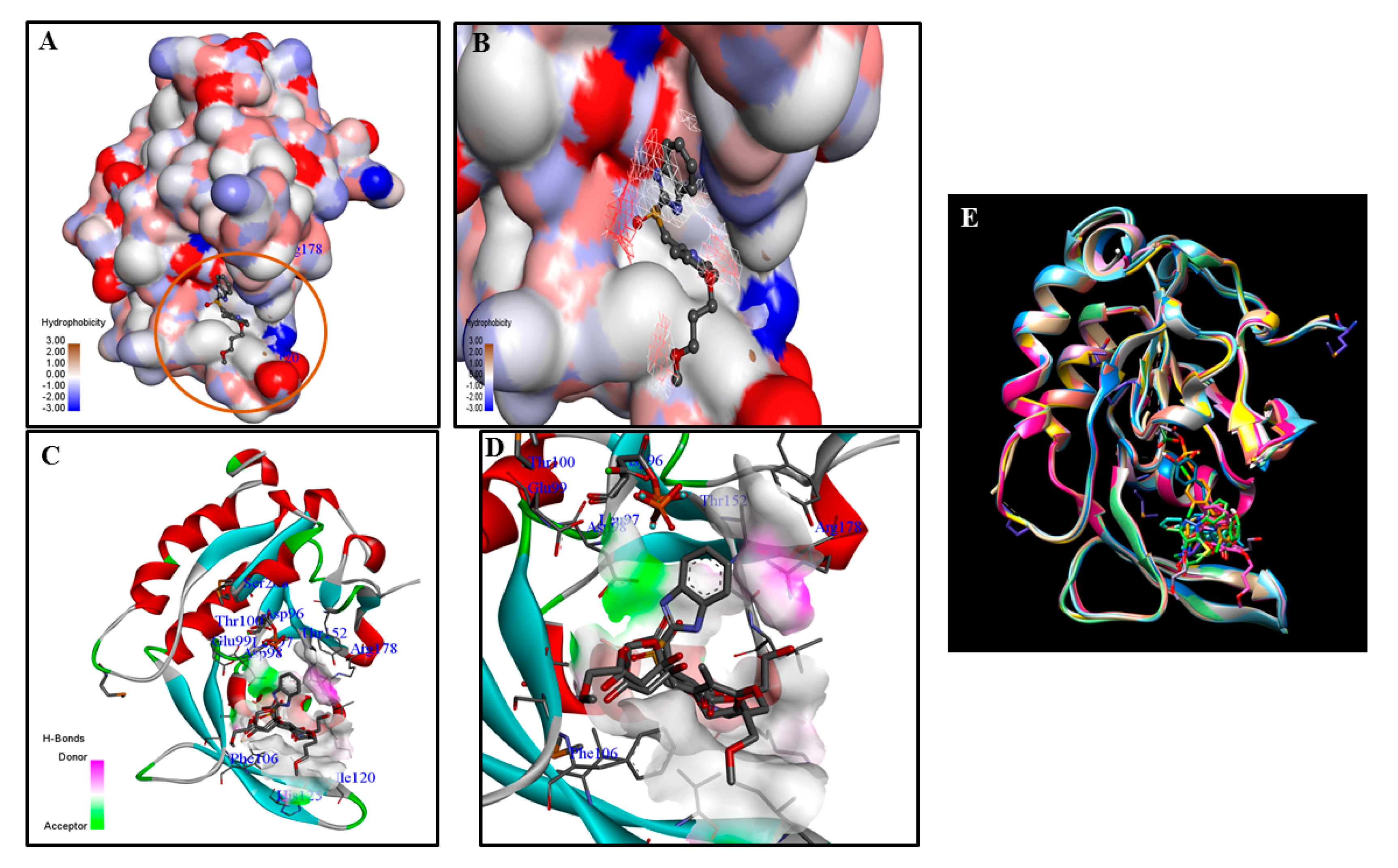
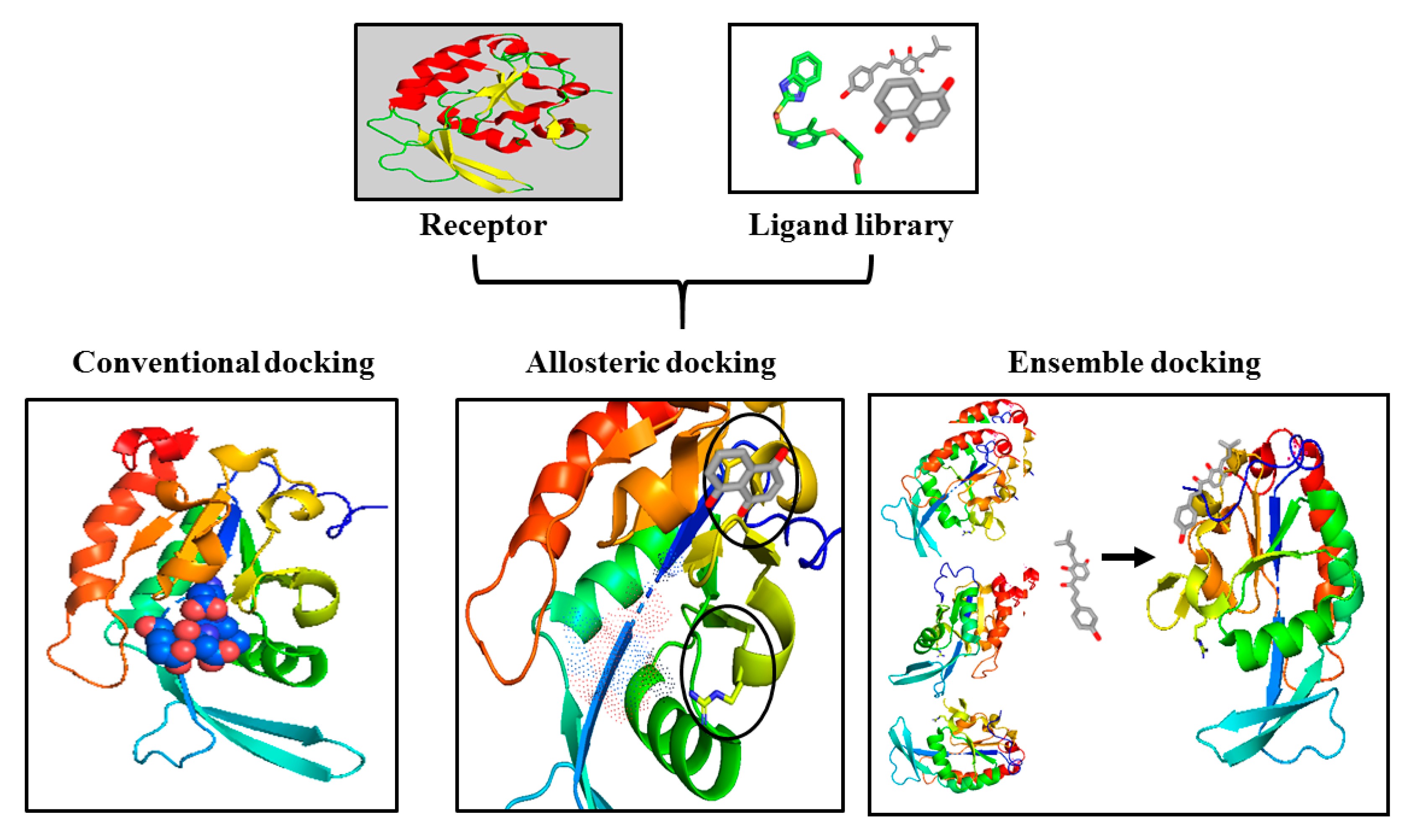
3. Challenges and Opportunities in Targeting CTDSP1
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harlen, K.M.; Churchman, L.S. The code and beyond: Transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 2017, 18, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, C.; Collin, P.; Robert, F. The RNA Polymerase II CTD: The Increasing Complexity of a Low-Complexity Protein Domain. J. Mol. Biol. 2016, 428, 2607–2622. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.S.; Cho, H.; Kim, S.; Cabane, K.; Reinberg, D. FCP1, a phosphatase specific for the heptapeptide repeat of the largest subunit of RNA polymerase II, stimulates transcription elongation. Mol. Cell. Biol. 2002, 22, 7543–7552. [Google Scholar] [CrossRef] [PubMed]
- Archambault, J.; Pan, G.; Dahmus, G.K.; Cartier, M.; Marshall, N.; Zhang, S.; Dahmus, M.E.; Greenblatt, J. FCP1, the RAP74-interacting subunit of a human protein phosphatase that dephosphorylates the carboxyl-terminal domain of RNA polymerase IIO. J. Biol. Chem. 1998, 273, 27593–27601. [Google Scholar] [CrossRef]
- Yeo, M.; Lin, P.S.; Dahmus, M.E.; Gill, G.N. A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J. Biol. Chem. 2003, 278, 26078–26085. [Google Scholar] [CrossRef]
- Zhang, Y.; Kim, Y.; Genoud, N.; Gao, J.; Kelly, J.W.; Pfaff, S.L.; Gill, G.N.; Dixon, J.E.; Noel, J.P. Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol. Cell 2006, 24, 759–770. [Google Scholar] [CrossRef]
- Kamenski, T.; Heilmeier, S.; Meinhart, A.; Cramer, P. Structure and mechanism of RNA polymerase II CTD phosphatases. Mol. Cell 2004, 15, 399–407. [Google Scholar] [CrossRef]
- R, H.R.; Kim, H.; Noh, K.; Kim, Y.J. The diverse roles of RNA polymerase II C-terminal domain phosphatase SCP1. BMB Rep. 2014, 47, 192–196. [Google Scholar]
- Nesti, E.; Corson, G.M.; McCleskey, M.; Oyer, J.A.; Mandel, G. C-terminal domain small phosphatase 1 and MAP kinase reciprocally control REST stability and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, E3929–E3936. [Google Scholar] [CrossRef]
- Yeo, M.; Lee, S.K.; Lee, B.; Ruiz, E.C.; Pfaff, S.L.; Gill, G.N. Small CTD phosphatases function in silencing neuronal gene expression. Science 2005, 307, 596–600. [Google Scholar] [CrossRef]
- Burkholder, N.T.; Mayfield, J.E.; Yu, X.; Irani, S.; Arce, D.K.; Jiang, F.; Matthews, W.L.; Xue, Y.; Zhang, Y.J. Phosphatase activity of small C-terminal domain phosphatase 1 (SCP1) controls the stability of the key neuronal regulator RE1-silencing transcription factor (REST). J. Biol. Chem. 2019, 293, 16851–16861. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, G.; Knockaert, M.; Alarcon, C.; Montalvo, E.; Brivanlou, A.H.; Massague, J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J. Biol. Chem. 2006, 281, 40412–40419. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liao, P.; Shen, M.; Chen, T.; Chen, Y.; Li, Y.; Lin, X.; Ge, X.; Wang, P. SCP1 regulates c-Myc stability and functions through dephosphorylating c-Myc Ser62. Oncogene 2015, 35, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, W.B.; Liu, K.J.; Ao, L.; Cao, J.; Zhong, J.L.; Liu, J.Y. Overexpression of miR-26b-5p regulates the cell cycle by targeting CCND2 in GC-2 cells under exposure to extremely low frequency electromagnetic fields. Cell Cycle 2015, 15, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Bahk, Y.Y. A study of substrate specificity for a CTD phosphatase, SCP1, by proteomic screening of binding partners. Biochem. Biophys. Res. Commun. 2014, 448, 189–194. [Google Scholar] [CrossRef]
- Sun, T.; Fu, J.; Shen, T.; Lin, X.; Liao, L.; Feng, X.H.; Xu, J. The Small C-terminal Domain Phosphatase 1 Inhibits Cancer Cell Migration and Invasion by Dephosphorylating Ser(P)68-Twist1 to Accelerate Twist1 Protein Degradation. J. Biol. Chem. 2016, 291, 11518–11528. [Google Scholar] [CrossRef]
- Zhang, M.; Cho, E.J.; Burstein, G.; Siegel, D.; Zhang, Y. Selective inactivation of a human neuronal silencing phosphatase by a small molecule inhibitor. ACS Chem. Biol. 2011, 6, 511–519. [Google Scholar] [CrossRef]
- Park, H.; Lee, H.S.; Ku, B.; Lee, S.R.; Kim, S.J. Two-track virtual screening approach to identify both competitive and allosteric inhibitors of human small C-terminal domain phosphatase 1. J. Comput. Aided Mol. Des. 2017, 31, 743–753. [Google Scholar] [CrossRef]
- Yoshida, T.; Yamazaki, K.; Imai, S.; Banno, A.; Kaneko, A.; Furukawa, K.; Chuman, Y. Identification of a Specific Inhibitor of Human Scp1 Phosphatase Using the Phosphorylation Mimic Phage Display Method. Catalysts 2019, 9, 842. [Google Scholar] [CrossRef]
- Bera, I.; Payghan, P.V. Use of Molecular Dynamics Simulations in Structure-Based Drug Discovery. Curr. Pharm. Des. 2019, 25, 3339–3349. [Google Scholar] [CrossRef]
- Lane, J.R.; Chubukov, P.; Liu, W.; Canals, M.; Cherezov, V.; Abagyan, R.; Stevens, R.C.; Katritch, V. Structure-based ligand discovery targeting orthosteric and allosteric pockets of dopamine receptors. Mol. Pharm. 2013, 84, 794–807. [Google Scholar] [CrossRef]
- Majewski, M.; Ruiz-Carmona, S.; Barril, X. Dynamic Undocking: A Novel Method for Structure-Based Drug Discovery. Methods Mol. Biol. 2018, 1824, 195–215. [Google Scholar]
- Morra, G.; Genoni, A.; Neves, M.A.; Merz, K.M., Jr.; Colombo, G. Molecular recognition and drug-lead identification: What can molecular simulations tell us? Curr. Med. Chem. 2009, 17, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Ferenczy, G.G.; Keseru, G.M. Structure-based Virtual Screening Approaches in Kinase-directed Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 2235–2259. [Google Scholar] [CrossRef]
- Barril, X.; Hubbard, R.E.; Morley, S.D. Virtual screening in structure-based drug discovery. Mini Rev. Med. Chem. 2004, 4, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Nie, A.; An, J.; Huang, Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr. Opin. Chem. Biol. 2006, 10, 194–202. [Google Scholar] [CrossRef]
- Tavousi, P.; Amin, R.; Shahbazmohamadi, S. Assemble-And-Match: A Novel Hybrid Tool for Enhancing Education and Research in Rational Structure Based Drug Design. Sci. Rep. 2018, 8, 849. [Google Scholar] [CrossRef]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Verlinde, C.L.; Hol, W.G. Structure-based drug design: Progress, results and challenges. Structure 1994, 2, 577–587. [Google Scholar] [CrossRef]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-Based Drug Design Strategies and Challenges. Curr. Top. Med. Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Grey, J.L.; Thompson, D.H. Challenges and opportunities for new protein crystallization strategies in structure-based drug design. Expert Opin. Drug Discov. 2010, 5, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Lounnas, V.; Ritschel, T.; Kelder, J.; McGuire, R.; Bywater, R.P.; Foloppe, N. Current progress in Structure-Based Rational Drug Design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 2013, 5, e201302011. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. Aaps J. 2012, 14, 133–141. [Google Scholar] [CrossRef]
- Van Montfort, R.L.M.; Workman, P. Structure-based drug design: Aiming for a perfect fit. Essays Biochem. 2017, 61, 431–437. [Google Scholar] [CrossRef]
- Bajusz, D.; Ferenczy, G.G.; Keseru, G.M. Ensemble docking-based virtual screening yields novel spirocyclic JAK1 inhibitors. J. Mol. Graph. Model. 2016, 70, 275–283. [Google Scholar] [CrossRef]
- Cavasotto, C.N. Normal mode-based approaches in receptor ensemble docking. Methods Mol. Biol. 2012, 819, 157–168. [Google Scholar]
- Cowan-Jacob, S.W.; Jahnke, W.; Knapp, S. Novel approaches for targeting kinases: Allosteric inhibition, allosteric activation and pseudokinases. Future Med. Chem. 2014, 6, 541–561. [Google Scholar] [CrossRef]
- Ellingson, S.R.; Miao, Y.; Baudry, J.; Smith, J.C. Multi-conformer ensemble docking to difficult protein targets. J. Phys. Chem. B 2014, 119, 1026–1034. [Google Scholar] [CrossRef]
- Da, C.; Mooberry, S.L.; Gupton, J.T.; Kellogg, G.E. How to deal with low-resolution target structures: Using SAR, ensemble docking, hydropathic analysis, and 3D-QSAR to definitively map the alphabeta-tubulin colchicine site. J. Med. Chem. 2013, 56, 7382–7395. [Google Scholar] [CrossRef]
- Erol, I.; Aksoydan, B.; Kantarcioglu, I.; Salmas, R.E.; Durdagi, S. Identification of novel serotonin reuptake inhibitors targeting central and allosteric binding sites: A virtual screening and molecular dynamics simulations study. J. Mol. Graph. Model. 2017, 74, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, W.; Weir, R.L.; Ellingson, S.R.; Harris, J.B.; Kapoor, K.; Smith, J.C.; Baudry, J. Ensemble-based docking: From hit discovery to metabolism and toxicity predictions. Bioorg. Med. Chem. 2016, 24, 4928–4935. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, S.M.; Rajanikant, G.K. Ensemble pharmacophore meets ensemble docking: A novel screening strategy for the identification of RIPK1 inhibitors. J. Comput. Aided Mol. Des. 2014, 28, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Narasimhan, S.; Rodrigues, J.P.; Bonvin, A.M. Information-Driven, Ensemble Flexible Peptide Docking Using HADDOCK. Methods Mol. Biol. 2017, 1561, 109–138. [Google Scholar] [PubMed]
- Henriksen, S.T.; Liu, J.; Estiu, G.; Oltvai, Z.N.; Wiest, O. Identification of novel bacterial histidine biosynthesis inhibitors using docking, ensemble rescoring, and whole-cell assays. Bioorg. Med. Chem. 2010, 18, 5148–5156. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins 2007, 66, 399–421. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kokubo, H.; Tanaka, T. Ligand docking simulations by generalized-ensemble algorithms. Adv. Protein Chem. Struct. Biol. 2013, 92, 63–91. [Google Scholar]
- Zhang, Z.; Ehmann, U.; Zacharias, M. Monte Carlo replica-exchange based ensemble docking of protein conformations. Proteins 2017, 85, 924–937. [Google Scholar] [CrossRef]
- Xu, M.; Lill, M.A. Utilizing experimental data for reducing ensemble size in flexible-protein docking. J. Chem. Inf. Model. 2012, 52, 187–198. [Google Scholar] [CrossRef]
- Lam, P.C.; Abagyan, R.; Totrov, M. Ligand-biased ensemble receptor docking (LigBEnD): A hybrid ligand/receptor structure-based approach. J. Comput. Aided Mol. Des. 2018, 32, 187–198. [Google Scholar] [CrossRef]
- Lape, M.; Elam, C.; Paula, S. Comparison of current docking tools for the simulation of inhibitor binding by the transmembrane domain of the sarco/endoplasmic reticulum calcium ATPase. Biophys. Chem. 2010, 150, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Westhead, D.R.; Clark, D.E.; Murray, C.W. A comparison of heuristic search algorithms for molecular docking. J. Comput. Aided Mol. Des. 1997, 11, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef]
- Schnecke, V.; Swanson, C.A.; Getzoff, E.D.; Tainer, J.A.; Kuhn, L.A. Screening a peptidyl database for potential ligands to proteins with side-chain flexibility. Proteins 1998, 33, 74–87. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, N.; Martinez-Arias, A.; Blundell, T.L. Probing the druggability of protein-protein interactions: Targeting the Notch1 receptor ankyrin domain using a fragment-based approach. Biochem. Soc. Trans. 2011, 39, 1327–1333. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, Z.; Zhu, L.; Cao, Y.; Wu, G.; Liu, X.; Chen, Y.; Wang, Q.; Shi, T.; Zhao, Y.; Wang, Y.; et al. ASD: A comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011, 39, D663–D669. [Google Scholar] [CrossRef] [PubMed]
- Amor, B.R.; Schaub, M.T.; Yaliraki, S.N.; Barahona, M. Prediction of allosteric sites and mediating interactions through bond-to-bond propensities. Nat. Commun. 2016, 7, 12477. [Google Scholar] [CrossRef]
- Cosconati, S.; Marinelli, L.; Di Leva, F.S.; La Pietra, V.; De Simone, A.; Mancini, F.; Andrisano, V.; Novellino, E.; Goodsell, D.S.; Olson, A.J. Protein flexibility in virtual screening: The BACE-1 case study. J. Chem. Inf. Model. 2012, 52, 2697–2704. [Google Scholar] [CrossRef]
- Fan, H.; Irwin, J.J.; Sali, A. Virtual ligand screening against comparative protein structure models. Methods Mol. Biol. 2011, 819, 105–126. [Google Scholar]
- Xie, L.; Bourne, P.E. Structure-based systems biology for analyzing off-target binding. Curr. Opin. Struct. Biol. 2011, 21, 189–199. [Google Scholar] [CrossRef]
- Chartier, M.; Morency, L.P.; Zylber, M.I.; Najmanovich, R.J. Large-scale detection of drug off-targets: Hypotheses for drug repurposing and understanding side-effects. BMC Pharm. Toxicol. 2017, 18, 18. [Google Scholar] [CrossRef]
- Trofimov, V.A.; Varentsova, S.A. Essential Limitations of the Standard THz TDS Method for Substance Detection and Identification and a Way of Overcoming Them. Sensors 2016, 16, 502. [Google Scholar] [CrossRef]
- Law, S.; Panwar, P.; Li, J.; Aguda, A.H.; Jamroz, A.; Guido, R.V.C.; Bromme, D. A composite docking approach for the identification and characterization of ectosteric inhibitors of cathepsin K. PLoS ONE 2017, 12, e0186869. [Google Scholar] [CrossRef] [PubMed]
- Pabon, N.A.; Xia, Y.; Estabrooks, S.K.; Ye, Z.; Herbrand, A.K.; Suss, E.; Biondi, R.M.; Assimon, V.A.; Gestwicki, J.E.; Brodsky, J.L.; et al. Predicting protein targets for drug-like compounds using transcriptomics. PLoS Comput. Biol. 2018, 14, e1006651. [Google Scholar] [CrossRef]
- Bull, S.C.; Doig, A.J. Properties of protein drug target classes. PLoS ONE 2015, 10, e0117955. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.; Caricasole, A.; Gaviraghi, G.; Pollio, G.; Robertson, G.; Terstappen, G.C.; Salerno, M.; Tunici, P. How to achieve confidence in drug discovery and development: Managing risk (from a reductionist to a holistic approach). ChemMedChem 2009, 4, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Krieger, J.M.; Li, H.; Bahar, I. Pharmmaker: Pharmacophore modeling and hit identification based on druggability simulations. Protein Sci. 2019, 29, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef]
- Jain, S.S.; Luiken, J.J.; Snook, L.A.; Han, X.X.; Holloway, G.P.; Glatz, J.F.; Bonen, A. Fatty acid transport and transporters in muscle are critically regulated by Akt2. FEBS Lett. 2015, 589, 2769–2775. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, M.; He, J.; Zhu, M.; Wang, J.C.; Jin, L.; Wang, X.F.; Yang, Y.J.; Xiang, J.Q.; Wei, Q. Polymorphisms in the AKT1 and AKT2 genes and oesophageal squamous cell carcinoma risk in an Eastern Chinese population. J. Cell. Mol. Med. 2016, 20, 666–677. [Google Scholar] [CrossRef]
- Liao, P.; Wang, W.; Li, Y.; Wang, R.; Jin, J.; Pang, W.; Chen, Y.; Shen, M.; Wang, X.; Jiang, D.; et al. Palmitoylated SCP1 is targeted to the plasma membrane and negatively regulates angiogenesis. Elife 2017, 6, e22058. [Google Scholar] [CrossRef]
- Hellesoy, M.; Lorens, J.B. Cellular context-mediated Akt dynamics regulates MAP kinase signaling thresholds during angiogenesis. Mol. Biol. Cell 2015, 26, 2698–2711. [Google Scholar] [CrossRef]
- Huang, J.J.; Shi, Y.Q.; Li, R.L.; Hu, A.; Lu, Z.Y.; Weng, L.; Wang, S.Q.; Han, Y.P.; Zhang, L.; Li, B.; et al. Angiogenesis effect of therapeutic ultrasound on HUVECs through activation of the PI3K-Akt-eNOS signal pathway. Am. J. Transl. Res. 2015, 7, 1106–1115. [Google Scholar] [PubMed]
- Tandon, M.; Chen, Z.; Othman, A.H.; Pratap, J. Role of Runx2 in IGF-1Rbeta/Akt- and AMPK/Erk-dependent growth, survival and sensitivity towards metformin in breast cancer bone metastasis. Oncogene 2016, 35, 4730–4740. [Google Scholar] [CrossRef]
- Tandon, M.; Chen, Z.; Pratap, J. Role of Runx2 in crosstalk between Mek/Erk and PI3K/Akt signaling in MCF-10A cells. J. Cell. Biochem. 2014, 115, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Tandon, M.; Chen, Z.; Pratap, J. Runx2 activates PI3K/Akt signaling via mTORC2 regulation in invasive breast cancer cells. Breast Cancer Res. 2014, 16, R16. [Google Scholar] [CrossRef] [PubMed]
- Tandon, M.; Othman, A.H.; Ashok, V.; Stein, G.S.; Pratap, J. The role of Runx2 in facilitating autophagy in metastatic breast cancer cells. J. Cell. Physiol. 2017, 233, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.I.; Maniar, K.; Lydon, J.P.; Kim, J.J. Akt regulates progesterone receptor B-dependent transcription and angiogenesis in endometrial cancer cells. Oncogene 2016, 35, 5191–5201. [Google Scholar] [CrossRef]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Puzanov, G.A.; Afanasyeva, M.A.; Dashinimaev, E.B.; Vishnyakova, K.S.; Beniaminov, A.D.; Adzhubei, A.A.; Kondratieva, T.T.; Yegorov, Y.E.; Senchenko, V.N. Tumor suppressor properties of the small C-terminal domain phosphatases in non-small cell lung cancer. Biosci. Rep. 2019, 39, BSR20193094. [Google Scholar] [CrossRef]
- Dai, M.; Al-Odaini, A.A.; Arakelian, A.; Rabbani, S.A.; Ali, S.; Lebrun, J.J. A novel function for p21Cip1 and acetyltransferase p/CAF as critical transcriptional regulators of TGFbeta-mediated breast cancer cell migration and invasion. Breast Cancer Res. 2012, 14, R127. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lu, L.T.; Chen, H.Y.; Duan, X.; Lin, X.; Feng, X.H.; Tang, M.J.; Chen, R.H. SCP phosphatases suppress renal cell carcinoma by stabilizing PML and inhibiting mTOR/HIF signaling. Cancer Res. 2014, 74, 6935–6946. [Google Scholar] [CrossRef]
- Khan, M.A.; Tania, M.; Wei, C.; Mei, Z.; Fu, S.; Cheng, J.; Xu, J.; Fu, J. Thymoquinone inhibits cancer metastasis by downregulating TWIST1 expression to reduce epithelial to mesenchymal transition. Oncotarget 2015, 6, 19580–19591. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y. Emerging Roles of CTD Phosphatases. J. Life Sci. 2017, 27, 370–381. [Google Scholar] [CrossRef]
- Yeo, M.; Lin, P.S. Functional characterization of small CTD phosphatases. Methods Mol. Biol. 2007, 365, 335–346. [Google Scholar] [PubMed]
- Thompson, J.; Lepikhova, T.; Teixido-Travesa, N.; Whitehead, M.A.; Palvimo, J.J.; Janne, O.A. Small carboxyl-terminal domain phosphatase 2 attenuates androgen-dependent transcription. EMBO J. 2006, 25, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Kloet, D.E.; Polderman, P.E.; Eijkelenboom, A.; Smits, L.M.; van Triest, M.H.; van den Berg, M.C.; Groot Koerkamp, M.J.; van Leenen, D.; Lijnzaad, P.; Holstege, F.C.; et al. FOXO target gene CTDSP2 regulates cell cycle progression through Ras and p21(Cip1/Waf1). Biochem. J. 2015, 469, 289–298. [Google Scholar] [CrossRef]
- Kim, Y.; Gentry, M.S.; Harris, T.E.; Wiley, S.E.; Lawrence, J.C., Jr.; Dixon, J.E. A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6596–6601. [Google Scholar] [CrossRef]
- Ghosh, A.; Shuman, S.; Lima, C.D. The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol. Cell 2008, 32, 478–490. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Kim, Y.; Dixon, J.E.; Pfaff, S.L.; Gill, G.N.; Noel, J.P.; Zhang, Y. Structural and functional analysis of the phosphoryl transfer reaction mediated by the human small C-terminal domain phosphatase, Scp1. Protein Sci. 2010, 19, 974–986. [Google Scholar] [CrossRef]
- Almo, S.C.; Bonanno, J.B.; Sauder, J.M.; Emtage, S.; Dilorenzo, T.P.; Malashkevich, V.; Wasserman, S.R.; Swaminathan, S.; Eswaramoorthy, S.; Agarwal, R.; et al. Structural genomics of protein phosphatases. J. Struct. Funct. Genom. 2007, 8, 121–140. [Google Scholar] [CrossRef]
- Schwer, B.; Ghosh, A.; Sanchez, A.M.; Lima, C.D.; Shuman, S. Genetic and structural analysis of the essential fission yeast RNA polymerase II CTD phosphatase Fcp1. RNA 2015, 21, 1135–1146. [Google Scholar] [CrossRef]
- Yun, J.H.; Ko, S.; Lee, C.K.; Cheong, H.K.; Cheong, C.; Yoon, J.B.; Lee, W. Solution structure and Rpn1 interaction of the UBL domain of human RNA polymerase II C-terminal domain phosphatase. PLoS ONE 2013, 8, e62981. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Engel, J.L.; Xiao, J.; Tagliabracci, V.S.; Wang, X.; Huang, L.; Dixon, J.E. UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc. Natl. Acad. Sci. USA 2011, 108, 18649–18654. [Google Scholar] [CrossRef] [PubMed]
- McConnell, J.L.; Wadzinski, B.E. Targeting protein serine/threonine phosphatases for drug development. Mol. Pharm. 2009, 75, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.V.; Singh, S.K. Identification of Potential Dual Negative Allosteric Modulators of Group I mGluR Family: A Shape Based Screening, ADME Prediction, Induced Fit Docking and Molecular Dynamics Approach Against Neurodegenerative Diseases. Curr. Top. Med. Chem. 2019, 19, 2687–2707. [Google Scholar] [CrossRef] [PubMed]
- SarathKumar, B.; Lakshmi, B.S. In silico investigations on the binding efficacy and allosteric mechanism of six different natural product compounds towards PTP1B inhibition through docking and molecular dynamics simulations. J. Mol. Model. 2019, 25, 272. [Google Scholar] [CrossRef]
- Ivasiv, V.; Albertini, C.; Goncalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, S.; Ying, T. From therapeutic antibodies to chimeric antigen receptors (CARs): Making better CARs based on antigen-binding domain. Expert Opin. Biol. 2016, 16, 1469–1478. [Google Scholar] [CrossRef]
- Fraczek, J.; Vanhaecke, T.; Rogiers, V. Toxicological and metabolic considerations for histone deacetylase inhibitors. Expert Opin. Drug Metab. Toxicol. 2013, 9, 441–457. [Google Scholar] [CrossRef]
- Kumar, S.; Tiwari, M. Variable selection based QSAR modeling on Bisphenylbenzimidazole as Inhibitor of HIV-1 reverse transcriptase. Med. Chem. 2012, 9, 955–967. [Google Scholar] [CrossRef]
- Faivre, S.; Sablin, M.P.; Dreyer, C.; Raymond, E. Novel anticancer agents in clinical trials for well-differentiated neuroendocrine tumors. Endocrinol. Metab. Clin. N. Am. 2010, 39, 811–826. [Google Scholar] [CrossRef]
- Selz, K.A.; Samoylova, T.I.; Samoylov, A.M.; Vodyanoy, V.J.; Mandell, A.J. Designing allosteric peptide ligands targeting a globular protein. Biopolymers 2007, 85, 38–59. [Google Scholar] [CrossRef] [PubMed]
- Stains, C.I.; Mondal, K.; Ghosh, I. Molecules that target beta-amyloid. ChemMedChem 2007, 2, 1674–1692. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.D.; Clarke, W.P.; von Zastrow, M.; Nichols, D.E.; Kobilka, B.; Weinstein, H.; Javitch, J.A.; Roth, B.L.; Christopoulos, A.; Sexton, P.M.; et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharm. Exp. 2007, 320, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Leibovici, L. Combination antimicrobial treatment versus monotherapy: The contribution of meta-analyses. Infect. Dis. Clin. N. Am. 2009, 23, 277–293. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, W.; Zhang, L.; Ying, Z.; Sha, O.; Li, C.; Lu, L.; Chen, X.; Li, Z.; Niu, F.; et al. The novel targets of DL-3-n-butylphthalide predicted by similarity ensemble approach in combination with molecular docking study. Quant. Imaging Med. Surg. 2017, 7, 532–536. [Google Scholar] [CrossRef]
- Ramirez, D.; Caballero, J. Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Coates, A.S.; Gelber, R.D.; Glick, J.H.; Thurlimann, B.; Senn, H.J. First—Select the target: Better choice of adjuvant treatments for breast cancer patients. Ann. Oncol. 2006, 17, 1772–1776. [Google Scholar] [CrossRef]
- Evers, A.; Klabunde, T. Structure-based drug discovery using GPCR homology modeling: Successful virtual screening for antagonists of the alpha1A adrenergic receptor. J. Med. Chem. 2005, 48, 1088–1097. [Google Scholar] [CrossRef]
- Arrondeau, J.; Huillard, O.; Tlemsani, C.; Cessot, A.; Boudou-Rouquette, P.; Blanchet, B.; Thomas-Schoemann, A.; Vidal, M.; Tigaud, J.M.; Durand, J.P.; et al. Investigational therapies up to Phase II which target PDGF receptors: Potential anti-cancer therapeutics. Expert Opin. Investig. Drugs 2015, 24, 673–687. [Google Scholar] [CrossRef]
- Chaput, L.; Martinez-Sanz, J.; Quiniou, E.; Rigolet, P.; Saettel, N.; Mouawad, L. vSDC: A method to improve early recognition in virtual screening when limited experimental resources are available. J. Cheminform. 2016, 8, 1. [Google Scholar] [CrossRef]
- Manning, A.; Highland, H.M.; Gasser, J.; Sim, X.; Tukiainen, T.; Fontanillas, P.; Grarup, N.; Rivas, M.A.; Mahajan, A.; Locke, A.E.; et al. A Low-Frequency Inactivating AKT2 Variant Enriched in the Finnish Population Is Associated With Fasting Insulin Levels and Type 2 Diabetes Risk. Diabetes 2017, 66, 2019–2032. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Tanaka, S. Cosolvent-Based Molecular Dynamics for Ensemble Docking: Practical Method for Generating Druggable Protein Conformations. J. Chem. Inf. Model. 2017, 57, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.P.; de Denus, S.; Tardif, J.C. Pharmacogenomics to Revive Drug Development in Cardiovascular Disease. Cardiovasc. Drugs 2016, 30, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2010, 25, 13–19. [Google Scholar] [CrossRef]
- Mbonye, A.K.; Magnussen, P.; Chandler, C.I.; Hansen, K.S.; Lal, S.; Cundill, B.; Lynch, C.A.; Clarke, S.E. Introducing rapid diagnostic tests for malaria into drug shops in Uganda: Design and implementation of a cluster randomized trial. Trials 2014, 15, 303. [Google Scholar] [CrossRef]
- Boucherit, H.; Chikhi, A.; Bensegueni, A.; Merzoug, A.; Bolla, J.M. The Research of New Inhibitors of Bacterial Methionine Aminopeptidase by Structure Based Virtual Screening Approache OF ZINC DATABASE and in vitro Validation. Curr. Comput. Aided Drug Des. 2019. (Epub ahead of print). [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Chessari, G.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Nissink, J.W.; Taylor, R.D.; Taylor, R. Modeling water molecules in protein-ligand docking using GOLD. J. Med. Chem. 2005, 48, 6504–6515. [Google Scholar] [CrossRef]
- Zhang, H.; Liao, L.; Saravanan, K.M.; Yin, P.; Wei, Y. DeepBindRG: A deep learning based method for estimating effective protein-ligand affinity. Peer J. 2019, 7, e7362. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva-Jr, F.P. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef]
- Lindh, M.; Svensson, F.; Schaal, W.; Zhang, J.; Skold, C.; Brandt, P.; Karlen, A. Toward a Benchmarking Data Set Able to Evaluate Ligand- and Structure-based Virtual Screening Using Public HTS Data. J. Chem. Inf. Model. 2015, 55, 343–353. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Christopoulos, A.; Carmeliet, P. Allosteric targeting of receptor tyrosine kinases. Nat. Biotechnol. 2014, 32, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.M.; Lim, C.S.; Bishop, A.C. Targeting a cryptic allosteric site for selective inhibition of the oncogenic protein tyrosine phosphatase Shp2. Biochemistry 2014, 54, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Rooklin, D.; Yang, D.; Liang, X.; Li, K.; Lu, J.; Wang, C.; Xiao, P.; Zhang, Y.; Sun, J.P.; et al. Computational Strategy for Bound State Structure Prediction in Structure-Based Virtual Screening: A Case Study of Protein Tyrosine Phosphatase Receptor Type O Inhibitors. J. Chem. Inf. Model. 2018, 58, 2331–2342. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.H.; Kim, H.; Cha, S.; Lee, B.; Kim, Y.J. Structure-Based Virtual Screening of Protein Tyrosine Phosphatase Inhibitors: Significance, Challenges, and Solutions. J. Microbiol. Biotechnol. 2017, 27, 878–895. [Google Scholar] [PubMed]
- Lamba, V.; Ghosh, I. New directions in targeting protein kinases: Focusing upon true allosteric and bivalent inhibitors. Curr. Pharm. Des. 2012, 18, 2936–2945. [Google Scholar] [CrossRef]
- Cossins, B.P.; Lawson, A.D. Small Molecule Targeting of Protein-Protein Interactions through Allosteric Modulation of Dynamics. Molecules 2015, 20, 16435–16445. [Google Scholar] [CrossRef]
- Grigoriadis, D.E.; Hoare, S.R.; Lechner, S.M.; Slee, D.H.; Williams, J.A. Drugability of extracellular targets: Discovery of small molecule drugs targeting allosteric, functional, and subunit-selective sites on GPCRs and ion channels. Neuropsychopharmacology 2009, 34, 106–125. [Google Scholar] [CrossRef]
- Hogg, P.J. Targeting allosteric disulphide bonds in cancer. Nat. Rev. Cancer 2013, 13, 425–431. [Google Scholar] [CrossRef]
- Jadaun, A.; Subbarao, N.; Dixit, A. Allosteric inhibition of topoisomerase I by pinostrobin: Molecular docking, spectroscopic and topoisomerase I activity studies. J. Photochem. Photobiol. B 2017, 167, 299–308. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Therapy 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharm. Sin. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wang, Y.; Wang, M.; Wang, J.; Li, N. Computational investigation of imidazopyridine analogs as protein kinase B (Akt1) allosteric inhibitors by using 3D-QSAR, molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sheik Amamuddy, O.; Veldman, W.; Manyumwa, C.; Khairallah, A.; Agajanian, S.; Oluyemi, O.; Verkhivker, G.; Tastan Bishop, O. Integrated Computational Approaches and Tools forAllosteric Drug Discovery. Int. J. Mol. Sci. 2020, 21, 847. [Google Scholar] [CrossRef]
- Zhang, W.; Li, R.; Shin, R.; Wang, Y.; Padmalayam, I.; Zhai, L.; Krishna, N.R. Identification of the binding site of an allosteric ligand using STD-NMR, docking, and CORCEMA-ST calculations. ChemMedChem 2013, 8, 1629–1633. [Google Scholar] [CrossRef]
- Ramirez, U.D.; Myachina, F.; Stith, L.; Jaffe, E.K. Docking to large allosteric binding sites on protein surfaces. Adv. Exp. Med. Biol. 2010, 680, 481–488. [Google Scholar]
- Li, S.; Zhang, J.; Lu, S.; Huang, W.; Geng, L.; Shen, Q. The mechanism of allosteric inhibition of protein tyrosine phosphatase 1B. PLoS ONE 2014, 9, e97668. [Google Scholar] [CrossRef]
- Craig, I.R.; Essex, J.W.; Spiegel, K. Ensemble docking into multiple crystallographically derived protein structures: An evaluation based on the statistical analysis of enrichments. J. Chem. Inf. Model. 2010, 50, 511–524. [Google Scholar] [CrossRef]
- Rueda, M.; Bottegoni, G.; Abagyan, R. Recipes for the selection of experimental protein conformations for virtual screening. J. Chem. Inf. Model. 2009, 50, 186–193. [Google Scholar] [CrossRef]
- Rao, S.; Sanschagrin, P.C.; Greenwood, J.R.; Repasky, M.P.; Sherman, W.; Farid, R. Improving database enrichment through ensemble docking. J. Comput. Aided Mol. Des. 2008, 22, 621–627. [Google Scholar] [CrossRef]
- Campbell, A.J.; Lamb, M.L.; Joseph-McCarthy, D. Ensemble-based docking using biased molecular dynamics. J. Chem. Inf. Model. 2014, 54, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Polgar, T.; Keseru, G.M. Ensemble docking into flexible active sites. Critical evaluation of FlexE against JNK-3 and beta-secretase. J. Chem. Inf. Model. 2006, 46, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Rallabandi, H.R.; Lee, D.; Sung, J.; Kim, Y.J. Peripheral Inhibition of Small C-terminal Domain Phosphatase 1 with Napthoquinone Analogues. Bull. Korean Chem. Soc. 2020, in press. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTD Small Phosphatase | Aliases | Substrates | PDB IDs | Biological Roles [References] |
|---|---|---|---|---|
| CTDSP1 | SCP1 NIF3 | RNAP II CTD REST R-SMADs CDCA3, RB TWIST1, AKT PML, c-Myc | 4YGY, 4YH1, 3PGL, 3L0B, 3L0C, 3L0Y, 2GHQ, 2GHT, 1T9Z, 1TA0, 3PGL | Transcription factor recruitment [5,93] Neuronal gene silencing [9,10] Osteoblast differentiation [12] Cell cycle regulation [13,14] Tumor suppressor [88,89,90,91] |
| CTDSP2 | SCP2 OS4 NIF2 | RNAP II CTD R-SMADs PML | 2Q5E | Transcription factor recruitment [5,94] Promoter clearance [94] Ras activation [95] |
| CTDSPL | SCP3 HYA22 NIF1 | RNAP II CTD R-SMADs PML | 2HHL | Transcription factor recruitment [5] Tumor suppressor [90] |
| Docking Tool | Algorithm | Scoring Function | Representative References |
|---|---|---|---|
| Autodock | Genetic algorithm, Simulated annealing, Lamarckian algorithm | Force field, Empirical | [55] |
| DOCK 4.0 | Incremental search, Shape fitting | Force field | [54] |
| GOLD | Genetic algorithm | Force field | [57] |
| Flex-X | Multiple copy, Simultaneous search, Incremental search | Empirical | [58] |
| ICM | Monte Carlo sampling | Empirical | [59] |
| SLIDE | Incremental construction | Force field, Empirical | [60] |
| GLIDE | Monte Carlo sampling | Empirical | [61,62] |
| GOLD/ASP | Genetic algorithm | Knowledge-based | [127,128] |
| DeepBindRG | Deep learning algorithm | Machine learning | [129] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rallabandi, H.R.; Ganesan, P.; Kim, Y.J. Targeting the C-Terminal Domain Small Phosphatase 1. Life 2020, 10, 57. https://doi.org/10.3390/life10050057
Rallabandi HR, Ganesan P, Kim YJ. Targeting the C-Terminal Domain Small Phosphatase 1. Life. 2020; 10(5):57. https://doi.org/10.3390/life10050057
Chicago/Turabian StyleRallabandi, Harikrishna Reddy, Palanivel Ganesan, and Young Jun Kim. 2020. "Targeting the C-Terminal Domain Small Phosphatase 1" Life 10, no. 5: 57. https://doi.org/10.3390/life10050057
APA StyleRallabandi, H. R., Ganesan, P., & Kim, Y. J. (2020). Targeting the C-Terminal Domain Small Phosphatase 1. Life, 10(5), 57. https://doi.org/10.3390/life10050057