A Possible Primordial Acetyleno/Carboxydotrophic Core Metabolism




Abstract
1. Introduction
2. Materials and Methods
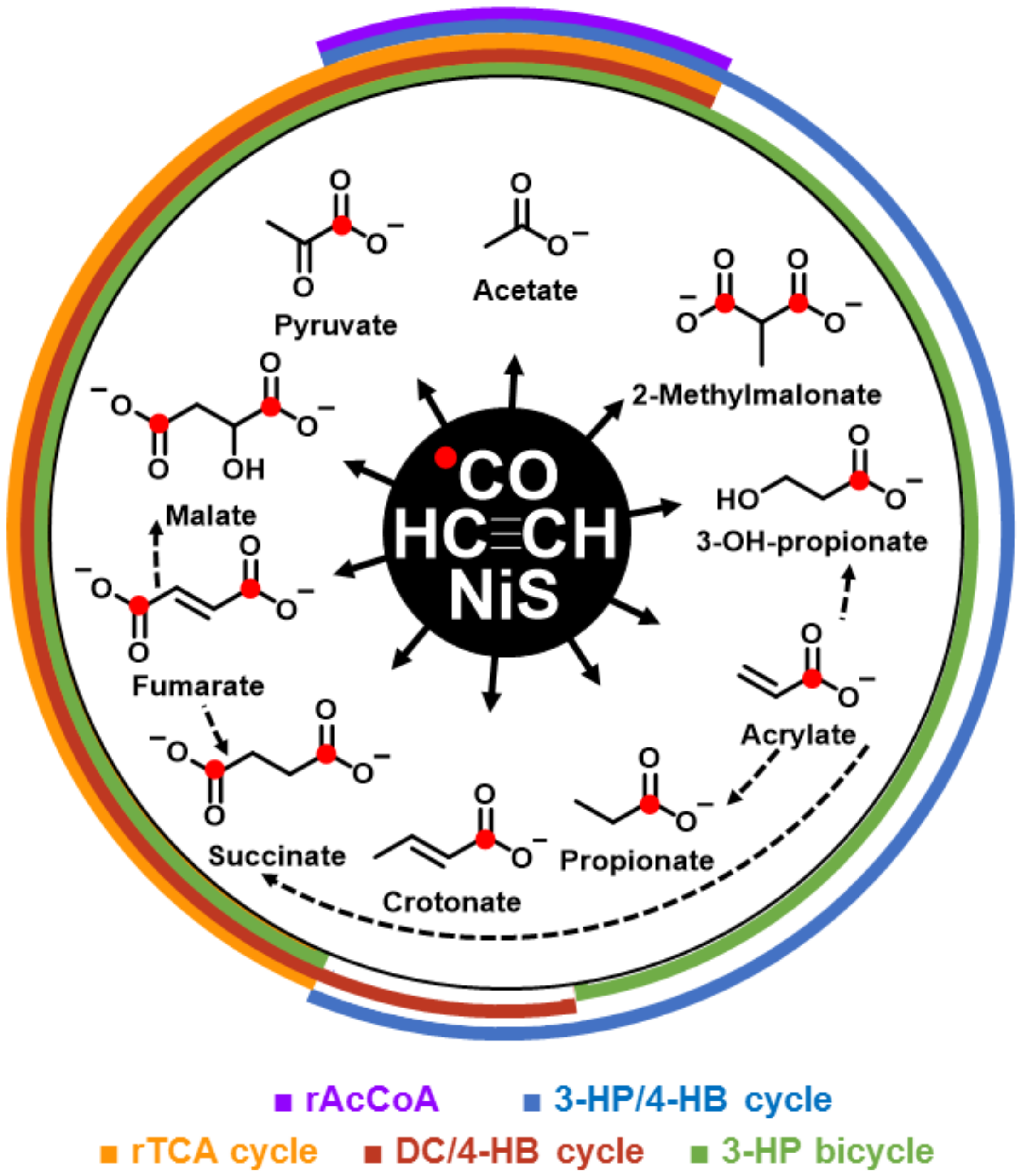
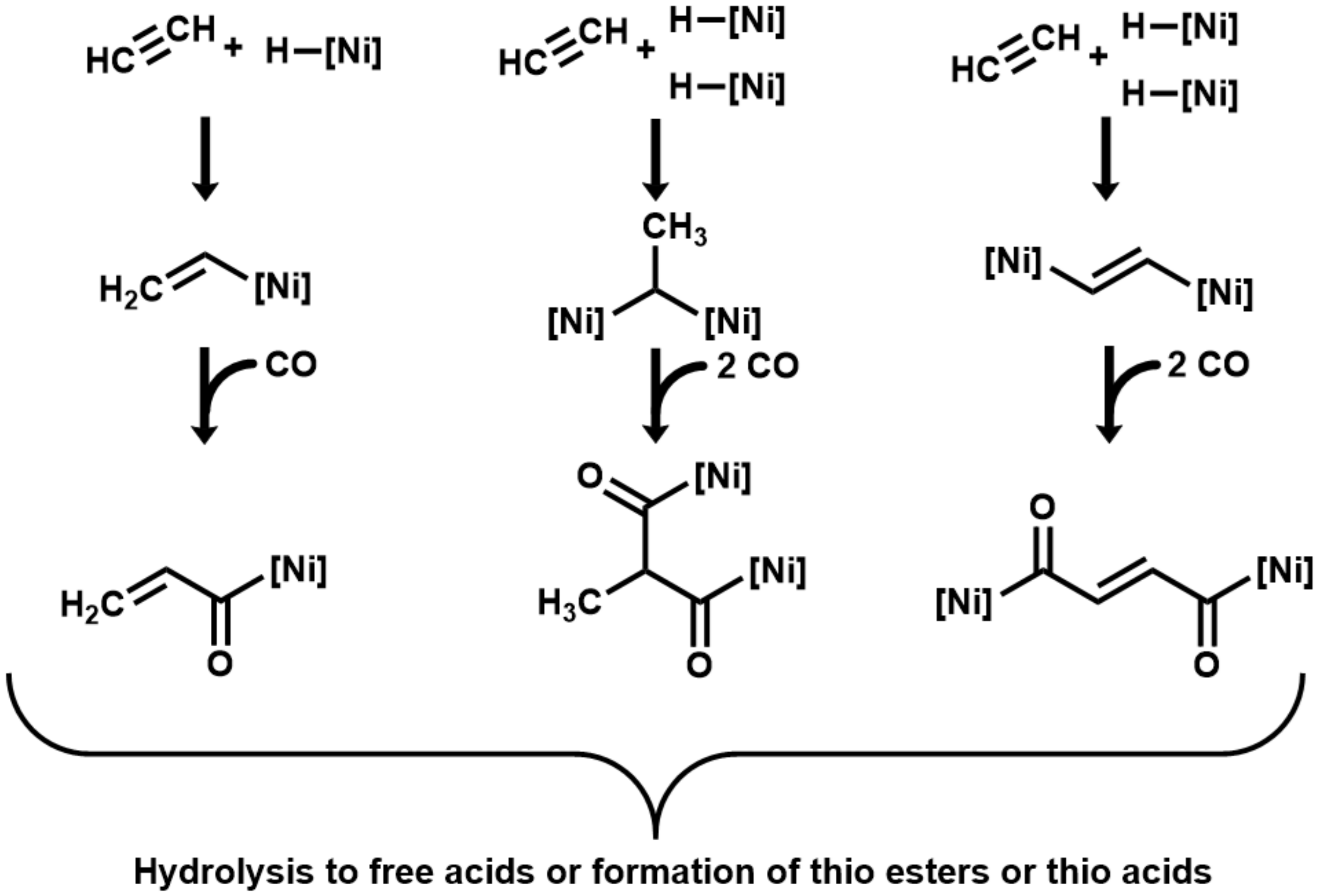
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Braakman, R.; Smith, E. The emergence and early evolution of biological carbon-fixation. PLoS Comput. Biol. 2012, 8, e1002455. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.J.; Muchowska, K.B.; Chatelain, P.; Moran, J. Native iron reduces CO2 to intermediates and end-products of the acetyl-CoA pathway. Nat. Ecol. Evol. 2018, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Preiner, M.; Igarashi, K.; Muchowska, K.B.; Yu, M.; Varma, S.J.; Kleinermanns, K.; Nobu, M.K.; Kamagata, Y.; Tüysüz, H.; Moran, J.; et al. A hydrogen-dependent geochemical analogue of primordial carbon and energy metabolism. Nat. Ecol. Evol. 2020, 4, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Mukhin, L.M. Volcanic processes and synthesis of simple organic compounds on primitive earth. Orig. Life Evol. Biosph. 1976, 7, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Igari, S.; Maekawa, T.; Sakata, S. Light hydrocarbons in fumarolic gases: A case study in the kakkonda geothermal area. Chikyukagau 2000, 34, 103–109. [Google Scholar]
- Oremland, R.S.; Voytek, M.A. Acetylene as fast food: Implications for development of life on anoxic primordial earth and in the outer solar system. Astrobiology 2008, 8, 45–58. [Google Scholar] [CrossRef]
- Hollemann, A.F.; Wiberg, E.; Wiberg, N. Lehrbuch der Anorganischen Chemie, 102nd ed.; Walter de Gryter: Berlin, Germany, 2007; pp. 1243–1247. [Google Scholar]
- Geisberger, T.; Diederich, P.; Steiner, T.; Eisenreich, W.; Schmitt-Kopplin, P.; Huber, C. Evolutionary steps in the analytics of primordial metabolic evolution. Life 2019, 9, 50. [Google Scholar] [CrossRef]
- Huber, C.; Kraus, F.; Hanzlik, M.; Eisenreich, W.; Wächtershäuser, G. Elements of metabolic evolution. Chemistry 2012, 18, 2063–2080. [Google Scholar] [CrossRef]
- Scheidler, C.; Sobotta, J.; Eisenreich, W.; Wächtershäuser, G.; Huber, C. Unsaturated C-3,5,7,9-monocarboxylic acids by aqueous, one-pot carbon fixation: Possible relevance for the origin of life. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Ljungdahl, L.G.; Wood, H.G. Total synthesis of acetate from co2 by heterotrophic bacteria. Annu. Rev. Microbiol. 1969, 23, 515–538. [Google Scholar] [CrossRef]
- Huber, C.; Wächtershäuser, G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 1997, 276, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Heinen, W.; Lauwers, A.M. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig. Life Evol. Biosph. 1996, 26, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Stupperich, E.; Eden, G. Autotrophic CO2 fixation in Chlorobium limicola. Evidence for the operation of a reductive tricarboxylic acid cycle in growing cells. Arch. Microbiol. 1980, 128, 64–71. [Google Scholar] [CrossRef]
- Kockelkorn, D.; Fuchs, G. Malonic semialdehyde reductase, succinic semialdehyde reductase, and succinyl-coenzyme a reductase from Metallosphaera sedula: Enzymes of the autotrophic 3-Hydroxypropionate/4-Hydroxybutyrate cycle in sulfolobales. J. Bacteriol. 2009, 191, 6352. [Google Scholar] [CrossRef][Green Version]
- Huber, H.; Gallenberger, M.; Jahn, U.; Eylert, E.; Berg, I.A.; Kockelkorn, D.; Eisenreich, W.; Fuchs, G. A dicarboxylate/4-hydroxybutyrate autotrophic carbon assimilation cycle in the hyperthermophilic Archaeum Ignicoccus hospitalis. Proc. Natl. Acad. Sci. USA 2008, 105, 7851–7856. [Google Scholar] [CrossRef]
- Strauss, G.; Fuchs, G. Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3-hydroxypropionate cycle. Eur. J. Biochem. 1993, 215, 633–643. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Evolution of the first metabolic cycles. Proc. Natl. Acad. Sci. USA 1990, 87, 200. [Google Scholar] [CrossRef]
- Fuchs, G. Alternative pathways of carbon dioxide fixation: Insights into the early evolution of life? Annu. Rev. Microbiol. 2011, 65, 631–658. [Google Scholar] [CrossRef]
- Berg, I.A. Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 2011, 77, 1925–1936. [Google Scholar] [CrossRef]
- Hügler, M.; Sievert, S.M. Beyond the calvin cycle: Autotrophic carbon fixation in the ocean. Annu. Rev. Mar. Sci. 2011, 3, 261–289. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Varma, S.J.; Chevallot-Beroux, E.; Lethuillier-Karl, L.; Li, G.; Moran, J. Metals promote sequences of the reverse krebs cycle. Nat. Ecol. Evol. 2017, 1, 1716–1721. [Google Scholar] [CrossRef]
- Keller, M.A.; Kampjut, D.; Harrison, S.A.; Ralser, M. Sulfate radicals enable a non-enzymatic krebs cycle precursor. Nat. Ecol. Evol. 2017, 1, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Synthesis and breakdown of universal metabolic precursors promoted by iron. Nature 2019, 569, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.M.; Thamdrup, B.; Dalsgaard, T. Effects of specific inhibitors on anammox and denitrification in marine sediments. Appl. Environ. Microbiol. 2007, 73, 3151–3158. [Google Scholar] [CrossRef] [PubMed]
- Schink, B.; Kroneck, P.M. Exploring the active site of the tungsten, iron-sulfur enzyme acetylene hydratase. J. Bacteriol. 2011, 193, 1229–1236. [Google Scholar] [CrossRef]
- Rosner, B.M.; Schink, B. Purification and characterization of acetylene hydratase of Pelobacter acetylenicus, a tungsten iron-sulfur protein. J. Bacteriol. 1995, 177, 5767. [Google Scholar] [CrossRef]
- Akob, D.M.; Sutton, J.M.; Fierst, J.L.; Haase, K.B.; Baesman, S.; Luther, G.W., III; Miller, L.G.; Oremland, R.S. Acetylenotrophy: A hidden but ubiquitous microbial metabolism? FEMS Microbiol. Ecol. 2018, 94, fiy103. [Google Scholar] [CrossRef]
- Holloway, J.R.; Blank, J.G. Application of experimental results to coh species in natural melts. Rev. Mineral. 1994, 30, 187. [Google Scholar]
- Wächtershäuser, G. On the chemistry and evolution of the pioneer organism. Chem. Biodivers. 2007, 4, 584–602. [Google Scholar] [CrossRef]
- Cox, P.A. The elements. Their origin, abundance, and distribution. In The Elements. Their Origin, Abundance, and Distribution; Cox, P.A., Ed.; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Hazen, R.M. Evolution of minerals. Sci. Am. 2010, 302, 58–65. [Google Scholar] [CrossRef]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Primordial reductive amination revisited. Tetrahedron Lett. 2003, 44, 1695–1697. [Google Scholar] [CrossRef]
- Bernardi, F.; Bottoni, A.; Nicastro, M.; Rossi, I.; Novoa, J.; Prat, X. Theoretical study of the mechanism of carbonyl insertion reactions catalyzed by nickel complexes. Organometallics 2000, 19, 2170–2178. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Runs | A | B | C | D | |
|---|---|---|---|---|---|
| NiSO4 • 6 H2O (mmol) | 1.0 | 0.5 | - | 0.5 | |
| FeSO4 • 7 H2O (mmol) | - | - | - | 0.5 | |
| β-Ni(OH)2 (mmol) | - | 0.5 | 1.0 | - | |
| Na2SO4 (mmol) | - | 0.5 | 1.0 | - | |
| Na2S • 9 H2O (mmol) | 1.0 | 0.5 | - | 1.0 | |
| NaOH (mmol) | 0.5 | 0.5 | 0.5 | 0.5 | |
| CO (ml) | 60 | 60 | 60 | 60 | |
| C2H2 (ml) | 60 | 60 | 60 | 60 | |
| pH end | 8.0 | 8.1 | 9.8 | 8.5 | |
| Products (µM) | Chemical formula | ||||
| C1 | |||||
| formate | HCOO- | 18983 | 24207 | 310 | 434 |
| C2 | |||||
| acetate | CH3COO- | 4358 | 3434 | 112 | 749 |
| glycolate | HOCH2COO- | 32 | 38 | n.d. | 11 |
| C3 | |||||
| acrylate | CH2CHCOO- | 9692 | 16874 | 243 | 763 |
| propionate | CH3CH2COO- | 10368 | 15021 | 171 | 339 |
| pyruvate | CH3COCOO- | 43 | 117 | n.d. | 4 |
| β-lactate | HOCH2CH2COO- | 273 | 793 | n.d. | n.d. |
| glycerate | HOCH2CH2(OH)COO- | 108 | 102 | n.d. | n.d. |
| C4 | |||||
| crotonate | CH3CHCHCOO- | 226 | 516 | n.d. | 22 |
| 2-methylmalonate | -OOCCH(CH3)COO- | 48 | 145 | n.d. | n.d. |
| maleate | -OOCCHCHCOO- | 72 | 585 | n.d. | 14 |
| succinate | -OOCCH2CH2COO- | 3964 | 4747 | 3 | 187 |
| fumarate | -OOCCHCHCOO- | 358 | 391 | n.d. | 12 |
| malate | -OOCCH(OH)CH2COO- | 17 | 85 | n.d. | n.d. |
| C5 | |||||
| (E)-2-methylbut-2-enoate | CH3CHC(CH3)COO- | 196 | 411 | n.d. | n.d. |
| ∑ C2–C5 | 29755 | 43259 | 358 | 2101 |
| Runs | A | B | Labelling in Characteristic Fragments | |
|---|---|---|---|---|
| Mass 1 | Mass 2 | |||
| NiSO4 • 6 H2O (mmol) | 2 | 2 | ||
| Na2S • 9 H2O (mmol) | 1.5 | 1.5 | ||
| NaOH (mmol) | 0.6 | 0.6 | ||
| C2H2 (ml) | 90 | 45 | ||
| CO (ml) | - | 45 | ||
| CH3SH (ml) | 25 | 25 | ||
| Methyl thioacetate (µM) | 2 | 4 | 90_3^0• | 43_3^0• |
| Runs | D | E | F | G | |
|---|---|---|---|---|---|
| Substrate | fumarate | malate | acrylate | succinate | |
| Product (%) | Chemical formula | ||||
| acrylate | CH2CHCOO- | n.d. | n.d. | 6.37 | n.d. |
| propionate | CH3CH2COO- | n.d. | n.d. | 6.80 | n.d. |
| β-lactate | HOCH2CH2COO- | n.d. | n.d. | 39.21 | n.d. |
| maleate | -OOCCHCHCOO- | 2.57 | 0.02 | 0.31 | n.d. |
| succinate | -OOCCH2CH2COO- | 71.67 | 1.13 | 43.83 | 99.82 |
| fumarate | -OOCCHCHCOO- | 21.94 | 0.24 | 1.14 | 0.14 |
| malate | -OOCCH(OH)CH2COO- | 3.70 | 98.58 | 2.36 | 0.04 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobotta, J.; Geisberger, T.; Moosmann, C.; Scheidler, C.M.; Eisenreich, W.; Wächtershäuser, G.; Huber, C. A Possible Primordial Acetyleno/Carboxydotrophic Core Metabolism. Life 2020, 10, 35. https://doi.org/10.3390/life10040035
Sobotta J, Geisberger T, Moosmann C, Scheidler CM, Eisenreich W, Wächtershäuser G, Huber C. A Possible Primordial Acetyleno/Carboxydotrophic Core Metabolism. Life. 2020; 10(4):35. https://doi.org/10.3390/life10040035
Chicago/Turabian StyleSobotta, Jessica, Thomas Geisberger, Carolin Moosmann, Christopher M. Scheidler, Wolfgang Eisenreich, Günter Wächtershäuser, and Claudia Huber. 2020. "A Possible Primordial Acetyleno/Carboxydotrophic Core Metabolism" Life 10, no. 4: 35. https://doi.org/10.3390/life10040035
APA StyleSobotta, J., Geisberger, T., Moosmann, C., Scheidler, C. M., Eisenreich, W., Wächtershäuser, G., & Huber, C. (2020). A Possible Primordial Acetyleno/Carboxydotrophic Core Metabolism. Life, 10(4), 35. https://doi.org/10.3390/life10040035