Abstract
Mutations in connexins are the most common causes of hearing impairment (HI) in many populations. Our aim was to review the global burden of pathogenic and likely pathogenic (PLP) variants in connexin genes associated with HI. We conducted a systematic review of the literature based on targeted inclusion/exclusion criteria of publications from 1997 to 2020. The databases used were PubMed, Scopus, Africa-Wide Information, and Web of Science. The protocol was registered on PROSPERO, the International Prospective Register of Systematic Reviews, with the registration number “CRD42020169697”. The data extracted were analyzed using Microsoft Excel and SPSS version 25 (IBM, Armonk, New York, United States). A total of 571 independent studies were retrieved and considered for data extraction with the majority of studies (47.8% (n = 289)) done in Asia. Targeted sequencing was found to be the most common technique used in investigating connexin gene mutations. We identified seven connexin genes that were associated with HI, and GJB2 (520/571 publications) was the most studied among the seven. Excluding PLP in GJB2, GJB6, and GJA1 the other connexin gene variants (thus GJB3, GJB4, GJC3, and GJC1 variants) had conflicting association with HI. Biallelic GJB2 PLP variants were the most common and widespread variants associated with non-syndromic hearing impairment (NSHI) in different global populations but absent in most African populations. The most common GJB2 alleles found to be predominant in specific populations include; p.Gly12ValfsTer2 in Europeans, North Africans, Brazilians, and Americans; p.V37I and p.L79Cfs in Asians; p.W24X in Indians; p.L56Rfs in Americans; and the founder mutation p.R143W in Africans from Ghana, or with putative Ghanaian ancestry. The present review suggests that only GJB2 and GJB3 are recognized and validated HI genes. The findings call for an extensive investigation of the other connexin genes in many populations to elucidate their contributions to HI, in order to improve gene-disease pair curations, globally.
1. Introduction
Hearing impairment (HI) is the most common sensorineural disability worldwide, with a global prevalence of 1.3 per 1000 population [1,2]. It occurs in about 1 per 1000 live births in high-income countries, with a much higher incidence of up to 6 per 1000 in the lower-income countries [3]. According to the World Health Organization, 466 million people are living with HI and about 900 people will be affected by the year 2050 [4]. Depending on the degree of severity, HI can be classified as mild, moderate, severe, or profound when the pure tone average ranges from 26 to 40 dB, 41 to 60 dB, 61 to 80 dB or is over 81 dB, respectively (Deafness and Hearing Loss, n.d.). It is estimated that approximately 50% of congenital profound HI cases are of genetic origin [5]. If there are no other distinguishing clinical findings, HI is classified as non-syndromic [6]. About 80% of non-syndromic HI (NSHI) cases are inherited in an autosomal recessive mode, while an autosomal dominant pattern of inheritance is observed in 18% of cases [7]. In the remaining 2% of cases, the mode of inheritance is either X-linked or mitochondrial [7].
Non-syndromic HI is extremely heterogeneous, with approximately 170 loci and 121 genes identified so far [8]. Studies in European and Asian populations have identified mutations in connexin genes as the major contributors to NSHI [9,10]. Connexins (Cx) are a homogeneous family of proteins expressed in a large variety of tissues in the human body and known for their assembly into intercellular channels, called gap junctions [11]. Twenty-one different human connexin genes have been reported so far, each coding for a transmembrane protein with the same protein topology [11]. Connexins have four transmembrane domains (TM), TM1–TM4, connected by two extracellular loops (E), E1, and E2, which mediate docking [12]. The N- and C-termini, and a loop connecting TM2 and TM3 are on the cytoplasmic side of the plasma membrane [12].
Connexins are synthesized in the endoplasmic reticulum (ER) and oligomerize in the ER/Golgi or trans-Golgi network to form hexameric hemichannels or connexons [11]. Connexons are transported to the plasma membrane, where they can act as functional channels by themselves, or move to regions of cell contact and find a partner hemichannel from an adjacent cell to form a complete gap junction channel [12]. Gap junctions play an important role in cell-cell communication and homeostasis in various tissues, by mediating a direct exchange of ions and other small molecules up to 1 kDa (including a variety of second messengers, metabolites, but also small linear peptides) between the cytoplasms of adjacent cells [11].
To date, mutations in four connexin genes including GJB2 (Cx26), GJB3 (Cx31), GJB4 (Cx30.3), and GJB6 (Cx30) have been associated with sensorineural HI [13,14,15]. These four connexins were shown to be expressed in the inner ear, and some studies supported their role in potassium removal and recycling in the ear, as well as a possible role for nutrient passage in the cochlea [16]. GJB2-related sensorineural HI can occur alone or in association with hyperproliferative skin disorders, as in the case in Keratitis-ichthyosis-deafness syndrome and Bart-Pumphrey syndrome [17,18,19]. It is has been shown that digenic inheritance of recessive deafness by mutations in GJB2 and GJB6, or GJB2 and GJB3 can occur [11]. In other words, deafness can be caused by the addition of a mutation in one allele of GJB2 and one allele of GJB6 or GJB3, indicating an interaction of these connexins in the cochlea [11]. GJB6 coding region variants have been proven not to cause HI using mouse models, however, the large deletions of the GJB6 gene especially GJB6-D13S1830 were implicated as causal factors of HI. The cis-acting element upstream of GJB2 and GJB6 gene is disrupted by the large genomic deletions abolishing the expression of GJB2 gene which is responsible for the development of HI [20].
GJB2 and GJB6 genes have been well studied in Europeans and Asians, with c.35delG identified as the most prevalent GJB2 mutations associated with NSHI [9]. However, the other NSHI-causing connexin genes (i.e., GJB3 and GJB4) have not been extensively studied [11,21]. Using a systematic review approach, we provided summary data on connexin gene variants associated with HI, and specifically the global contribution of connexin genes to NSHI.
2. Results
Of the 2592 studies that were screened, 571 articles were downloaded and analyzed. The 571 articles comprised publications that dated as far back as 1997 to recent publications in 2020 (Figure 1A). The analysis suggested that there were few studies on connexin gene variants association with HI in first the three years of the study timeframe (1997, 1998, and 1999), followed by a drastic increase in the last two decades. The year 2015 recorded the highest number of publications on connexin gene variants (Figure 1A).
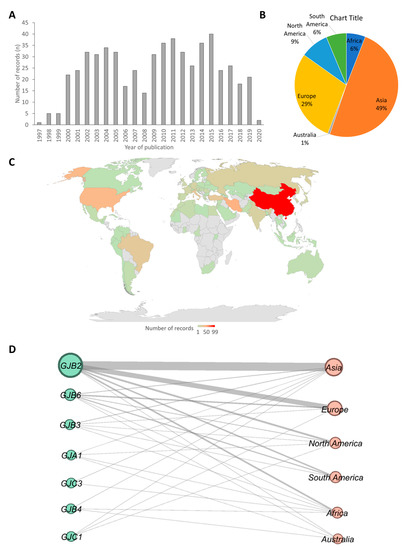
Figure 1.
Geographical distributions of the studies included in this review. (A) A bar chart showing frequency of articles by the year of publication. (B) A pie chart of distribution of articles from which data were extracted by continent. (C) A map of countries showing the number studies that reported at least one connexin gene variant. The gray regions have no record included in this study. Different shades of blue were used to represent the number of studies retrieved and reviewed per country with the darkest shade of blue as the highest number and the lightest as the smallest number. The number written on the map denotes the number of studies. The map was created in Microsoft Excel (Office 365 education license under the University of Cape Town, South Africa) (D) Network of connexin gene plotted against continents from which they were reported. The nodes on the left (green) and the right (pink) correspond to connexin genes and continents respectively. The size of the nodes and the thickness of the lines between nodes are proportional to the number of publications. The network was built using the open-source software Gephi [22]
Most of the articles retrieved were from Asia (47.8% (n = 289)) with China (99/289) recording the highest number of articles. Australia had the least number (0.7% (n = 4)) of retrieved articles (Figure 1B,C). There were relatively few studies from Africa (6% (n = 35), compared to other continents (Figure 1). Asia had reports on all connexins (7) found in this review, while Europe and Australia reported on 4 connexins. Africa, North America, and South America had reports of 6, 5, and 3 connexins respectively (Figure 1D). We identified GJB2 as the most widely studied connexin in all the continents (Figure 1D).
We identified a cocktail of methods used by the researchers to investigate connexin gene variants in hearing-impaired patient samples. Several studies employed two or more approaches while others depended on a single approach. Targeted sequencing was the most common method followed by polymerase chain reaction (PCR) techniques. The targeted sequencing studies were mostly by Sanger sequencing where one or more primer sets were used to amplify and sequence the coding regions of the connexins under investigation. The analysis of the primer sets used for sequencing revealed their uniqueness for each study (Table S1). Among the PCR techniques were amplification-refractory mutation system (ARMS), PACE™ (PCR Allele Competitive Extension), and multiplex PCR. A few studies used next-generation sequencing techniques (NGS) such as whole-exome sequencing (NGS) and NGS panel sequencing (Figure 2).
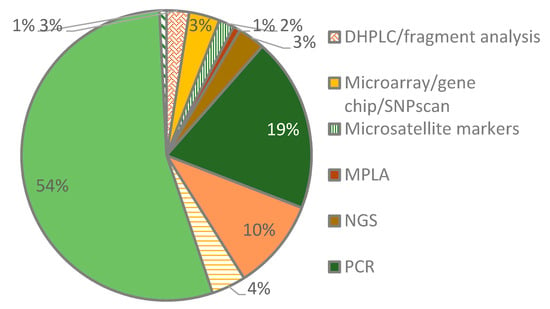
Figure 2.
Methods used to investigate connexin gene variants. Among the methods are denaturing high-performance liquid chromatography (DHPLC), multiplex ligation-dependent probe amplification (MLPA), polymerase chain reaction (PCR), next-generation sequencing (NGS), restriction fragment length polymorphism (RFLP), and single-strand conformational polymorphism (SSCP).
2.1. Connexin 26 (GJB2)
We identified 337 variants in GJB2 from the review of the publications included in this study. Analysis of the clinical significance of these variants gave 124 (37.2%), 53 (15.9%), 46 (13.8%), and 25 (7.5%) pathogenic, likely pathogenic, uncertain significance, and benign variants, respectively (Figure 3). Asia (n = 288; 48%) was the highest contributor of pathogenic (PLP) variants followed by Europe (n = 166; 28%), North America (n = 52; 9%), Africa (n = 48, 8%); South America (n = 37; 6%) and Australia (n = 4; 1%). The three databases used in this study did not have data on the clinical significance of 13 out of the 333 variants, implying that some of these variants may be novel (Table S2).
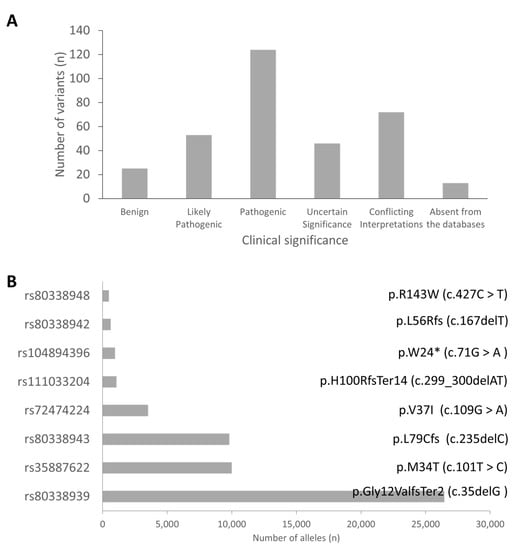
Figure 3.
Common GJB2 variants. (A) Clinical significance of identified variants. (B) The top eight GJB2 variants ranked based on the total number of alleles.
The pathogenic variants were sorted based on the number of reported alleles to identify the commonly reported variants. The variants with more than 450 reported alleles were considered as the commonly associated GJB2 variants. Based on the number of reported alleles, p.Gly12ValfsTer2 (c.35delG) was ranked as the most commonly reported GJB2 variant, found in 26,429 (15.1%) out of 175,491 investigated alleles. The frequencies of the other common GJB2 mutations were 10,009/82,805 (12.1%), 9813/277,116 (3.5%), 3520/127,802 (2.8%), 972/54,394 (1.8%), 641/54,279 (1.2%), 1080/10,7855 (1.0%), and 497/90,305 (0.6%) for p.M34T (c.101T > C), p.L79Cfs (c.235delC), p.V37I (c.109G > A), p.W24X (c.71G > A), p.L56Rfs (c.167delT), p.H100RfsTer14 (c.299_300delAT), and p.R143W (c.427C > T), respectively (Table S3). Further analysis of the p.Gly12ValfsTer2 (c.35delG) showed its widespread and high prevalence in European countries, and North African countries and some parts of Brazil and America. In Asia, p.V37I (c.109G > A and p.L79Cfs (c.235delC)) variants were the most frequently reported GJB2 variant. On a country-wise analysis, we observed that the highest number of p.Gly12ValfsTer2 (c.35delG) and p.M34T (c.101T > C) mutated alleles were from the United Kingdom (Figure 4). The highest number of mutated alleles for p.L79Cfs (c.235delC), p.V37I (c.109G > A), and p.H100RfsTer14 (c.299_300delAT) were recorded in China. India, the United States of America, and Ghana recorded the highest number of p.W24X (c.71G > A), p.L56Rfs (c.167delT), and p.R143W (c.427C > T) mutated alleles, respectively (Figure 4).
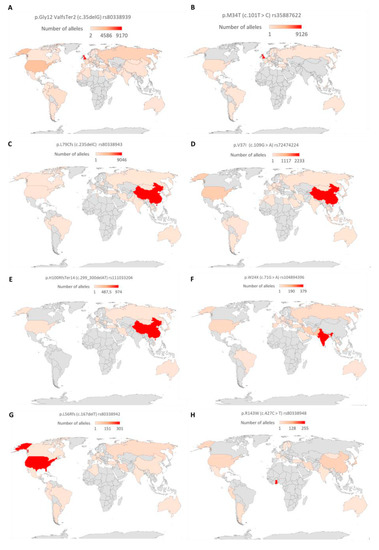
Figure 4.
Global distribution of common GJB2 variants. A graph showing the total number of reported alleles of (A) p.Gly12ValfsTer2 (c.35delG), (B) p.M34T (c.101T > C), (C) p.L79Cfs/c.235delC, (D) p.V37I/c.109G > A, (E) p.H100RfsTer14/c.299_300delAT, (F) p.W24X/c.71G > A, (G) p.L56Rfs/c.167delT, and (H) p.R143W/c.427C > T. The countries were colored with a gradient from red (highest number of alleles) to brown (lowest number of alleles). Countries shaded grey either had no reports or no alleles. The map was created by the authors in Microsoft Excel (Office 365 education license of the University of Cape Town, South Africa).
We extracted data on known PLP variants in GJB2 (p.W44*: c.131G > A (North America), c.IVS1 + 1G > A (Russia), p.W172*: c.516G > A (Siberia), p.W172C: c.516G > C (Siberia) p.W172R, and c.514T>A (Siberia) that are prevalent in isolated ethnic groups. The majority of mutated c.IVS1 + 1G > A variant was recorded in Asia with high frequencies from Yakutia, Siberia, and Russia (Figure S3). The other three rare GJB2 variants (p.W172*: c.516G > A, p.W172C: c.516G > C, p.W172R, and c.514T > A) which are at the same amino acid position were common in the Asian countries with Rusia having the highest frequency (Table S4).
2.2. Connexin 30 Gene (GJB6)
We selected reports of 18 variants in GJB6, including two large genomic deletions. Most coding region variants in GJB6 were predicted as benign or uncertain significance (n = 12 (85.7%)). Two variants were predicted as pathogenic; GJB6: p.A40V/c.119C > T/rs780320724 from Taiwan [23] and GJB6: p.T5M/c.14C > T/rs104894414 from Germany [24], and Iran [25], respectively (Table 1). The del(GJB6-D13S1830) was the most common GJB6 variant reported from 31 countries from all continents, with virtually no case from Africa. High allele frequencies were particularly reported in France and Spain (Figure 5). We found few studies that reported del(GJB6-D13S1854) variation among the hearing-impaired; these studies were from Argentina, Colombia, Portugal, and Brazil.

Table 1.
Global distribution of connexin 30 (GJB6) gene variants.
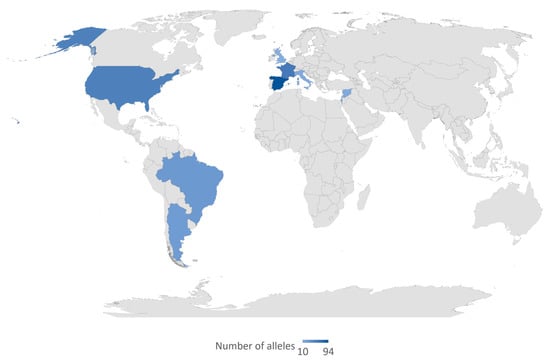
Figure 5.
Global distribution of del(GJB6-D13S1830). The variant del(GJB6-D13S1830) was reported in all the countries highlighted in blue color. The intensity of the blue color denotes the frequency of reported alleles. The map was created by the authors in Microsoft Excel (Office 365 education license of the University of Cape Town, South Africa).
2.3. Connexin 31 Gene (GJB3)
We identified reports of variants of GJB3 from nine countries with Korea having the highest number of reported alleles. The variant with the highest number of reported alleles was found to be synonymous and was predicted to be benign. None of the variants was predicted pathogenic although they were identified in hearing-impaired populations. Based on the databases used, we predicted the variants as benign, with uncertain significance or conflicting interpretations. Two variants (c.547G > A/rs74315318 and c.497A > G/rs121908851) were predicted as pathogenic by only one database with conflicting interpretations from the other databases; it was, therefore, difficult to conclude on their pathogenicity (Table 2).
Table 2.
Global distribution of connexin 31 (GJB3) gene variants.
2.4. Connexin 30.3 Gene (GJB4)
In this review, we identified studies from 5 countries that reported GJB4 variants. The majority of identified GJB4 variants had conflicting clinical significance since they had different interpretations in the databases used. VarSome predicted seven variants as pathogenic or likely pathogenic and InterVar predicted an additional variant as pathogenic. The pathogenic variants of VarSome were not predicted as pathogenic by InterVar and vice versa (Table 3).
Table 3.
Connexin 30.3 (GJB4) gene variants.
2.5. Connexin 29 Gene (GJC3)
We identified seven GJC3 variants from 4 countries (Table 4) which were predicted as benign or of uncertain significance with no PLP variant found.
Table 4.
Connexin 29 (GJC3) gene variants.
2.6. Connexin 43 Gene (GJA1)
Twelve (12) variants in GJA1 were reported by different researchers of which three were predicted to be pathogenic or likely pathogenic (PLP). The pathogenic variants were reported in hearing-impaired patients from America and Asia, the variants reported from Africa (South Africa) were predicted as benign or uncertain significance. GJA1 c.932delC variant had the highest number of alleles reported from Australia. Four (4) out of the 11 GJA1 variants were reported in a study from South Africa (Table 5).
Table 5.
Connexin 43 (GJA1) gene variants.
2.7. Connexin 45 (GJC1)
Connexin 45 was previously given the gene symbol GJA7 but the Human Genome Organization (HUGO) Gene Nomenclature Committee [51] has given it the symbol GJC1. We identified a multi-site study that reported GJC1 variants from three different populations: the USA, Turkey, and the UK [52]. None of the reported variants were predicted pathogenic although they were identified in hearing-impaired cohorts. We observed conflicting interpretations of the variants’ clinical significance based on the predictions from the databases used (Table 6).
Table 6.
Connexin 45 (GJC1) gene variants.
2.8. Summary of the Global Allele Frequencies of the Common Connexin Genes Pathogenic (PLP) Variants Associated to Hearing Impairment (HI)
We calculated the allele frequency of the reported PLP variants in connexin genes in patients and controls for each continent, in order to assess the global contribution of these variants associated with HI. Asia had the highest allele frequencies of the common GJB2 variants in the hearing controls compared to the other continents. GJB2:p.V37I:c.109G > A had the highest allele frequency (200/3478 (5.8%)) among the control group from Asia (200/3478 (5.8%)) and North America (11/588 (1.9%)). In Africa, GJB2:p.Gly12ValfsTer2:c.35delG and p.R143W:c.427C > T recorded the highest frequencies in Ghana. In Europe, among the eight common GJB2 variants, p.Gly12ValfsTer2:c.35delG (Table 7). We did not find reports of carriers of the GJB6 large deletions and GJA1 PLP variants.
Table 7.
Summary of the global allele frequencies of the common pathogenic (PLP) variants in connexin genes associated to hearing impairment (HI).
3. Discussion
To the best of our knowledge, this paper is, to date, the most comprehensive review on the contribution of connexin gene variants in HI, globally. Connexin channels regulate the transport of small signaling molecules between cells to aid the proper functioning of the tissue/organ systems in the body [53]. Our review found that more than 570 studies were conducted globally on connexin-related HI investigations with most studies performed in Asia, while relatively few have been done in Africa.
Most studies used targeted sequencing, but the decline in next-generation sequencing cost has accelerated the discovery of novel disease gene variants through available high-throughput targeted panels or whole-exome sequencing technologies investigating several gene targets in a single test [54,55,56]. Indeed, there was a clear migration from non-sequencing approaches such as denaturing high-performance liquid chromatography (DHPLC), multiplex ligation-dependent probe amplification (MLPA), PCR, restriction fragment length polymorphism (RFLP), and single-strand conformational polymorphism (SSCP) to sequencing techniques or a combination of sequencing and non-sequencing techniques.
Connexin 26 gene (GJB2, OMIM:121011) located on chromosome 13q12.11 is known to be expressed in different tissues including the cochlear of humans [57], mouse, and rat [58]. GJB2 gene variants were the most common genetic factors associated with NSHI among several populations [59,60], however, they are rare in African, and African American populations [61]. Similarly, it was clear from our review that GJB2 is the most investigated gene and had the highest number of pathogenic variants identified among hearing-impaired patients. The most common pathogenic variants (GJB2: p.Gly12ValfsTer2, p.M34T, p.L79Cfs, p.V37I, p.H100RfsTer14, p.W24X, p.L56Rfs, and p.R143W) appeared to be localized to specific populations, due to a founder effect [10,62].
In a previous review by Chan and Chang in 2014, 216 original GJB2 research articles reporting not less than 10 probands were retrieved and analyzed [10]. In our current review, 571 original research publications on connexins associated with HI were considered of which 566 articles reported on GJB2 associated HI. The previous report was from 63 countries [10], while in this study, we retrieved GJB2 publications from 106 countries. The differences in the number of publications and countries involved can be explained by the time difference between the previous report and the present study. Also, we did not exclude case reports, contrary to the previous report. In contrast to the report from Chan and Chang, Australia, and not Africa, had the lowest contribution of GJB2 variants. Moreover, Asia was identified as the highest contributor while the previous report had Europe as the highest contributor of GJB2 PLP to HI [10]; this can be attributed to the increasing interest and number of genetic researches in all parts of the world. Despite the above differences, the commonly reported PLP GJB2 variants were similar in both studies. Furthermore, our study and the studies from Chan and Chang and Tsukada reported a similar ethnic-specific spectrum of the common PLP variants in GJB2 [10,62].
The most common GJB2 variant is p.Gly12ValfsTer2 (c.35delG) which is frequently reported among populations in Europe, the Middle East, Australia, North, and South America [10]. We observed widespread of this variant across the globe but it was almost absent in sub-Saharan Africa although there were studies from Ghana [59,63], Cameroon [64,65] and South Africa [64] that investigated this variant in African populations. Morocco is an exception, where five independent studies identified biallelic c.35delG mutation in hearing-impaired patients [66,67,68,69,70]. The spread of the variant from Europe and North Africa to North and South America seems to follow migration patterns [10].
Second to GJB2: c.35delG is GJB2- p.M34T (c.101T > C) which was found to be most prevalent in the United Kingdom (UK). The carrier rate of GJB2: p.M34T was calculated at 2.69% in the UK, which was almost twice the carrier rate of GJB2-c.35delG (1.36%). In the United States of America, the carrier rate for the GJB2: p.M34T variant was found to be 2.3% [71]. The high carrier rates of variants suggested the possibility of heterozygous advantage. However, the audiometric characterization of GJB2-p.M34T carriers was not different from homozygous hearing individuals. Hence there is no effect on the hearing ability of the carriers [72].
In the present review, we identified three variants (GJB2: p.L79Cfs/c.235delC, p.V37I/c.109G > A, and p.H100RfsTer14/c.299_300delAT) with very high allele frequencies from Asia compared to other continents. These variants were absent in sub-Saharan African countries but were found in a few cases in some North African countries. The Chinese population was found to have a high prevalence of GJB2: c.235delC [73] with frequencies of about 14.7% homozygous among a hearing-impaired sub-population, and 16.1% heterozygous in the hearing population [74]. The carrier frequency of GJB2: c.235delC is similar to that of the entire Asian population [73,74], and a high prevalence of that variant was reported in Japan [75,76], Korea [77], and Taiwan [78].
The GJB2: p.V37I variant was described as a polymorphism by some researchers while others consider it a potential disease-causing missense mutation [79]. The high carrier frequency of the variant among hearing controls informs the polymorphism argument, however individual homozygous of the variant had HI [80]. Compound heterozygosity of the GJB2: p.V37I variant and other known GJB2 pathogenic variants produced mild to severe HI. It was proposed that the milder phenotype was due to the GJB2-p.V37I allele [81]. We have identified several independent studies that reported the variant in hearing-impaired individuals, implying that the variant is likely disease-causing. In addition, GJB2: p.V37I was predicted as pathogenic by CinVar, Varsome [82], and InterVar [83]. The majority of GJB2: p.V37I mutated alleles were identified among Asians and mostly Chinese [79,80]. The third most common GJB2 variant associated with HI in Asia was c.299_300delAT with an estimated allele frequency of 3.89% [76,84]. Although this variant is very prevalent in China, it appears that this variant is not common in other populations [85].
The truncating GJB2 mutation p.W24X is the predominant mutation among the Indian and European Gypsy populations [86,87,88]. The GJB2: p.W24X was the most commonly observed mutation and accounted for about 95% of all GJB2 mutations found in the Indian population with a carrier frequency of 2.4% [88]. The mutation was proposed to be a founder effect and confirmed through haplotype analysis of the flanking markers of the GJB2 gene [86,88].
GJB2: c.167delT was reported to be common in the Eurasian populations and postulated to have a single origin of allele due to the observed conserved haplotypes around the mutation [89]. Although the mutation was prevalent in the territories of the Middle East [89,90,91], we found a high number of alleles with PLP in the United States of America. The fourth most common GJB2 mutation in the American population is GJB2: c.167delT and was found to account for about 3.6% of cases. The variant was more prevalent in the White-American population compared to other populations [92].
As, an exception in populations of African ancestry, the GJB2: p.R143W is a founder mutation reported first in a Ghanaian village known for its extremely high number of deaf people. To date, GJB2: p.R143W mutation is still the most common HI gene in Ghana [59,63,93]. In 1998, there was a study that reported the homozygous form of the mutation in all 11 families investigated [94]. An update from the Ghanaian village in 2020 found 7 out of 8 families with the homozygous mutation, the 8th family had the heterozygous form [93]. The phenotype-genotype correlation from both studies showed that patients with biallelic GJB2: p.R143W had profound HI while no difference was observed between the carriers and normal hearing participants [93,94]. In our review, we identified studies from the United States of America and Asia that had reports of patients with the mutated GJB2: p.R143W. Considering that the p.R143W mutation has a high frequency in Ghana, it is likely that the mutation emerged in this population and was introduced in the other populations with African ancestry in the diaspora via ancient and/or recent migration events, specially through the Black African transatlantic slave trade.
In our current review, we identified some rare GJB2 variants that were absent from the databases used. Although these variants were identified in hearing-impaired participants, they may not be considered as HI variants since there are no functional and/or population studies on these variants. A recent report showed that rare variants within the coding region of GJB2 gene and other HI genes are associated with Meniere disease (MD) [95]. The signs and symptoms of MD are sensorineural hearing loss, tinnitus, and episodic vertigo which are sometimes common to patients with NSHI [96]. Meniere disease was reported to be extremely rare in individuals of African descent which supported the negligible contribution of GJB2 variants to HI in sub-Saharan Africa [97].
Rare known PLP variants in GJB2 were found to displace some degree of ethnic specificity in some populations. Similar to previous reports [98,99], we identified extremely high frequencies of GJB2: c.IVS1+1G > A from Yakutia and Russia; the high prevalence of the variant within the Yakutia population can be seen in the high carrier rate of 10% [99]. This is also suggestive of a potential accumulation of GJB2 pathogenic variants in this population and calls for public health attention [98]. The Siberian population which comprises Russia, Kazakhstan, boarders of Mongolia and China was found to have the rare GJB2: p.W172C (c.516G > C). We analyzed other variants (p.W172* (c.516G > A), p.W172R (c.514T > A)) at the same site of mutation, and similar to our observation, a previous study reported an extremely high frequency of 62.9% from these populations [100]. Other population-specific GJB2 rare variants found were p.W44X, p.Q7X, and p.S199F which are common to North America [73,92,101], Ecuador [102], and Colombia [103] respectively.
Although not in high numbers, we identified pathogenic GJB2 intronic variants that were reported in hearing-impaired patients. These variants were c.IVS1 − 1G > A [104], c.IVS1 − 15C > T [73], c.IVS1 + 12G > A [105], c.IVS1 + 27G > C [106], and c. − 23 + 1G > A. The −23 + 1G > A variant was reported in studies from Russia [107], Poland [108], China [109], India [88], United States of America [71], and with a high prevalence in Turkey [110].
GJB6 (OMIM:604418) encodes connexin 30, which is part of the family of proteins that form gap junction channels. The location of GJB6 in the human genome is chromosome 13q12.11 (the same as GJB2). The GJB6 gene is expressed mainly in the brain and skin [111,112] with about 76% protein identity when compared to human GJB2 [113]. The GJB6 gene has been associated with Clouston syndrome and hearing impairment. The association of GJB6 coding region variations to HI has recently been refuted [20]; however, previous reports from Taiwan [23] Germany [24], and Iran [25] identified pathogenic variants within the coding regions of the gene. Six GJB6 large genomic deletions have been found and previously reported, they are: >920kb deletion [114], 179 kb deletion [115], 131 kb deletion [116], del(GJB2-D13S175), del(GJB6-D13S1830) and del(GJB6-D13S1854) [117]. In addition to the six, there was a report of a seventh GJB6 deletion, del(GJB6-D13S1834), in February 2020 [118]. We identified the large genomic deletions, GJB6-D13S1830, and GJB6-D18S1854 as the frequently reported GJB6 variants. In many cases, the large deletions were in trans with pathogenic GJB2 variants and similar to previous observations [20]. Unlike the GJB6 coding region variations, the large deletions disrupt a 5′ cis-acting element upstream of both genes. The disruption of the cis-acting element abolishes the expression of GJB2 and hence is responsible for the phenotype [119,120]. The previously reported GJB6 knockout mice had significant reduction in the GJB2 expression which was the cause of HI [121]. Mice models with only the coding region of GJB6 deleted were found to have normal hearing which provided evidence that the coding region plays no role in the development of HI [115,116].
Our analysis of the global distribution of GJB6-D13S1830 confirmed a previous observation by del Castillo et al. [122,123] with a high prevalence of the variant in North America, South America, and Europe with no record from Asia, Australia, and Africa. This variant is known to be frequent in Spain, France, the United Kingdom, Israel, and Brazil [123], and similarly, we observed that the highest number of alleles were from Spain and France. The absence of GJB6-D13S1830 from the Asian, Australian, and African populations is indicative of a population-specific spread of the variant and it would inform future studies as well as inform public health policies.
Connexin 43 (GJA1, OMIM:121014) is located on chromosome 6 (6q21-q23.2) [50] of the human genome and has been implicated in a number of diseases but mainly in oculodentodigital dysplasia [124,125,126] with pleiotropic phenotypes [127]. It should be noted that numerous pathogenic variants that have been linked to HI were found in the GJA1 pseudogene, which could explain their lack of representation in HI-associated genes. Indeed, GJA1 pseudogene has the features of an expressed gene [128]. The messenger RNA of GJA1 gene was identified in tumor cells which was contrary to the characteristics of pseudogenes (inability to produce functional mRNA and proteins) [129]. Also, GJA1 is ubiquitously expressed in many human tissues and cells [130]. Although GJA1 has been associated with HI, only a few pathogenic variants (GJA1: p.L11Y, p.V24A, p.L181F, and p.S69P) were reported in deaf patients from the USA [50] and Taiwan [40]. This gene’s contribution to HI is not conclusive considering the low number of patients with the pathogenic variants, and more data from diverse populations are needed to refine the gene-disease pair curation. It is possible that the voltage-gating mechanism of connexin 43 may be affected by the pathogenic variants, culminating in defective gap junction channels. The expression pattern from the mouse genome database has shown that GJA1 is expressed in the auditory system and may play key roles in hearing and the functioning of the ear [50,131].
The connexin 45 gene, GJC1 (OMIM:608655), is located on chromosome 17q21.31. Connexin 45 is a candidate HI gene expressed in the auditory system as part of the connexin proteins; however, the mouse genome database has no report of its association with the HI phenotype [131]. Cardiovascular disorders are the most common phenotypes associated with GJC1 [132]. We found a research effort to identify gene variants in GJC1 among hearing-impaired participants [52]. The authors studied participants from 3 different populations but did not find any pathogenic variants from the HI cohorts studied. It is imperative to screen a larger population for GJC1 mutations to investigate the gene’s contribution to HI.
In humans, GJB3 (connexin 31 gene, OMIM:603324) found on chromosome 1p34.3, encodes gap junction beta 3 (protein [133]. The gene has been associated with two major conditions erythrokeratodermia variabilis et progressive (MIM:133200) and non-syndromic hearing loss. In our review, we found only a few hearing-impaired participants with mutations in GJB3, at low allele frequencies. However, only two likely pathogenic variants (GJB3-p.E183K/c.547G > A/rs74315318 and GJB3-p.N166S/c.497A > G/rs121908851) were reported from publications considered [134]. We could not conclude on the clinical significance of these variants since the 3 databases gave conflicting clinical significance [82,83,135]. Functional analysis suggested an expression overlap and possible interactions between connexin 26 and connexin 31 in the cells at the tip of the spiral limbus. It was further demonstrated that the two connexins formed heterotypic channels in the [38]. The data from the above functional studies confirmed a digenic form of NSHI. In addition, a biallelic mutation in any of the two connexins can result in NSHI. Confirming the digenic claim, GJB2 p.V37I/p.L213S/GJB3 p.V84I was found to co-segregate with NSHI in a family [42]. To fully understand the role of GJB3 in HI, there is a need to study many populations as well as conduct further functional assays to assess the ironic and biochemical functions of the gene and its mutant forms.
Like other connexins, GJB4 (OMIM:605425) encodes connexin 30.3 which oligomerizes to form gap junction channels. Unlike the common connexins, the contribution of GJB4 to HI remains unknown. There were considerable efforts by five groups of researchers identified in this review to investigate GJB4 mutations in deaf populations. The clinical significance of the identified variants of GJB4 had conflicting clinical significance; six variants were predicted as pathogenic or likely pathogenic on not more than one of the three databases used [82,83,135]. Functional genomics using mouse models showed that the auditory system of GJB4 null mice is unaffected, the mutant mice had normal hearing when assessed by brain stem-evoked potentials [136]. However, the gene was found to be expressed in the mouse auditory system [131] and rat cochlear [137] suggesting its role in hearing.
The association of GJC3 (a gene that encodes connexin 29, OMIM:611925) to NSHI remains unclear although a few studies have associated variations in connexin 29 to NSHI. These variants as presented in our results were predicted to have uncertain clinical significance. However, data from the Mouse Genome Informatics has shown that GJC3 is expressed in the auditory system and has hearing loss as one of its phenotypes [131]. Functional studies using GJC3 null mice showed reduced maturation of the hearing threshold. Noise-induced HI has been reported in GJC3 null mice [131], while another study conducted with adult mice did not find any difference in the hearing threshold of GJC3 null and wildtype mice [138]. To conclude on the contribution of GJC3 to HI, we recommend screening more deaf populations across the globe.
There was a limited number of publications retrieved on GJB3, GJB4, GJC3, GJA1, and GJC1 which was a major challenge encountered in this study. Therefore, it was difficult to determine whether these genes should be considered or catalogued as HI genes. Apart from GJB2, which is globally known to be associated with HI, there is a need for more studies from different populations as well as functional studies for a concluding decision to be made on GJB4, GJA1, GJC3, and GJC1. The Hearing Loss Home Page [8] has categorized GJB3 as an autosomal dominant HI gene. Although GJB6 was previously considered as a HI gene, recent studies have disproved its classification as a HI gene. The present review suggests that only GJB2 and GJB3 are recognized and validated HI genes.
4. Materials and Methods
4.1. Search Terms
We reviewed publications on connexin genes variants implicated in human HI and the distributions of the common variants across the globe. Also, we evaluated the methods used to investigate NSHI-implicated gene variants. The protocol was registered on PROSPERO, International Prospective Register of Systematic Reviews with the registration number “CRD42020169697”. Two independent reviewers conducted the literature search on PubMed, Scopus, Africa-Wide Information, and Web of Science databases. The search term used in the study comprised of three major components: the first component was (connexins OR “gap junction alpha protein” OR “Gap junction alpha proteins” OR “Gap junction beta-protein” OR “Gap junction proteins” OR connexin OR GJB OR GJA), the second component was (“hearing loss” OR “non-syndromic sensorineural hearing loss” OR “non-syndromic deafness” OR “non-syndromic hearing impairment” OR “hearing impairment” OR deafness) and the last component was (genetic OR gene OR genes) OR (“genetic loci” OR genes OR “genetic diseases” OR “genetic markers). Each component of the search term was joined with “AND” to obtain the resultant search term which was used to retrieve publications from the databases used (Figure 1).
4.2. Data Extraction
Two independent reviewers conducted the literature search between 1 April to 31 May 2020 and 2592 full-length articles were selected based on the inclusion and exclusion criteria outlined below.
Inclusion criteria:
- Publications on human hearing impairment;
- Publications on the genetics of non-syndromic hearing impairment;
- Publications reporting on connexins association with NSHI.
Exclusion criteria:
- Studies that are not on human hearing impairment;
- Review or meta-analysis publications;
- Policy papers;
- Publications that are not on connexin hearing impairment;
- Publications on environmental and/or syndromic hearing impairment;
- Publications focusing on in silico analysis.
The search with the keywords gave 874, 992, 31, and 695 results from PubMed, Scopus, Africa-Wide Information, and Web of Science databases, respectively. A blinded screening was undertaken by the two reviewers (SMA, and EWT, first and second authors of this manuscript) using the titles as the first-level screening followed by the abstracts. The search results were downloaded into EndNote referencing software and duplicates removed (Figure 6). The data extraction was conducted independently by the two reviewers and compared to remove any form of bias. The following data elements were extracted: (1) location and date; (2) connexin genes investigated; (3) the number of mutant alleles; (4) methods for genetic screening. The data extracted were manually captured onto Microsoft Excel sheets and analyzed using Microsoft Excel (Office 365 education license under the University of Cape Town, South Africa) and SPSS version 25 (IBM, Armonk, New York, United States). A third person (AW) who is an expert in the field was consulted in times of disagreement between the individual judgments during the screening and data extraction process.
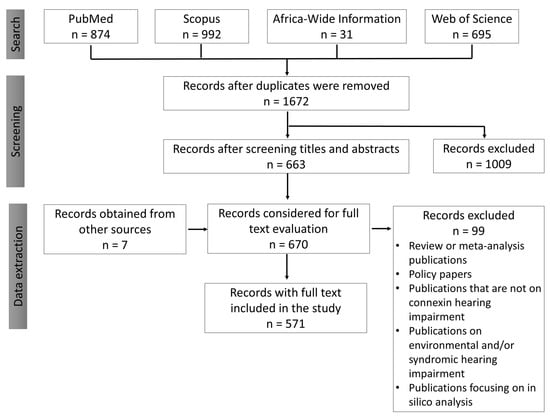
Figure 6.
Flow diagram illustrating the screening of articles obtained after the literature search.
4.3. Quality Assessment
To avoid any form of bias, two independent reviewers (SMA and EWT) synthesized the data and assessed the quality of the documents included, using the quality of genetic studies (Q-Genie) tool developed by Sohani et al. [139] for genetic studies and the risk of bias assessment tool for prevalence studies developed by Hoy et al. [140] for the other studies. Discrepancies were solved by discussion and consensus. An expert (AW) was consulted to resolve disagreements between the judgment of the reviewers, by discussion and consensus. The quality assessment was conducted at the outcome level of each study. The studies were assessed for selective outcome reporting and whenever there was evidence of this, the effect of the selective outcome reporting on the study results was further analyzed.
4.4. Clinical Significance
The clinical significance of the identified variants was assessed on three databases InterVar [83], VarSome [82], and ClinVar [135]. Both VarSome and InterVar are bioinformatic web-based tools build on the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) 2015 guidelines and are useful for clinical interpretation of human genetic variants [82,83]. ClinVar is also a web-based database that provides evidence and relationships between human variants (found in biological sample) and phenotypes which serve as strong evidence for clinical significance interpretation [135]. Our judgement on the clinical significance of each variant was based on the prediction from the 3 databases mentioned above.
5. Conclusions
The present comprehensive review on the contribution of connexins genes in HI, globally, found most investigations performed in populations from China, with relatively few studies from African populations. In most populations, except Africans, common GJB2 pathogenic variants that were found were p.Gly12ValfsTer2 and p.M34T (commonly among Europeans), p.L79Cfs, p.V37I, and p.H100RfsTer14 (mostly in Asians), p.W24X among Indians, p.L56Rfs among Americans, and p.R143W particularly in Ghanaians, the African exception for GJB2 variants. These GJB2 variants exhibited population-specific prevalence due to founder effects. The second most common HI-associated connexin was GJB6. We identified two main deletions in GJB6 (GJB6-D13S1830 and GJB6-D13S1854), that were predicted to be pathogenic, however, the coding region variants of GJB6 are no longer considered as causes of HI. From the review, we identified 11 GJA1 variants of which 3 were predicted to be pathogenic but their pathogenicity needs to be confirmed with more data from multiple populations. The GJB4 variants found from the reports that were used for this review mostly had conflicting clinical significance, but the majority were predicted pathogenic by one of the 3 databases used. None of the GJC1, GJB3, and GJC3 variants were predicted pathogenic. Most researchers used targeted sequencing approaches to investigate connexin genes associated with HI. The present review suggests that only GJB2 and GJB3 are recognized and validated HI genes. It is likely that the wide use of whole-exome sequencing, particularly in understudied African populations, will rapidly increase the identification of novel HI-associated gene variants and improve disease-gene pairs curation, globally.
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-1729/10/11/258/s1, Table S1: Connexin genes primers sets used for targeted sequencing, Table S2: GJB2 variants with no clinical significance data from the three databases used, Table S3: Eight commonly reported connexin 26 gene (GJB2) variants, Table S4: Known but rare PLP GJB2 variants common in isolated populations.
Author Contributions
Conceptualization, A.W., and S.M.A.; literature such, S.M.A., E.W.-T., E.T.A., D.W.N.-G. and M.B.A.; data extraction and original draft preparation, S.M.A., E.W.-T. and A.W.; writing-review and editing, S.M.A., E.W.-T., E.T.A., D.W.N.-G., M.B.A., O.Q., G.A.A. and A.W.; supervision, A.W., G.A.A. and O.Q.; funding acquisition, A.W., and G.A.A. All authors contributed important intellectual content presented and have read and agreed to the published version of the manuscript.
Funding
This work was supported by funds from the World Bank African Centres of Excellence grant (ACE02-WACCBIP: Awandare) and a DELTAS Africa grant (DEL-15-007 to G Awandare). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s with funding from the Wellcome Trust (107755/Z/15/Z, to GAwandare) and the UK government. The work was also funded by funds from the National Institutes of Health (NIH), USA, grant number U01-HG-009716 to AW; and the African Academy of Science/Wellcome Trust, grant number H3A/18/001 to A.W. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Samuel Mawuli Adadey is supported by WACCBIP DELTAS PhD fellowship and Africa Regional International Staff/Student Exchange (ARISE II) mobility fund.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
Abbreviations
| GJB2 | Gap junction beta-2 |
| GJB6 | Gap junction beta-6 |
| GJB3 | Gap junction beta-3 |
| GJB3 | Gap junction beta-4 |
| GJC3 | Gap junction gamma-3 |
| GJA1 | Gap Junction Alpha 1 |
| GJC1 | Gap junction gamma 1 |
References
- Bitner-Glindzicz, M. Hereditary deafness and phenotyping in humans. Br. Med. Bull. 2002, 63, 73–94. [Google Scholar] [CrossRef] [PubMed]
- James, M.; Kumar, P.; Ninan, P. A study on prevalence and risk factors of hearing impairment among newborns. Int. J. Contemp. Pediatr 2018, 5, 304–309. [Google Scholar] [CrossRef][Green Version]
- Olusanya, B.O.; Neumann, K.J.; Saunders, J.E. The global burden of disabling hearing impairment: A call to action. Bull. World Health Organ. 2014, 92, 367–373. [Google Scholar] [CrossRef] [PubMed]
- WHO. Prevention of Blindness and Deafness. Available online: https://www.who.int/pbd/deafness/hearing_impairment_grades/en/ (accessed on 30 March 2019).
- Bayazit, Y.A.; Yılmaz, M. An overview of hereditary hearing loss. ORL 2006, 68, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. Rev. Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary hearing loss and deafness overview. In GeneReviews®[Internet]; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Van Camp, G.; Smith, R. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 31 March 2020).
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Wohlwend, A.; Kwak, B.R. Mutations in connexin genes and disease. Eur. J. Clin. Investig. 2011, 41, 103–116. [Google Scholar] [CrossRef]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 2018, 1860, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, F.J.; del Castillo, I. Genetics of isolated auditory neuropathies. Front. Biosci. Landmark 2012, 17, 1251–1265. [Google Scholar] [CrossRef]
- Mikstiene, V.; Jakaitiene, A.; Byckova, J.; Gradauskiene, E.; Preiksaitiene, E.; Burnyte, B.; Tumiene, B.; Matuleviciene, A.; Ambrozaityte, L.; Uktveryte, I.; et al. The high frequency of GJB2 gene mutation c.313_326del14 suggests its possible origin in ancestors of Lithuanian population. Bmc Genet. 2016, 17, 45. [Google Scholar] [CrossRef]
- Li, H.; Wang, B.; Liu, D.; Wang, T.; Li, Q.; Wang, W.; Li, H. SNPscan as a high-performance screening tool for mutation hotspots of hearing loss-associated genes. Genomics 2015, 106, 83–87. [Google Scholar] [CrossRef]
- Xu, J.; Nicholson, B.J. The role of connexins in ear and skin physiology—Functional insights from disease-associated mutations. Biochim. Biophys. Acta 2013, 1828, 167–178. [Google Scholar] [CrossRef]
- Wonkam, A.; Noubiap, J.J.N.; Djomou, F.; Fieggen, K.; Njock, R.; Toure, G.B. Aetiology of childhood hearing loss in Cameroon (sub-Saharan Africa). Eur. J. Med. Genet. 2013, 56, 20–25. [Google Scholar] [CrossRef]
- Richard, G.; Brown, N.; Ishida-Yamamoto, A.; Krol, A. Expanding the phenotypic spectrum of Cx26 disorders: Bart–Pumphrey syndrome is caused by a novel missense mutation in GJB2. J. Investig. Dermatol. 2004, 123, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Barruet, K.; Saka, B.; Kombate, K.; Mouhari-Toure, A.; Nguepmeni, N.J.; Akakpo, S.; Tchangai-Walla, K.; Pitche, P. Keratitis-ichthyosis-deafness (KID) syndrome: An observation in a child in sub-Saharan Africa. In Proceedings of the Annales de Dermatologie et de Venereologie, Chamonix, France, 2–5 February 2011; p. 453. [Google Scholar]
- DiStefano, M.T.; Hemphill, S.E.; Oza, A.M.; Siegert, R.K.; Grant, A.R.; Hughes, M.Y.; Cushman, B.J.; Azaiez, H.; Booth, K.T.; Chapin, A. ClinGen expert clinical validity curation of 164 hearing loss gene–disease pairs. Genet. Med. 2019, 21, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Adadey, S.M.; Esoh, K.K.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. GJB4 and GJC3 variants in non-syndromic hearing impairment in Ghana. Exp. Biol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. Icwsm 2009, 8, 361–362. [Google Scholar]
- Yang, J.J.; Huang, S.H.; Chou, K.H.; Liao, P.J.; Su, C.C.; Li, S.Y. Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audiol. Neuro Otol. 2007, 12, 198–208. [Google Scholar] [CrossRef]
- Grifa, A.; Wagner, C.A.; D’Ambrosio, L.; Melchionda, S.; Bernardi, F.; Lopez-Bigas, N.; Rabionet, R.; Arbones, M.; Della Monica, M.; Estivill, X.; et al. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat. Genet. 1999, 23, 16–18. [Google Scholar] [CrossRef]
- Ghasemnejad, T.; Shekari Khaniani, M.; Zarei, F.; Farbodnia, M.; Mansoori Derakhsahan, S. An update of common autosomal recessive non-syndromic hearing loss genes in Iranian population. Int. J. Pediatric Otorhinolaryngol. 2017, 97, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Asma, A.; Ashwaq, A.; Norzana, A.G.; Atmadini, A.M.; Ruszymah, B.H.; Saim, L.; Wahida, I.F. The association between GJB2 mutation and GJB6 gene in non syndromic hearing loss school children. Med. J. Malays. 2011, 66, 124–128. [Google Scholar]
- Beck, C.; Perez-Alvarez, J.C.; Sigruener, A.; Haubner, F.; Seidler, T.; Aslanidis, C.; Strutz, J.; Schmitz, G. Identification and genotype/phenotype correlation of mutations in a large German cohort with hearing loss. Eur. Arch. Oto-Rhino-Laryngol. 2015, 272, 2765–2776. [Google Scholar] [CrossRef] [PubMed]
- Javidnia, H.; Carson, N.; Awubwa, M.; Byaruhanga, R.; Mack, D.; Vaccani, J.P. Connexin Gene Mutations Among Ugandan Patients With Nonsyndromic Sensorineural Hearing Loss. Laryngoscope 2014, 124, E373–E376. [Google Scholar] [CrossRef]
- Battelino, S.; Repic Lampret, B.; Zargi, M.; Podkrajsek, K.T. Novel connexin 30 and connexin 26 mutational spectrum in patients with progressive sensorineural hearing loss. J. Laryngol. Otol. 2012, 126, 763–769. [Google Scholar] [CrossRef]
- Oh, S.K.; Choi, S.Y.; Yu, S.H.; Lee, K.Y.; Hong, J.H.; Hur, S.W.; Kim, S.J.; Jeon, C.J.; Kim, U.K. Evaluation of the pathogenicity of GJB3 and GJB6 variants associated with nonsyndromic hearing loss. Biochim. Biophys. Acta 2013, 1832, 285–291. [Google Scholar] [CrossRef]
- Alkowari, M.K.; Vozzi, D.; Bhagat, S.; Krishnamoorthy, N.; Morgan, A.; Hayder, Y.; Logendra, B.; Najjar, N.; Gandin, I.; Gasparini, P.; et al. Targeted sequencing identifies novel variants involved in autosomal recessive hereditary hearing loss in Qatari families. Mutat. Res. 2017, 800, 29–36. [Google Scholar] [CrossRef]
- Belguith, H.; Tlili, A.; Dhouib, H.; Ben Rebeh, I.; Lahmar, I.; Charfeddine, I.; Driss, N.; Ghorbel, A.; Ayadi, H.; Masmoudi, S. Mutation in gap and tight junctions in patients with non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 2009, 385, 1–5. [Google Scholar] [CrossRef]
- Frei, K.; Ramsebner, R.; Hamader, G.; Lucas, T.; Schoefer, C.; Baumgartner, W.D.; Wachtler, F.J.; Kirschhofer, K. Lack of association between Connexin 31 (GJB3) alterations and sensorineural deafness in Austria. Hear. Res. 2004, 194, 81–86. [Google Scholar] [CrossRef]
- Mhatre, A.N.; Weld, E.; Lalwani, A.K. Mutation analysis of Connexin 31 (GJB3) in sporadic non-syndromic hearing impairment. Clin. Genet. 2003, 63, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Alexandrino, F.; Oliveira, C.A.; Reis, F.C.; Maciel-Guerra, A.T.; Sartorato, E.L. Screening for mutations in the GJB3 gene in Brazilian patients with nonsyndromic deafness. J. Appl. Genet. 2004, 45, 249–254. [Google Scholar] [PubMed]
- Li, Y.H.; Jiang, H.; Yang, L.J.; Xu, H.X.; Li, H.; Li, H.W.; Luo, Y.H.; Wang, C.W.; Zou, G.H. Study of mtDNA 12S rRNA A1555G, GJB2, GJB3 gene mutation in Uighur and Han people with hereditary nonsyndromic hearing loss in Xinjiang. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2010, 45, 645–651. [Google Scholar] [PubMed]
- Nahili, H.; Ridal, M.; Boulouiz, R.; Abidi, O.; Imken, L.; Rouba, H.; Alami, M.N.; Chafik, A.; Hassar, M.; Barakat, A. Absence of GJB3 and GJB6 mutations in Moroccan familial and sporadic patients with autosomal recessive non-syndromic deafness. Int. J. Pediatric Otorhinolaryngol. 2008, 72, 1633–1636. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Z.; Yuan, Y.; Yan, D.; Ding, E.H.; Ouyang, X.M.; Fei, Y.; Tang, W.; Yuan, H.; Chang, Q.; Du, L.L.; et al. Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum. Genet. 2009, 125, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, A.R.; Kim, N.K.; Lee, C.; Kim, M.Y.; Jeon, E.H.; Park, W.Y.; Choi, B.Y. Unraveling of Enigmatic Hearing-Impaired GJB2 Single Heterozygotes by Massive Parallel Sequencing: DFNB1 or Not? Medicine 2016, 95, e3029. [Google Scholar] [CrossRef]
- Yang, J.J.; Wang, W.H.; Lin, Y.C.; Weng, H.H.; Yang, J.T.; Hwang, C.F.; Wu, C.M.; Li, S.Y. Prospective variants screening of connexin genes in children with hearing impairment: Genotype/phenotype correlation. Hum. Genet. 2010, 128, 303–313. [Google Scholar] [CrossRef]
- Gao, W.H.; Ke, X.M.; Liu, Y.H.; Zhu, P.; Pan, K.F. Study of the relation between Cx31 gene and hereditary hearing impairment. Zhonghua Er Bi Yan Hou Ke Za Zhi 2004, 39, 344–348. [Google Scholar]
- Chen, K.; Wu, X.; Zong, L.; Jiang, H. GJB3/GJB6 screening in GJB2 carriers with idiopathic hearing loss: Is it necessary? J. Clin. Lab. Anal. 2018, 32, e22592. [Google Scholar] [CrossRef]
- Choi, B.Y.; Park, G.; Gim, J.; Kim, A.R.; Kim, B.J.; Kim, H.S.; Park, J.H.; Park, T.; Oh, S.H.; Han, K.H.; et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS ONE 2013, 8, e68692. [Google Scholar] [CrossRef]
- Xia, J.H.; Liu, C.Y.; Tang, B.S.; Pan, Q.; Huang, L.; Dai, H.P.; Zhang, B.R.; Xie, W.; Hu, D.X.; Zheng, D. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat. Genet. 1998, 20, 370–373. [Google Scholar] [CrossRef]
- de Oliveira, C.A.; Alexandrino, F.; Christiani, T.V.; Steiner, C.E.; Cunha, J.L.; Guerra, A.T.; Sartorato, E.L. Molecular genetics study of deafness in Brazil: 8-year experience. Am. J. Med. Genet. Part A 2007, 143, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Laleh, M.A.; Naseri, M.; Zonouzi, A.A.P.; Zonouzi, A.P.; Masoudi, M.; Ahangari, N.; Shams, L.; Nejatizadeh, A. Diverse pattern of gap junction beta-2 and gap junction beta-4 genes mutations and lack of contribution of DFNB21, DFNB24, DFNB29, and DFNB42 loci in autosomal recessive nonsyndromic hearing loss patients in Hormozgan, Iran. J. Res. Med. Sci. 2017, 22, 1–11. [Google Scholar] [CrossRef]
- Ramchander, P.V.; Panda, K.C.; Panda, A.K. Mutations in the connexin 29 gene are not a major cause of nonsyndromic hearing impairment in India. Genet. Test. Mol. Biomark. 2010, 14, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Yang, J.J.; Lin, Y.C.; Yang, J.T.; Chan, C.H.; Li, S.Y. Identification of novel variants in the Cx29 gene of nonsyndromic hearing loss patients using buccal cells and restriction fragment length polymorphism method. Audiol. Neuro Otol. 2010, 15, 81–87. [Google Scholar] [CrossRef]
- Bosch, J.; Lebeko, K.; Nziale, J.J.; Dandara, C.; Makubalo, N.; Wonkam, A. In search of genetic markers for nonsyndromic deafness in Africa: A study in Cameroonians and Black South Africans with the GJB6 and GJA1 candidate genes. OMICS 2014, 18, 481–485. [Google Scholar] [CrossRef]
- Liu, X.Z.; Xia, X.J.; Adams, J.; Chen, Z.Y.; Welch, K.O.; Tekin, M.; Ouyang, X.M.; Kristiansen, A.; Pandya, A.; Balkany, T.; et al. Mutations in GJA1 (connexin 43) are associated with non-syndromic autosomal recessive deafness. Hum. Mol. Genet. 2001, 10, 2945–2951. [Google Scholar] [CrossRef]
- HGNC. Symbol Report for GJC1. Available online: https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:4280 (accessed on 1 June 2020).
- Ouyang, X.M.; Yan, D.; Aslan, I.; Du, L.L.; Tekin, M.; Liu, X.Z. Mutation screening of the GJA7 (Cx45) gene in a large international series of probands with nonsyndromic hearing impairment. Genet. Test. Mol. Biomark. 2011, 15, 333–336. [Google Scholar] [CrossRef]
- Srinivas, M.; Jannace, T.F.; Cocozzelli, A.G.; Li, L.; Slavi, N.; Sellitto, C.; White, T.W. Connexin43 mutations linked to skin disease have augmented hemichannel activity. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef]
- Liu, W. Journal of Translational Medicine Advances in Translational Genomics and Genetics Era; BioMed Central: London, UK, 2019. [Google Scholar]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers, Inc.: New York, NY, USA, 2019; pp. 661–673. [Google Scholar]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Rabionet, R.; Gasparini, P.; Estivill, X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum. Mutat. 2000, 16, 190–202. [Google Scholar] [CrossRef]
- Adadey, S.M.; Manyisa, N.; Mnika, K.; de Kock, C.; Nembaware, V.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. GJB2 and GJB6 Mutations in Non-Syndromic Childhood Hearing Impairment in Ghana. Front. Genet. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, H.; Kupsch, P.; Sudendey, J.; Winterhager, E.; Jahnke, K.; Lautermann, J. Mutations in the connexin26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum. Mutat. 2001, 17, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Samanich, J.; Lowes, C.; Burk, R.; Shanske, S.; Lu, J.; Shanske, A.; Morrow, B.E. Mutations in GJB2, GJB6, and mitochondrial DNA are rare in African American and Caribbean Hispanic individuals with hearing impairment. Am. J. Med. Genet. Part A 2007, 143, 830–838. [Google Scholar] [CrossRef]
- Tsukada, K.; Nishio, S.-Y.; Hattori, M.; Usami, S.-I. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124, 61S–76S. [Google Scholar] [CrossRef]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef]
- Bosch, J.; Noubiap, J.J.; Dandara, C.; Makubalo, N.; Wright, G.; Entfellner, J.B.; Tiffin, N.; Wonkam, A. Sequencing of GJB2 in Cameroonians and Black South Africans and comparison to 1000 Genomes Project Data Support Need to Revise Strategy for Discovery of Nonsyndromic Deafness Genes in Africans. OMICS 2014, 18, 705–710. [Google Scholar] [CrossRef]
- Tingang Wonkam, E.; Chimusa, E.; Noubiap, J.J.; Adadey, S.M.; F Fokouo, J.V.; Wonkam, A. GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon. Genes 2019, 10, 844. [Google Scholar] [CrossRef]
- Abidi, O.; Boulouiz, R.; Nahili, H.; Ridal, M.; Alami, M.N.; Tlili, A.; Rouba, H.; Masmoudi, S.; Chafik, A.; Hassar, M. GJB2 (connexin 26) gene mutations in Moroccan patients with autosomal recessive non-syndromic hearing loss and carrier frequency of the common GJB2–35delG mutation. Int. J. Pediatric Otorhinolaryngol. 2007, 71, 1239–1245. [Google Scholar] [CrossRef]
- Bakhchane, A.; Bousfiha, A.; Charoute, H.; Salime, S.; Detsouli, M.; Snoussi, K.; Nadifi, S.; Kabine, M.; Rouba, H.; Dehbi, H.; et al. Update of the spectrum of GJB2 gene mutations in 152 Moroccan families with autosomal recessive nonsyndromic hearing loss. Eur. J. Med. Genet. 2016, 59, 325–329. [Google Scholar] [CrossRef]
- Gazzaz, B.; Weil, D.; Rais, L.; Akhyat, O.; Azeddoug, H.; Nadifi, S. Autosomal recessive and sporadic deafness in Morocco: High frequency of the 35delG GJB2 mutation and absence of the 342-kb GJB6 variant. Hear. Res. 2005, 210, 80–84. [Google Scholar] [CrossRef]
- Moctar, E.C.; Riahi, Z.; El Hachmi, H.; Veten, F.; Meiloud, G.; Bonnet, C.; Abdelhak, S.; Errami, M.; Houmeida, A. Etiology and associated GJB2 mutations in Mauritanian children with non-syndromic hearing loss. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 3693–3698. [Google Scholar] [CrossRef]
- Ratbi, I.; Hajji, S.; Ouldim, K.; Aboussair, N.; Feldmann, D.; Sefiani, A. The mutation 35delG of the gene of the connexin 26 is a frequent cause of autosomal-recessive non-syndromic hearing loss in Morocco. Arch. Pediatrie 2007, 14, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Green, G.E.; Scott, D.A.; McDonald, J.M.; Woodworth, G.G.; Sheffield, V.C.; Smith, R.J. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA 1999, 281, 2211–2216. [Google Scholar] [CrossRef]
- Hall, A.; Pembrey, M.; Lutman, M.; Steer, C.; Bitner-Glindzicz, M. Prevalence and audiological features in carriers of GJB2 mutations, c.35delG and c.101T>C (p.M34T), in a UK population study. BMJ Open 2012, 2, e001238. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Fang, P.; Ward, P.A.; Schmitt, E.; Darilek, S.; Manolidis, S.; Oghalai, J.S.; Roa, B.B.; Alford, R.L. DNA sequence analysis of GJB2, encoding connexin 26: Observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am. J. Med. Genet. Part A 2006, 140, 2401–2415. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Yu, F.; Han, B.; Yuan, Y.; Li, Q.; Wang, G.; Liu, X.; He, J.; Huang, D.; Kang, D.; et al. The prevalence of the 235delC GJB2 mutation in a Chinese deaf population. Genet. Med. 2007, 9, 283–289. [Google Scholar] [CrossRef]
- Ohtsuka, A.; Yuge, I.; Kimura, S.; Namba, A.; Abe, S.; Van Laer, L.; Van Camp, G.; Usami, S. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum. Genet. 2003, 112, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Usami, S.; Shinkawa, H.; Kelley, P.M.; Kimberling, W.J. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J. Med. Genet. 2000, 37, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Hahn, S.H.; Chun, Y.M.; Park, K.; Kim, H.N. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope 2000, 110, 1535–1538. [Google Scholar] [CrossRef]
- Hwa, H.L.; Ko, T.M.; Hsu, C.J.; Huang, C.H.; Chiang, Y.L.; Oong, J.L.; Chen, C.C.; Hsu, C.K. Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genet. Med. 2003, 5, 161–165. [Google Scholar] [CrossRef]
- Bason, L.; Dudley, T.; Lewis, K.; Shah, U.; Potsic, W.; Ferraris, A.; Fortina, P.; Rappaport, E.; Krantz, I.D. Homozygosity for the V37I Connexin 26 mutation in three unrelated children with sensorineural hearing loss. Clin. Genet. 2002, 61, 459–464. [Google Scholar] [CrossRef]
- Wattanasirichaigoon, D.; Limwongse, C.; Jariengprasert, C.; Yenchitsomanus, P.T.; Tocharoenthanaphol, C.; Thongnoppakhun, W.; Thawil, C.; Charoenpipop, D.; Pho-iam, T.; Thongpradit, S.; et al. High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing-impaired and control Thai individuals. Clin. Genet. 2004, 66, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Huang, B.Q.; Wang, G.J.; Yuan, Y.Y.; Dai, P. The Relationship between the p.V37I Mutation in GJB2 and Hearing Phenotypes in Chinese Individuals. PLoS ONE 2015, 10, e0129662. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Liu, X.-W.; Wang, J.-C.; Wang, S.-Y.; Li, S.-J.; Zhu, Y.-M.; Ding, W.-J.; Xu, C.-Y.; Duan, L.; Xu, B.-C.; Guo, Y.-F. The mutation frequencies of GJB2, GJB3, SLC26A4 and MT-RNR1 of patients with severe to profound sensorineural hearing loss in northwest China. Int. J. Pediatric Otorhinolaryngol. 2020, 110143. [Google Scholar] [CrossRef]
- Liu, Y.; Ke, X.; Qi, Y.; Li, W.; Zhu, P. Connexin26 gene (GJB2): Prevalence of mutations in the Chinese population. J. Hum. Genet. 2002, 47, 0688–0690. [Google Scholar] [CrossRef]
- Alvarez, A.; del Castillo, I.; Villamar, M.; Aguirre, L.A.; Gonzalez-Neira, A.; Lopez-Nevot, A.; Moreno-Pelayo, M.A.; Moreno, F. High prevalence of the W24X mutation in the gene encoding connexin-26 (GJB2) in Spanish Romani (gypsies) with autosomal recessive non-syndromic hearing loss. Am. J. Med. Genet. Part A 2005, 137, 255–258. [Google Scholar] [CrossRef]
- Radulescu, L.; Martu, C.; Birkenhager, R.; Cozma, S.; Ungureanu, L.; Laszig, R. Prevalence of mutations located at the dfnb1 locus in a population of cochlear implanted children in eastern Romania. Int. J. Pediatric Otorhinolaryngol. 2012, 76, 90–94. [Google Scholar] [CrossRef]
- RamShankar, M.; Girirajan, S.; Dagan, O.; Ravi Shankar, H.M.; Jalvi, R.; Rangasayee, R.; Avraham, K.B.; Anand, A. Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J. Med. Genet. 2003, 40, e68. [Google Scholar] [CrossRef] [PubMed]
- Dzhemileva, L.U.; Barashkov, N.A.; Posukh, O.L.; Khusainova, R.I.; Akhmetova, V.L.; Kutuev, I.A.; Gilyazova, I.R.; Tadinova, V.N.; Fedorova, S.A.; Khidiyatova, I.M.; et al. Carrier frequency of GJB2 gene mutations c.35delG, c.235delC and c.167delT among the populations of Eurasia. J. Hum. Genet. 2010, 55, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Mahasneh, A.; Battah, R. Prevalence of connexin 26 mutations in patients from Jordan with non syndromic hearing loss. Int. J. Hum. Genet. 2006, 6, 119–124. [Google Scholar] [CrossRef]
- Niceta, M.; Fabiano, C.; Sammarco, P.; Piccione, M.; Antona, V.; Giuffre, M.; Corsello, G. Epidemiological study of nonsyndromic hearing loss in Sicilian newborns. Am. J. Med. Genet. Part A 2007, 143, 1666–1670. [Google Scholar] [CrossRef]
- Putcha, G.V.; Bejjani, B.A.; Bleoo, S.; Booker, J.K.; Carey, J.C.; Carson, N.; Das, S.; Dempsey, M.A.; Gastier-Foster, J.M.; Greinwald, J.H., Jr.; et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet. Med. 2007, 9, 413–426. [Google Scholar] [CrossRef]
- Adadey, S.M.; Tingang Wonkam, E.; Twumasi Aboagye, E.; Quansah, D.; Asante-Poku, A.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. Enhancing Genetic Medicine: Rapid and Cost-Effective Molecular Diagnosis for a GJB2 Founder Mutation for Hearing Impairment in Ghana. Genes 2020, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Brobby, G.W.; Muller-Myhsok, B.; Horstmann, R.D. Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N. Engl. J. Med. 1998, 338, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Martinez, A.; Requena, T.; Roman-Naranjo, P.; Lopez-Escamez, J.A. Excess of rare missense variants in hearing loss genes in sporadic Meniere disease. Front. Genet. 2019, 10, 76. [Google Scholar] [CrossRef]
- Oosterveld, W. Meniere’s disease, signs and symptoms. J. Laryngol. Otol. 1980, 94, 885–892. [Google Scholar] [CrossRef]
- Patel, V.; Oberman, B.; Zacharia, T.; Isildak, H. Magnetic resonance imaging findings in Ménière’s disease. J. Laryngol. Otol. 2017, 131, 602. [Google Scholar] [CrossRef]
- Romanov, G.; Barashkov, N.; Teryutin, F.; Lashin, S.; Solovyev, A.; Pshennikova, V.; Bondar, A.; Morozov, I.; Sazonov, N.; Tomsky, M. Marital Structure, Genetic Fitness, and the GJB2 Gene Mutations among Deaf People in Yakutia (Eastern Siberia, Russia). Russ. J. Genet. 2018, 54, 554–561. [Google Scholar] [CrossRef]
- Barashkov, N.A.; Pshennikova, V.G.; Posukh, O.L.; Teryutin, F.M.; Solovyev, A.V.; Klarov, L.A.; Romanov, G.P.; Gotovtsev, N.N.; Kozhevnikov, A.A.; Kirillina, E.V. Spectrum and frequency of the GJB2 gene pathogenic variants in a large cohort of patients with hearing impairment living in a subarctic region of Russia (the Sakha Republic). PLoS ONE 2016, 11, e0156300. [Google Scholar] [CrossRef] [PubMed]
- Posukh, O.L.; Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Bondar, A.A.; Morozov, I.V.; Maximov, V.N.; Voevoda, M.I. Unique mutational spectrum of the GJB2 gene and its pathogenic contribution to deafness in tuvinians (Southern siberia, russia): A high prevalence of rare variant c. 516G>C (p. trp172Cys). Genes 2019, 10, 429. [Google Scholar] [CrossRef] [PubMed]
- Carranza, C.; Menendez, I.; Herrera, M.; Castellanos, P.; Amado, C.; Maldonado, F.; Rosales, L.; Escobar, N.; Guerra, M.; Alvarez, D.; et al. A Mayan founder mutation is a common cause of deafness in Guatemala. Clin. Genet. 2016, 89, 461–465. [Google Scholar] [CrossRef]
- Paz-y-Miño, C.; Beaty, D.; López-Cortés, A.; Proaño, I. Frequency of GJB2 and del(GJB6-D13S1830) mutations among an Ecuadorian mestizo population. Int. J. Pediatric Otorhinolaryngol. 2014, 78, 1648–1654. [Google Scholar] [CrossRef]
- Tamayo, M.; Olarte, M.; Gelvez, N.; Gómez, M.; Frías, J.; Bernal, J.; Florez, S.; Medina, D. Molecular studies in the GJB2 gene (Cx26) among a deaf population from Bogotá, Colombia: Results of a screening program. Int. J. Pediatric Otorhinolaryngol. 2009, 73, 97–101. [Google Scholar] [CrossRef]
- Azadegan-Dehkordi, F.; Bahrami, T.; Shirzad, M.; Karbasi, G.; Yazdanpanahi, N.; Farrokhi, E.; Koohiyan, M.; Tabatabaiefar, M.A.; Hashemzadeh-Chaleshtori, M. Mutations in GJB2 as Major Causes of Autosomal Recessive Non-Syndromic Hearing Loss: First Report of c.299-300delAT Mutation in Kurdish Population of Iran. J. Audiol. Otol. 2019, 23, 20–26. [Google Scholar] [CrossRef]
- Khalifa Alkowari, M.; Girotto, G.; Abdulhadi, K.; Dipresa, S.; Siam, R.; Najjar, N.; Badii, R.; Gasparini, P. GJB2 and GJB6 genes and the A1555G mitochondrial mutation are only minor causes of nonsyndromic hearing loss in the Qatari population. Int. J. Audiol. 2012, 51, 181–185. [Google Scholar] [CrossRef]
- Posukh, O.; Pallares-Ruiz, N.; Tadinova, V.; Osipova, L.; Claustres, M.; Roux, A.F. First molecular screening of deafness in the Altai Republic population. BMC Med. Genet. 2005, 6, 12. [Google Scholar] [CrossRef]
- Bliznetz, E.A.; Galkina, V.A.; Matyushchenko, G.N.; Kisina, A.G.; Markova, T.G.; Polyakov, A.V. Changes in the connexin 26 gene (GJB2) in Russian patients with hearing loss: Results of long-term molecular diagnostics of hereditary nonsyndromic hearing loss. Russ. J. Genet. 2012, 48, 101–112. [Google Scholar] [CrossRef]
- Pollak, A.; Skorka, A.; Mueller-Malesinska, M.; Kostrzewa, G.; Kisiel, B.; Waligora, J.; Krajewski, P.; Oldak, M.; Korniszewski, L.; Skarzynski, H.; et al. M34T and V37I mutations in GJB2 associated hearing impairment: Evidence for pathogenicity and reduced penetrance. Am. J. Med. Genet. Part A 2007, 143, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yu, F.; Wang, G.; Huang, S.; Yu, R.; Zhang, X.; Huang, D.; Han, D.; Dai, P. Prevalence of the GJB2 IVS1+1G>A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2. J. Transl. Med. 2010, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Akcayoz-Duman, D.; Tekin, M. The c. IVS1+ 1G> A mutation inthe GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J. Genet. 2006, 85, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Hennemann, H.; Suchyna, T.; Lichtenberg-Fraté, H.; Jungbluth, S.; Dahl, E.; Schwarz, J.; Nicholson, B.J.; Willecke, K. Molecular cloning and functional expression of mouse connexin40, a second gap junction gene preferentially expressed in lung. J. Cell Biol. 1992, 117, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Eiberger, J.; Jung, Y.T.; Kozak, C.A.; Willecke, K. The mouse gap junction gene connexin29 is highly expressed in sciatic nerve and regulated during brain development. Biol. Chem. 2001, 382, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Lautermann, J.; Frank, H.G.; Jahnke, K.; Traub, O.; Winterhager, E. Developmental expression patterns of connexin26 and-30 in the rat cochlea. Dev. Genet. 1999, 25, 306–311. [Google Scholar] [CrossRef]
- Feldmann, D.; Le Maréchal, C.; Jonard, L.; Thierry, P.; Czajka, C.; Couderc, R.; Ferec, C.; Denoyelle, F.; Marlin, S.; Fellmann, F. A new large deletion in the DFNB1 locus causes nonsyndromic hearing loss. Eur. J. Med. Genet. 2009, 52, 195–200. [Google Scholar] [CrossRef]
- Tayoun, A.N.A.; Mason-Suares, H.; Frisella, A.L.; Bowser, M.; Duffy, E.; Mahanta, L.; Funke, B.; Rehm, H.L.; Amr, S.S. Targeted droplet-digital PCR as a tool for novel deletion discovery at the DFNB1 locus. Hum. Mutat. 2016, 37, 119–126. [Google Scholar] [CrossRef]
- Wilch, E.; Azaiez, H.; Fisher, R.A.; Elfenbein, J.; Murgia, A.; Birkenhäger, R.; Bolz, H.; Da Silva-Costa, S.; Del Castillo, I.; Haaf, T. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin. Genet. 2010, 78, 267–274. [Google Scholar] [CrossRef]
- Bliznetz, E.A.; Lalayants, M.R.; Markova, T.G.; Balanovsky, O.P.; Balanovska, E.V.; Skhalyakho, R.A.; Pocheshkhova, E.A.; Nikitina, N.V.; Voronin, S.V.; Kudryashova, E.K.; et al. Update of the GJB2/DFNB1 mutation spectrum in Russia: A founder Ingush mutation del(GJB2-D13S175) is the most frequent among other large deletions. J. Hum. Genet. 2017, 62, 789–795. [Google Scholar] [CrossRef][Green Version]
- Pandya, A.; O’Brien, A.; Kovasala, M.; Bademci, G.; Tekin, M.; Arnos, K.S. Analyses of del (GJB6-D13S1830) and del (GJB6-D13S1834) deletions in a large cohort with hearing loss: Caveats to interpretation of molecular test results in multiplex families. Mol. Genet. Genom. Med. 2020, 8, e1171. [Google Scholar] [CrossRef]
- Rodriguez-Paris, J.; Schrijver, I. The digenic hypothesis unraveled: The GJB6 del (GJB6-D13S1830) mutation causes allele-specific loss of GJB2 expression in cis. Biochem. Biophys. Res. Commun. 2009, 389, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Tang, W.; Chang, Q.; Qu, Y.; Hibshman, J.; Li, Y.; Söhl, G.; Willecke, K.; Chen, P.; Lin, X. Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc. Natl. Acad. Sci. USA 2007, 104, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Teubner, B.; Michel, V.; Pesch, J.; Lautermann, J.; Cohen-Salmon, M.; Söhl, G.; Jahnke, K.; Winterhager, E.; Herberhold, C.; Hardelin, J.-P. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 2003, 12, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, I.; Moreno-Pelayo, M.A.; Del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; Chamberlin, G.P.; et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: A multicenter study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef]
- Kabahuma, R.I.; Ouyang, X.; Du, L.L.; Yan, D.; Hutchin, T.; Ramsay, M.; Penn, C.; Liu, X.-Z. Absence of GJB2 gene mutations, the GJB6 deletion (GJB6-D13S1830) and four common mitochondrial mutations in nonsyndromic genetic hearing loss in a South African population. Int. J. Pediatric Otorhinolaryngol. 2011, 75, 611–617. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tumminelli, G.; Di Donato, I.; Guida, V.; Rufa, A.; De Luca, A.; Federico, A. Oculodentodigital dysplasia with massive brain calcification and a new mutation of GJA1 gene. J. Alzheimer’s Dis. 2016, 49, 27–30. [Google Scholar] [CrossRef]
- Kjaer, K.W.; Hansen, L.; Eiberg, H.; Leicht, P.; Opitz, J.M.; Tommerup, N. Novel Connexin 43 (GJA1) mutation causes oculo–dento–digital dysplasia with curly hair. Am. J. Med. Genet. Part A 2004, 127, 152–157. [Google Scholar] [CrossRef]
- Paznekas, W.A.; Karczeski, B.; Vermeer, S.; Lowry, R.B.; Delatycki, M.; Laurence, F.; Koivisto, P.A.; Van Maldergem, L.; Boyadjiev, S.A.; Bodurtha, J.N. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 2009, 30, 724–733. [Google Scholar] [CrossRef]
- Paznekas, W.A.; Boyadjiev, S.A.; Shapiro, R.E.; Daniels, O.; Wollnik, B.; Keegan, C.E.; Innis, J.W.; Dinulos, M.B.; Christian, C.; Hannibal, M.C. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 2003, 72, 408–418. [Google Scholar] [CrossRef]
- Hong, H.-M.; Yang, J.-J.; Shieh, J.-C.; Li, M.-L.; Li, S.-Y. Novel mutations in the connexin43 (GJA1) and GJA1 pseudogene may contribute to nonsyndromic hearing loss. Hum. Genet. 2010, 127, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Kandouz, M.; Bier, A.; Carystinos, G.D.; Alaoui-Jamali, M.A.; Batist, G. Connexin43 pseudogene is expressed in tumor cells and inhibits growth. Oncogene 2004, 23, 4763–4770. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Kerrebrouck, M.; Wang, N.; Leybaert, L. Neurological manifestations of oculodentodigital dysplasia: A Cx43 channelopathy of the central nervous system? Front. Pharmacol. 2013, 4, 120. [Google Scholar] [CrossRef]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E. Mouse genome database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef]
- Vozzi, C.; Dupont, E.; Coppen, S.R.; Yeh, H.-I.; Severs, N.J. Chamber-related differences in connexin expression in the human heart. J. Mol. Cell. Cardiol. 1999, 31, 991–1003. [Google Scholar] [CrossRef]
- National_Library_of_Medicine (US). Genetics Home Reference [Internet] Bethesda (MD): The Library. 16 September 2013. Available online: https://ghr.nlm.nih.gov/ (accessed on 11 August 2020).
- Huang, S.; Huang, B.; Wang, G.; Kang, D.Y.; Zhang, X.; Meng, X.; Dai, P. The relationship between the GJB3 c.538C>T variant and hearing phenotype in the Chinese population. Int. J. Pediatric Otorhinolaryngol. 2017, 102, 67–70. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Zheng-Fischhöfer, Q.; Schnichels, M.; Dere, E.; Strotmann, J.; Loscher, N.; McCulloch, F.; Kretz, M.; Degen, J.; Reucher, H.; Nagy, J.I. Characterization of connexin30. 3-deficient mice suggests a possible role of connexin30. 3 in olfaction. Eur. J. Cell Biol. 2007, 86, 683–700. [Google Scholar]
- Wang, W.-H.; Yang, J.-J.; Lin, Y.-C.; Yang, J.-T.; Li, S.-Y. Novel expression patterns of connexin 30.3 in adult rat cochlea. Hear. Res. 2010, 265, 77–82. [Google Scholar] [CrossRef]
- Eiberger, J.; Kibschull, M.; Strenzke, N.; Schober, A.; Büssow, H.; Wessig, C.; Djahed, S.; Reucher, H.; Koch, D.A.; Lautermann, J. Expression pattern and functional characterization of connexin29 in transgenic mice. GLIA 2006, 53, 601–611. [Google Scholar] [CrossRef]
- Sohani, Z.; Sarma, S.; Alyass, A.; De Souza, R.; Robiou-du-Pont, S.; Li, A.; Mayhew, A.; Yazdi, F.; Reddon, H.; Lamri, A. Empirical evaluation of the Q-Genie tool: A protocol for assessment of effectiveness. BMJ Open 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Hoy, D.; Brooks, P.; Woolf, A.; Blyth, F.; March, L.; Bain, C.; Baker, P.; Smith, E.; Buchbinder, R. Assessing risk of bias in prevalence studies: Modification of an existing tool and evidence of interrater agreement. J. Clin. Epidemiol. 2012, 65, 934–939. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).