Ultradeep Microbial Communities at 4.4 km within Crystalline Bedrock: Implications for Habitability in a Planetary Context

 ,
, 
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Descriptions
2.2. Sampling
2.3. Sample Preparation for Molecular Biology Analyses
2.4. Sequencing of the Microbial Community
2.5. Quantification of Taxonomic and Functional Marker Genes
2.6. Sequence Data Analyses
2.7. Diversity Indices
2.8. Data Deposition
3. Results
3.1. Geochemistry
3.2. Low Biomass Environment
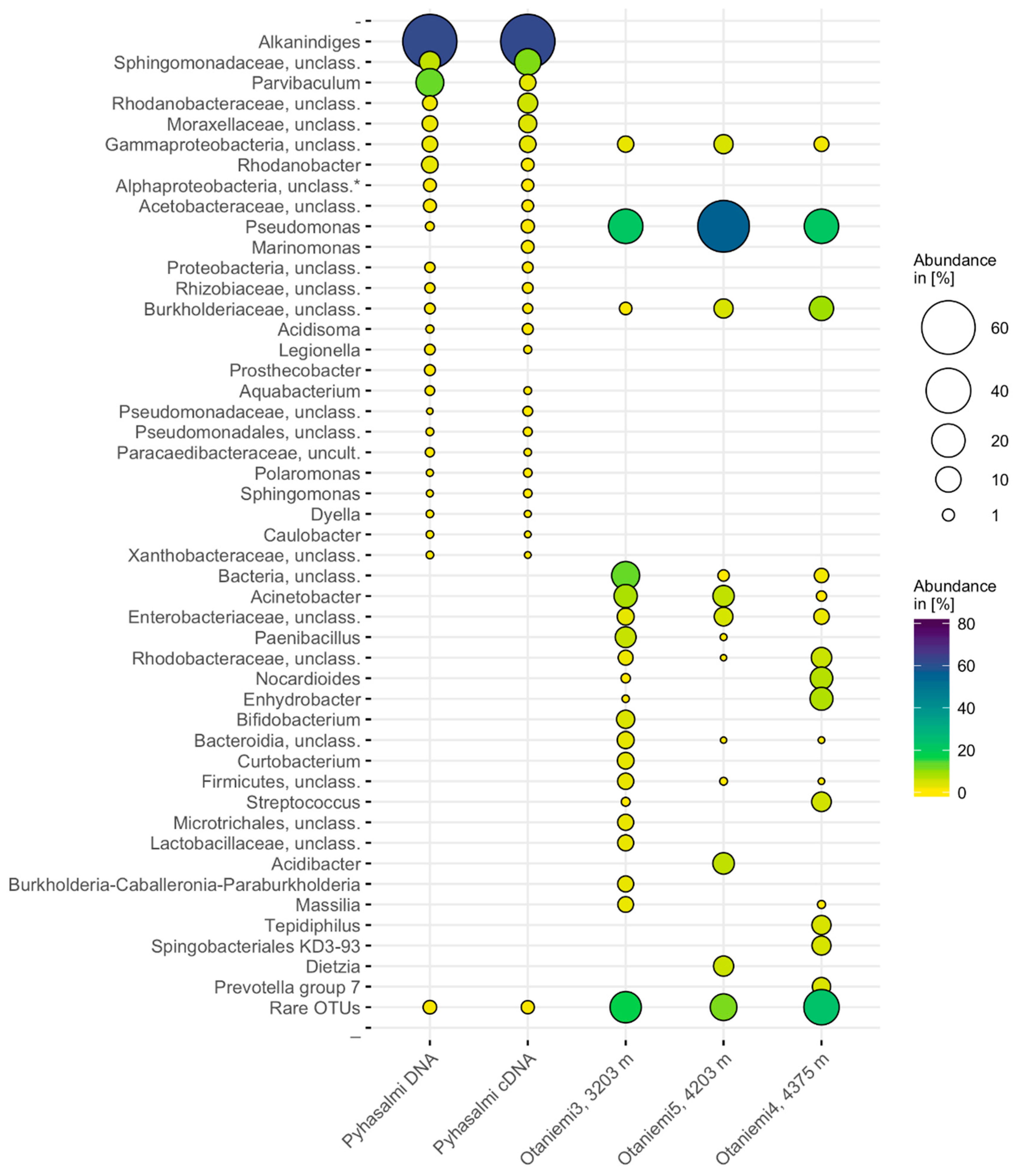
3.3. Microbial Community Composition
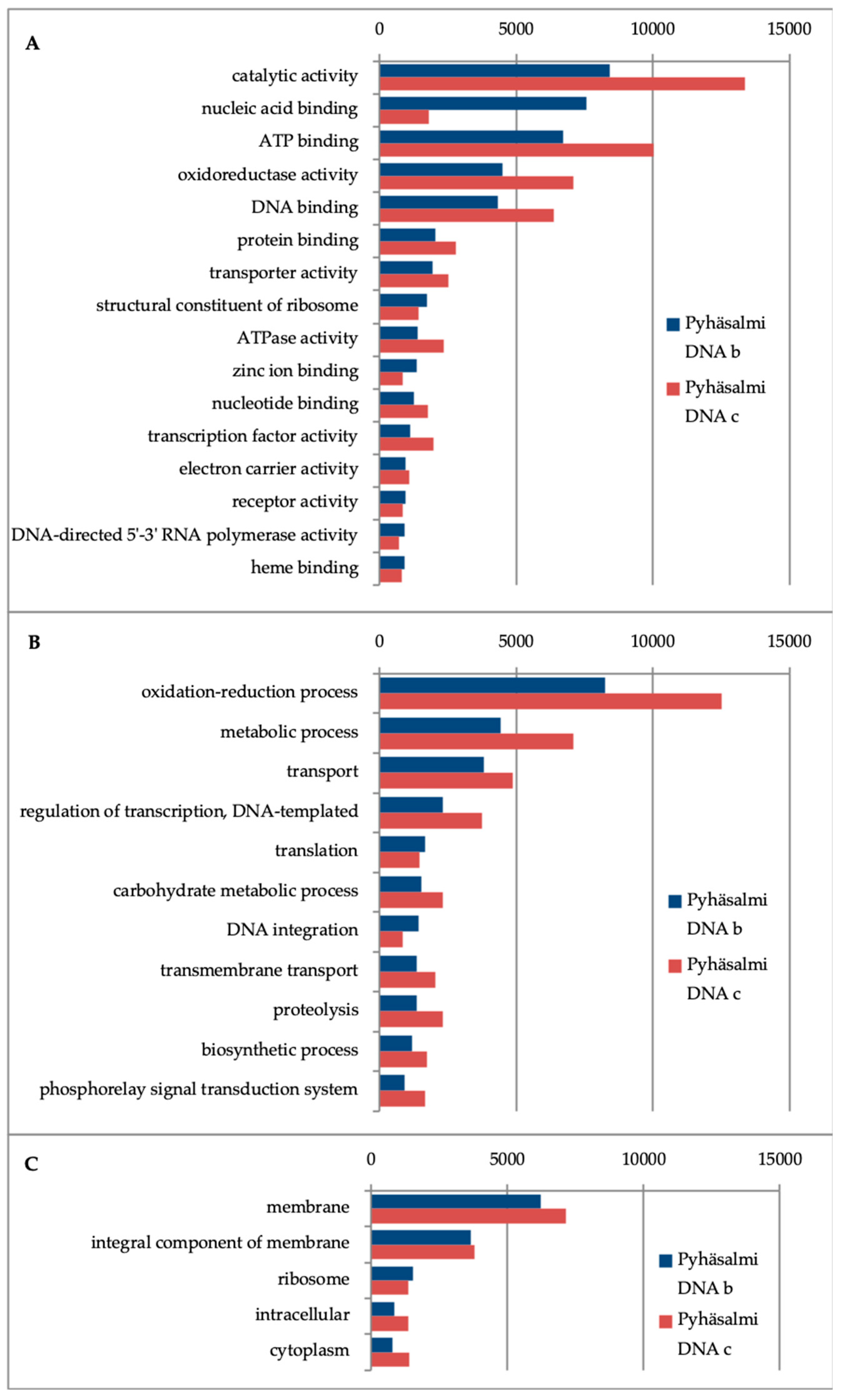
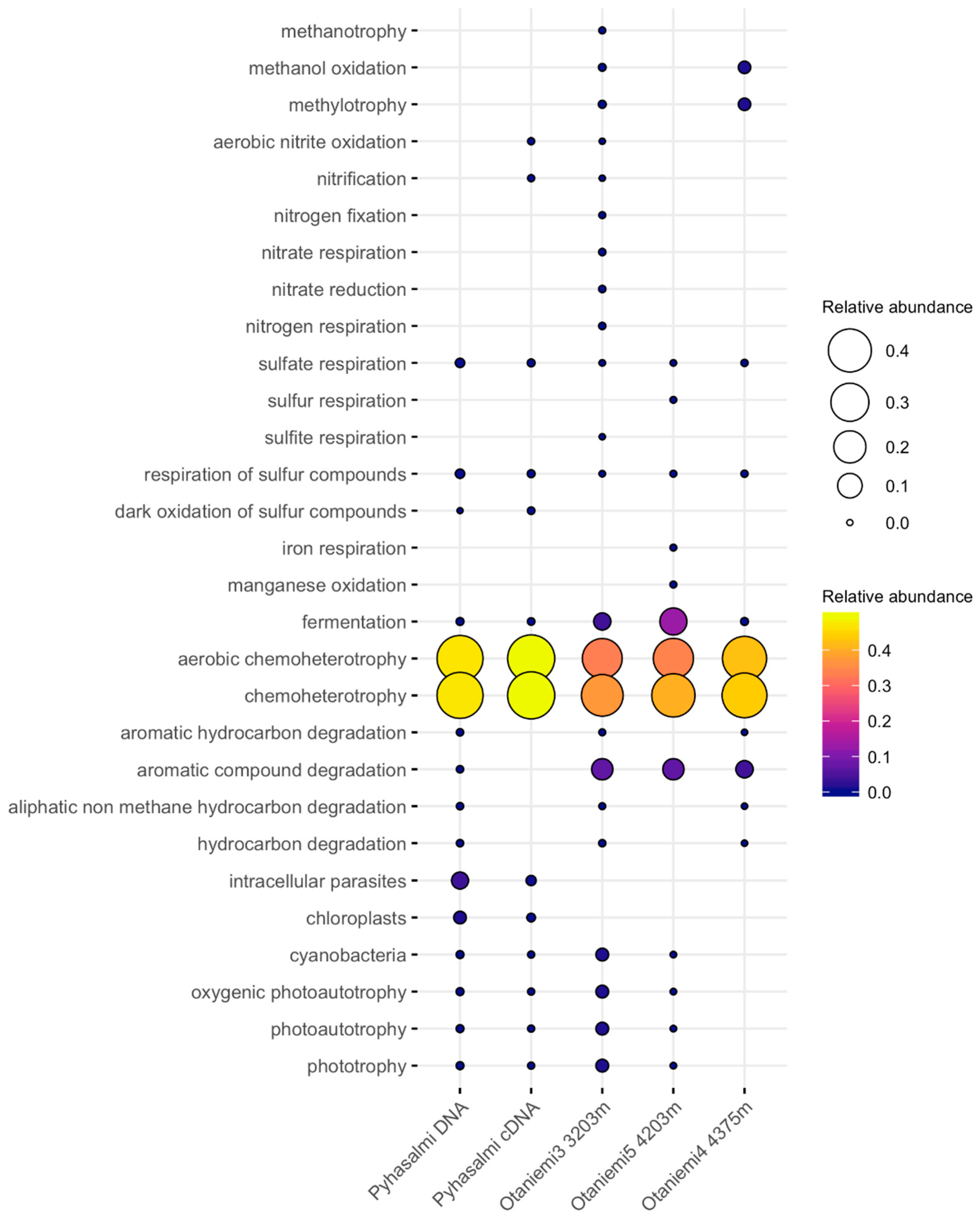
3.4. Microbial Functionality
4. Discussion
4.1. Habitats Hosting Low Biomass
4.2. Microbial Community Structure
4.3. Metabolic Capacities of Microbial Communities
4.4. Considerations on Contamination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rekola, R.T.F. Life and habitable zones in the Universe. Planet. Space Sci. 2009, 57, 430–433. [Google Scholar] [CrossRef]
- Cockell, C.S.; Bush, T.; Bryce, C.; Direito, S.; Fox-Powell, M.; Harrison, J.P.; Lammer, H.; Landenmark, H.; Martin-Torres, J.; Nicholson, N.; et al. Habitability: A Review. Astrobiology 2016, 16, 89–117. [Google Scholar] [CrossRef] [PubMed]
- Scharf, C.; Cronin, L. Quantifying the origins of life on a planetary scale. Proc. Natl. Acad. Sci. USA 2016, 113, 8127–8132. [Google Scholar] [CrossRef]
- Harrison, J.P.; Gheeraert, N.; Tsigelnitskiy, D.; Cockell, C.S. The limits for life under multiple extremes. Trends Microbiol. 2013, 21, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Goordial, J.M.; Orcutt, B.N. Low energy subsurface environments as extraterrestrial analogs. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Preston, L.J.; Dartnell, L.R. Planetary habitability: Lessons learned from terrestrial analogues. Int. J. Astrobiol. 2014, 13, 81–98. [Google Scholar] [CrossRef]
- Escudero, C.; Oggerin, M.; Amils, R. The deep continental subsurface: The dark biosphere. Int. Microbiol. 2018, 21, 3–14. [Google Scholar] [CrossRef]
- Stotler, R.L.; Frape, S.K.; Ruskeeniemi, T.; Ahonen, L.; Onstott, T.C.; Hobbs, M.Y. Hydrogeochemistry of groundwaters in and below the base of thick permafrost at Lupin, Nunavut, Canada. J. Hydrol. 2009, 373, 80–95. [Google Scholar] [CrossRef]
- Onstott, T.C.; Lin, L.-H.H.; Davidson, M.; Mislowack, B.; Borcsik, M.; Hall, J.; Slater, G.; Ward, J.; Lollar, B.S.; Lippmann-Pipke, J.; et al. The origin and age of biogeochemical trends in deep fracture water of the Witwatersrand Basin, South Africa. Geomicrobiol. J. 2006, 23, 369–414. [Google Scholar] [CrossRef]
- Haveman, S.A.; Pedersen, K. Microbially mediated redox processes in natural analogues for radioactive waste. J. Contam. Hydrol. 2002, 55, 161–174. [Google Scholar] [CrossRef]
- Haveman, S.A.; Pedersen, K.; Ruotsalainen, P. Distribution and metabolic diversity of microorganisms in deep igneous rock aquifers of Finland. Geomicrobiol. J. 1999, 16, 277–294. [Google Scholar] [CrossRef]
- Dutta, A.; Dutta Gupta, S.; Gupta, A.; Sarkar, J.; Roy, S.; Mukherjee, A.; Sar, P. Exploration of deep terrestrial subsurface microbiome in Late Cretaceous Deccan traps and underlying Archean basement, India. Sci. Rep. 2018, 8, 17459. [Google Scholar] [CrossRef] [PubMed]
- Holland, G.; Lollar, B.S.; Li, L.; Lacrampe-Couloume, G.; Slater, G.F.; Ballentine, C.J. Deep fracture fluids isolated in the crust since the Precambrian era. Nature 2013, 497, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Chivian, D.; Brodie, E.L.; Alm, E.J.; Culley, D.E.; Dehal, P.S.; DeSantis, T.Z.; Gihring, T.M.; Lapidus, A.; Lin, L.-H.H.; Lowry, S.R.; et al. Environmental genomics reveals a single-species ecosystem deep within Earth. Science 2008, 322, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Borgonie, G.; García-Moyano, A.; Litthauer, D.; Bert, W.; Bester, A.; van Heerden, E.; Möller, C.; Erasmus, M.; Onstott, T.C. Nematoda from the terrestrial deep subsurface of South Africa. Nature 2011, 474, 79–82. [Google Scholar] [CrossRef]
- Baker, B.J.; Saw, J.H.; Lind, A.E.; Lazar, C.S.; Hinrichs, K.-U.; Teske, A.P.; Ettema, T.J.G. Genomic inference of the metabolism of cosmopolitan subsurface Archaea, Hadesarchaea. Nat. Microbiol. 2016, 1, 16002. [Google Scholar] [CrossRef]
- Takai, K.; Moser, D.P.; DeFlaun, M.; Onstott, T.C.; Fredrickson, J.K. Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol. 2001, 67, 5750–5760. [Google Scholar] [CrossRef]
- Gihring, T.M.; Moser, D.P.; Lin, L.H.; Davidson, M.; Onstott, T.C.; Morgan, L.; Milleson, M.; Kieft, T.L.; Trimarco, E.; Balkwill, D.L.; et al. The distribution of microbial taxa in the subsurface water of the Kalahari Shield, South Africa. Geomicrobiol. J. 2006, 23, 415–430. [Google Scholar] [CrossRef]
- Shimizu, S.; Akiyama, M.; Ishijima, Y.; Hama, K.; Kunimaru, T.; Naganuma, T. Molecular characterization of microbial communities in fault-bordered aquifers in the Miocene formation of northernmost Japan. Geobiology 2006, 4, 203–213. [Google Scholar] [CrossRef]
- Fukuda, A.; Hagiwara, H.; Ishimura, T.; Kouduka, M.; Ioka, S.; Amano, Y.; Tsunogai, U.; Suzuki, Y.; Mizuno, T. Geomicrobiological properties of ultra-deep granitic groundwater from the Mizunami Underground Research Laboratory (MIU), central Japan. Microb. Ecol. 2010, 60, 214–225. [Google Scholar] [CrossRef]
- Purkamo, L.; Bomberg, M.; Kietäväinen, R.; Salavirta, H.; Nyyssönen, M.; Nuppunen-Puputti, M.; Ahonen, L.; Kukkonen, I.; Itävaara, M. Microbial co-occurrence patterns in deep Precambrian bedrock fracture fluids. Biogeosciences 2016, 13. [Google Scholar] [CrossRef]
- Magnabosco, C.; Ryan, K.; Lau, M.C.Y.; Kuloyo, O.; Sherwood Lollar, B.; Kieft, T.L.; van Heerden, E.; Onstott, T.C. A metagenomic window into carbon metabolism at 3 km depth in Precambrian continental crust. ISME J. 2016, 10, 730. [Google Scholar] [CrossRef] [PubMed]
- Simkus, D.N.; Slater, G.F.; Lollar, B.S.; Wilkie, K.; Kieft, T.L.; Magnabosco, C.; Lau, M.C.Y.Y.; Pullin, M.J.; Hendrickson, S.B.; Wommack, K.E.; et al. Variations in microbial carbon sources and cycling in the deep continental subsurface. Geochim. Cosmochim. Acta 2016, 173, 264–283. [Google Scholar] [CrossRef]
- Wu, X.; Holmfeldt, K.; Hubalek, V.; Lundin, D.; Åström, M.; Bertilsson, S.; Dopson, M. Microbial metagenomes from three aquifers in the Fennoscandian shield terrestrial deep biosphere reveal metabolic partitioning among populations. ISME J. 2016, 1192–1203. [Google Scholar] [CrossRef]
- Bomberg, M.; Nyyssönen, M.; Pitkänen, P.; Lehtinen, A.; Itävaara, M. Active Microbial Communities Inhabit Sulphate-Methane Interphase in Deep Bedrock Fracture Fluids in Olkiluoto, Finland. Biomed Res. Int. 2015, 2015, 979530. [Google Scholar] [CrossRef]
- Pedersen, K. Microbial life in deep granitic rock. FEMS Microbiol. Rev. 1997, 20, 399–414. [Google Scholar] [CrossRef]
- Kotelnikova, S.; Pedersen, K. Evidence for methanogenic Archaea and homoacetogenic Bacteria in deep granitic rock aquifers. FEMS Microbiol. Rev. 1997, 20, 339–349. [Google Scholar] [CrossRef]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef]
- Hug, L.A.; Thomas, B.C.; Sharon, I.; Brown, C.T.; Sharma, R.; Hettich, R.L.; Wilkins, M.J.; Williams, K.H.; Singh, A.; Banfield, J.F. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ. Microbiol. 2016, 18, 159–173. [Google Scholar] [CrossRef]
- Richter, A. Final Phase Launched of Drilling the World’s Deepest Geothermal Heat Wells in Otaniemi, Finland. Available online: http://www.thinkgeoenergy.com/final-phase-launched-of-drilling-the-worlds-deepest-geothermal-heat-wells-in-otaniemi-finland (accessed on 10 December 2019).
- Abramov, O.; Mojzsis, S.J. Microbial habitability of the Hadean Earth during the late heavy bombardment. Nature 2009, 459, 419–422. [Google Scholar] [CrossRef]
- Ivarsson, M.; Lindgren, P. The search for sustainable subsurface habitats on mars, and the sampling of impact ejecta. Sustainability 2010, 2, 1969–1990. [Google Scholar] [CrossRef]
- Rull, F.; Sansano, A.; Díaz, E.; Canora, C.P.; Moral, A.G.; Tato, C.; Colombo, M.; Belenguer, T.; Fernández, M.; Manfredi, J.A.R.; et al. ExoMars Raman laser spectrometer for Exomars. In Instruments, Methods, and Missions for Astrobiology XIV; Proc. SPIE: Bellingham, WD, USA, 2011; Volume 8152, p. 81520J. [Google Scholar]
- Vago, J.L.; Westall, F.; Coates, A.J.; Jaumann, R.; Korablev, O.; Ciarletti, V.; Mitrofanov, I.; Josset, J.L.; De Sanctis, M.C.; Bibring, J.P.; et al. Habitability on Early Mars and the Search for Biosignatures with the ExoMars Rover. Astrobiology 2017, 17, 471–510. [Google Scholar] [CrossRef] [PubMed]
- Osburn, M.R.; LaRowe, D.E.; Momper, L.M.; Amend, J.P. Chemolithotrophy in the continental deep subsurface: Sanford Underground Research Facility (SURF), USA. Front. Microbiol. 2014, 5, 610. [Google Scholar] [CrossRef] [PubMed]
- Colman, D.R.; Poudel, S.; Stamps, B.W.; Boyd, E.S.; Spear, J.R. The deep, hot biosphere: Twenty-five years of retrospection. Proc. Natl. Acad. Sci. USA 2017, 114, 6895–6903. [Google Scholar] [CrossRef]
- Miettinen, H.; Kietäväinen, R.; Sohlberg, E.; Numminen, M.; Ahonen, L.; Itävaara, M. Microbiome composition and geochemical characteristics of deep subsurface high-pressure environment, Pyhäsalmi mine Finland. Front. Microbiol. 2015, 6, 1203. [Google Scholar] [CrossRef] [PubMed]
- Leary, P.; Malin, P.; Saarno, T.; Kukkonen, I. Prospects for assessing enhanced geothermal system (EGS) basement rock flow stimulation by wellbore temperature data. Energies 2017, 10, 1979. [Google Scholar] [CrossRef]
- Pajunen, M.; Airo, M.L.; Elminen, T.; Niemelä, R.; Salmelainen, J.; Vaarma, M.; Wasenius, P.; Wennerström, M. Construction suitability of bedrock in the Helsinki area based on the tectonic structure of the Svecofennian crust of southern Finland. Spec. Pap. Geol. Surv. Finl. 2008, 47, 309–326. [Google Scholar]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O.; Klindworth, A.; Pruesse, E.; Schweer, T.; et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, 1–11. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, S.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phyologenetics.pdf. PCR Protoc. A Guid. Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lever, M.A.; Torti, A.; Eickenbusch, P.; Michaud, A.B.; Šantl-Temkiv, T.; Jørgensen, B.B. A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, M.; Kudo, T. Phylogenetic analysis of the symbiotic intestinal microflora of the termite Cryptotermes domesticus. FEMS Microbiol. Lett. 1998, 164, 389–399. [Google Scholar] [CrossRef][Green Version]
- Cadillo-Quiroz, H.; Bräuer, S.; Yashiro, E.; Sun, C.; Yavitt, J.; Zinder, S. Vertical profiles of methanogenesis and methanogens in two contrasting acidic peatlands in central New York State, USA. Environ. Microbiol. 2006, 8, 1428–1440. [Google Scholar] [CrossRef]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef]
- Wagner, M.; Roger, A.J.; Flax, J.L.; Gregory, A.; Stahl, D.A.; Wagner, M.; Roger, A.J.; Flax, J.L.; Brusseau, G.A.; Stahl, D.A. Phylogeny of Dissimilatory Sulfite Reductases Supports an Early Origin of Sulfate Respiration Phylogeny of Dissimilatory Sulfite Reductases Supports an Early Origin of Sulfate Respiration. J. Bacteriol. 1998, 180, 2975–2982. [Google Scholar] [CrossRef]
- Geets, J.; Borremans, B.; Diels, L.; Springael, D.; Vangronsveld, J.; Van Der Lelie, D.; Vanbroekhoven, K. DsrB gene-based DGGE for community and diversity surveys of sulfate-reducing bacteria. J. Microbiol. Methods 2006, 66, 194–205. [Google Scholar] [CrossRef]
- Steinberg, L.M.; Regan, J.M. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl. Environ. Microbiol. 2008, 74, 6663–6671. [Google Scholar] [CrossRef]
- López-Gutiérrez, J.C.; Henry, S.; Hallet, S.; Martin-Laurent, F.; Catroux, G.; Philippot, L. Quantification of a novel group of nitrate-reducing bacteria in the environment by real-time PCR. J. Microbiol. Methods 2004, 57, 399–407. [Google Scholar] [CrossRef]
- Hales, B.A.; Edwards, C.; Ritchie, D.A.; Hall, G.; Pickup, R.W.; Saunders, J.R. Isolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis. Appl. Environ. Microbiol. 1996, 62, 668–675. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Purkamo, L.; Bomberg, M.; Nyyssönen, M.; Ahonen, L.; Kukkonen, I.; Itävaara, M.; Palmer, K. Response of Deep Subsurface Microbial Community to Different Carbon Sources and Electron Acceptors during 2 months Incubation in Microcosms. Front. Microbiol. 2017, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Cochrane, G.; Qureshi, M.; Scheremetjew, M.; Potter, S.; Mitchell, A.L.; Amid, C.; ten Hoopen, P.; Boland, M.A.; Wilkinson, D.J.; et al. EBI Metagenomics in 2017: Enriching the analysis of microbial communities, from sequence reads to assemblies. Nucleic Acids Res. 2017, 46, D726–D735. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. MSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. R package Version 0.2. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 15 September 2019).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Glud, R.N.; Wenzhöfer, F.; Middelboe, M.; Oguri, K.; Turnewitsch, R.; Canfield, D.E.; Kitazato, H. High rates of microbial carbon turnover in sediments in the deepest oceanic trench on Earth. Nat. Geosci. 2013. [Google Scholar] [CrossRef]
- Hernsdorf, A.W.; Amano, Y.; Miyakawa, K.; Ise, K.; Suzuki, Y.; Anantharaman, K.; Probst, A.; Burstein, D.; Thomas, B.C.; Banfield, J.F. Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments. ISME J. 2017, 11, 1915–1929. [Google Scholar] [CrossRef] [PubMed]
- Breuker, A.; Köweker, G.; Blazejak, A.; Schippers, A.; Koweker, G.; Blazejak, A.; Schippers, A. The deep biosphere in terrestrial sediments in the Chesapeake Bay area, Virginia, USA. Front. Microbiol. 2011, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mcmahon, S.; Parnell, J. Weighing the deep continental biosphere. FEMS Microbiol. Ecol. 2014, 87, 113–120. [Google Scholar] [CrossRef]
- Magnabosco, C.; Lin, L.H.; Dong, H.; Bomberg, M.; Ghiorse, W.; Stan-Lotter, H.; Pedersen, K.; Kieft, T.L.; van Heerden, E.; Onstott, T.C. The biomass and biodiversity of the continental subsurface. Nat. Geosci. 2018, 11, 707–717. [Google Scholar] [CrossRef]
- Jørgensen, B.B. Shrinking majority of the deep biosphere. Proc. Natl. Acad. Sci. USA 2012, 109, 15976–15977. [Google Scholar] [CrossRef]
- Kallmeyer, J.; Pockalny, R.; Adhikari, R.R.; Smith, D.C.; D’Hondt, S. From the Cover: Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. USA 2012, 109, 16213–16216. [Google Scholar] [CrossRef]
- Bar-On, Y.M.; Phillips, R.; Milo, R. The biomass distribution on Earth. Proc. Natl. Acad. Sci. USA 2018, 115, 6506–6511. [Google Scholar] [CrossRef] [PubMed]
- Fairén, A.G.; Davila, A.F.; Lim, D.; Bramall, N.; Bonaccorsi, R.; Zavaleta, J.; Uceda, E.R.; Stoker, C.; Wierzchos, J.; Dohm, J.M.; et al. Astrobiology through the ages of Mars: The study of terrestrial analogues to understand the habitability of Mars. Astrobiology 2010, 10, 821–843. [Google Scholar] [CrossRef] [PubMed]
- Neveu, M.; Hays, L.E.; Voytek, M.A.; New, M.H.; Schulte, M.D. The ladder of life detection. Astrobiology 2018, 18, 1375–1402. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.E.; Dorazio, R.M.; Butterfield, J.S.S.; Meigs-Friend, G.; Nico, L.G.; Ferrante, J.A. Detection limits of quantitative and digital PCR assays and their influence in presence–absence surveys of environmental DNA. Mol. Ecol. Resour. 2017, 17, 221–229. [Google Scholar] [CrossRef]
- Bogan, B.W.; Sullivan, W.R.; Kayser, K.J.; Derr, K.D.; Aldrich, H.C.; Paterek, J.R. Alkanindiges illinoisensis gen. nov., sp. nov., an obligately hydrocarbonoclastic, aerobic squalane-degrading bacterium isolated from oilfield soils. Int. J. Syst. Evol. Microbiol. 2003, 53, 1389–1395. [Google Scholar] [CrossRef]
- Pedersen, K.; Nilsson, E.; Arlinger, J.; Hallbeck, L.; O’Neill, A. Distribution, diversity and activity of microorganisms in the hyper-alkaline spring waters of Maqarin in Jordan. Extremophiles 2004, 8, 151–164. [Google Scholar] [CrossRef]
- Purkamo, L.; Bomberg, M.; Kietäväinen, R.; Salavirta, H.; Nyyssönen, M.; Nuppunen-Puputti, M.; Ahonen, L.; Kukkonen, I.; Itävaara, M. The keystone species of Precambrian deep bedrock biosphere belong to Burkholderiales and Clostridiales. Biogeosciences Discuss. 2015, 12, 18103–18150. [Google Scholar] [CrossRef]
- Soares, A.; Edwards, A.; An, D.; Bagnoud, A.; Bomberg, M.; Budwill, K.; Caffrey, S.M.; Fields, M.; Gralnick, J.; Kadnikov, V.; et al. A global perspective on microbial diversity in the terrestrial deep subsurface. bioRxiv 2019, 602672. [Google Scholar] [CrossRef]
- Hubalek, V.; Wu, X.; Eiler, A.; Buck, M.; Heim, C.; Dopson, M.; Bertilsson, S.; Ionescu, D. Connectivity to the surface determines diversity patterns in subsurface aquifers of the Fennoscandian shield. ISME J. 2016, 10, 2447. [Google Scholar] [CrossRef]
- Sheik, C.S.; Reese, B.K.; Twing, K.I.; Sylvan, J.B.; Grim, S.L.; Schrenk, M.O.; Sogin, M.L.; Colwell, F.S. Identification and removal of contaminant sequences from ribosomal gene databases: Lessons from the Census of Deep Life. Front. Microbiol. 2018, 9, 840. [Google Scholar] [CrossRef]
- Bomberg, M.; Raulio, M.; Jylhä, S.; Mueller, C.W.; Höschen, C.; Rajala, P.; Purkamo, L.; Kietäväinen, R.; Ahonen, L.; Itävaara, M. CO2 and carbonate as substrate for the activation of the microbial community in 180 m deep bedrock fracture fluid of Outokumpu Deep Drill Hole, Finland. AIMS Microbiol. 2017, 3, 846–871. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.R.B.; Tindall, B.J.; Martins Dos Santos, V.A.P.; Pieper, D.H.; Ramos, J.; Palleroni, N.J. Nonmedical: Pseudomonas. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.S., Stackebrandt, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Teixeira, L.M.; Merquior, V.L.C. The Family Moraxellaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Palleroni, N.J. Introduction to the Family Pseudomonadaceae. In The Prokaryotes; Starr, M.P., Stolp, H., Trüper, H.G., Balows, A., Schlegel, H.G., Eds.; Springer: Berlin/Heidelberg, Germany, 1981. [Google Scholar]
- Moser, D.P.; Gihring, T.M.; Brockman, F.J.; Fredrickson, J.K.; Balkwill, D.L.; Dollhopf, M.E.; Lollar, B.S.; Pratt, L.M.; Boice, E.; Southam, G.; et al. Desulfotomaculum and Methanobacterium spp. dominate a 4- to 5-kilometer-deep fault. Appl. Environ. Microbiol. 2005, 71, 8773–8783. [Google Scholar] [CrossRef] [PubMed]
- Purkamo, L.; Bomberg, M.; Nyyssönen, M.; Kukkonen, I.; Ahonen, L.; Itävaara, M. Heterotrophic Communities Supplied by Ancient Organic Carbon Predominate in Deep Fennoscandian Bedrock Fluids. Microb. Ecol. 2015, 69, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Comolli, L.R.; Baker, B.J.; Downing, K.H.; Siegerist, C.E.; Banfield, J.F. Three-dimensional analysis of the structure and ecology of a novel, ultra-small archaeon. ISME J. 2009. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Wegner, C.E.; Taubert, M.; Geesink, P.; Lehmann, K.; Yan, L.; Lehmann, R.; Totsche, K.U.; Küsel, K. Predominance of Cand. Patescibacteria in groundwater is caused by their preferential mobilization from soils and flourishing under oligotrophic conditions. Front. Microbiol. 2019, 10, 1407. [Google Scholar] [CrossRef]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef]
- Nuppunen-Puputti, M.; Purkamo, L.; Kietäväinen, R.; Nyyssönen, M.; Itävaara, M.; Ahonen, L.; Kukkonen, I.; Bomberg, M. Rare biosphere archaea assimilate acetate in Precambrian terrestrial subsurface at 2.2 km depth. Geosciences 2018, 8, 418. [Google Scholar] [CrossRef]
- Sohlberg, E.; Bomberg, M.; Miettinen, H.; Nyyssönen, M.; Salavirta, H.; Vikman, M.; Itävaara, M. Revealing the unexplored fungal communities in deep groundwater of crystalline bedrock fracture zones in Olkiluoto, Finland. Front. Microbiol. 2015, 6, 573. [Google Scholar] [CrossRef]
- Ivarsson, M.; Bengtson, S.; Drake, H.; Francis, W. Fungi in Deep Subsurface Environments, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 1877081116. [Google Scholar]
- Rummel, J.D.; Beaty, D.W.; Jones, M.A.; Bakermans, C.; Barlow, N.G.; Boston, P.J.; Chevrier, V.F.; Clark, B.C.; De Vera, J.P.P.; Gough, R.V.; et al. A new analysis of mars “Special Regions”: Findings of the second MEPAG special regions science analysis group (SR-SAG2). Astrobiology 2014, 14, 1227. [Google Scholar] [CrossRef]
- Seto, M.; Noguchi, K.; Van Cappellen, P. Potential for Aerobic Methanotrophic Metabolism on Mars. Astrobiology 2019. [Google Scholar] [CrossRef]
- Onstott, T.C.; Ehlmann, B.L.; Sapers, H.; Coleman, M.; Ivarsson, M.; Marlow, J.J.; Neubeck, A.; Niles, P. Paleo-Rock-Hosted Life on Earth and the Search on Mars: A Review and Strategy for Exploration. Astrobiology 2019. [Google Scholar] [CrossRef]
- Eigenbrode, J.L.; Summons, R.E.; Steele, A.; Freissinet, C.; Millan, M.; Navarro-González, R.; Sutter, B.; McAdam, A.C.; Franz, H.B.; Glavin, D.P.; et al. Organic matter preserved in 3-billion-year-old mudstones at Gale crater, Mars. Science 2018, 360, 1096–1101. [Google Scholar] [CrossRef]
- Waite, J.H.; Combi, M.R.; Ip, W.H.; Cravens, T.E.; McNutt, R.L.; Kasprzak, W.; Yelle, R.; Luhmann, J.; Niemann, H.; Gell, D.; et al. Cassini ion and neutral mass spectrometer: Enceladus plume composition and structure. Science 2006. [Google Scholar] [CrossRef]
- Postberg, F.; Khawaja, N.; Abel, B.; Choblet, G.; Glein, C.R.; Gudipati, M.S.; Henderson, B.L.; Hsu, H.W.; Kempf, S.; Klenner, F.; et al. Macromolecular organic compounds from the depths of Enceladus. Nature 2018. [Google Scholar] [CrossRef]
- Berg, I.A. Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 2011, 77, 1925–1936. [Google Scholar] [CrossRef]
- Stern, J.C.; Sutter, B.; Freissinet, C.; Navarro-González, R.; McKay, C.P.; Archer, P.D.; Buch, A.; Brunner, A.E.; Coll, P.; Eigenbrode, J.L.; et al. Evidence for indigenous nitrogen in sedimentary and aeolian deposits from the Curiosity rover investigations at Gale crater, Mars. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef]
- Franz, H.B.; Trainer, M.G.; Malespin, C.A.; Mahaffy, P.R.; Atreya, S.K.; Becker, R.H.; Benna, M.; Conrad, P.G.; Eigenbrode, J.L.; Freissinet, C.; et al. Initial SAM calibration gas experiments on Mars: Quadrupole mass spectrometer results and implications. Planet. Space Sci. 2017. [Google Scholar] [CrossRef]
- Könneke, M.; Bernhard, A.E.; de la Torre, J.R.; Walker, C.B.; Waterbury, J.B.; Stahl, D. A Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 2005, 437, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Heal, K.R.; Ramdasi, R.; Kobelt, J.N.; Martens-Habbena, W.; Bertagnolli, A.D.; Amin, S.A.; Walker, C.B.; Urakawa, H.; Könneke, M.; et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammoniaoxidizing archaea of the phylum thaumarchaeo. Int. J. Syst. Evol. Microbiol. 2017, 67, 5067–5079. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zerkle, A.; Claire, M.; Stueeken, E. Atmospheric Nitrate as a Potential Nutrient for Life on Mars. Extremophiles 2019. [Google Scholar] [CrossRef]
- Cockell, C.S. Trajectories of martian habitability. Astrobiology 2014, 14, 182–203. [Google Scholar] [CrossRef] [PubMed]
- Gudelj, I.; Beardmore, R.E.; Arkin, S.S.; Maclean, R.C. Constraints on microbial metabolism drive evolutionary diversification in homogeneous environments. J. Evol. Biol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalez, N.; Huber, J.A.; Vallino, J.J. Microbial Communities are Well Adapted to Disturbances in Energy Input Nuria Fernandez-Gonzalez. MSystems 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.E.; Mojarro, A.; Tani, J.; Bhattaru, S.A.; Zuber, M.T.; Doebler, R.; Brown, M.; Herrington, K.; Talbot, R.; Fuller, C.W.; et al. Advancing the search for extra-terrestrial genomes. In Proceedings of the 2016 IEEE Aerospace Conference, Big Sky, MT, USA, 5–12 March 2016; pp. 1–15. [Google Scholar]
- Carr, C.E.; Mojarro, A.; Hachey, J.; Saboda, K.; Tani, J.; Bhattaru, S.A.; Smith, A.; Pontefract, A.; Zuber, M.T.; Doebler, R.; et al. Towards in situ sequencing for life detection. In Proceedings of the 2017 IEEE Aerospace Conference, Big Sky, MT, USA, 4–11 March 2016; pp. 1–18. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geohemistry Variable | units | Sample a | Sample b |
|---|---|---|---|
| Alkalinity | mmol/L | 0.16 | 0.17 |
| Total dissolved solids | g/L | 77 | 81 |
| Cations: | |||
| Al | µg/L | 5.76 | 6.50 |
| As | µg/L | 0.94 | 0.61 |
| B | µg/L | 728 | 678 |
| Ba | µg/L | 902 | 928 |
| Be | µg/L | 0.16 | 0.12 |
| Co | µg/L | 0.41 | 0.40 |
| Cr | µg/L | 6.46 | 6.28 |
| Cu | µg/L | 0.62 | 0.58 |
| K | mg/L | 57.4 | 56.1 |
| Mn | µg/L | 15.2 | 14.1 |
| Mo | µg/L | 1.89 | 1.89 |
| Ni | µg/L | 4.77 | 26.6 |
| P | µg/L | 47.0 | 60.2 |
| Pb | µg/L | 0.42 | 0.20 |
| Rb | µg/L | 283 | 282 |
| Se | µg/L | 0.41 | 0.73 |
| V | µg/L | 2.39 | 2.28 |
| Zn | µg/L | 2.47 | 1.63 |
| Ca | mg/L | 20500 | 21500 |
| Fe | mg/L | 0.14 | 0.21 |
| Li | mg/L | 0.337 | 0.322 |
| Mg | mg/L | 3.22 | 3.44 |
| Na | mg/L | 7190 | 7420 |
| S | mg/L | 88.7 | 87.9 |
| Si | mg/L | 2.60 | 2.99 |
| Sr | mg/L | 254 | 266 |
| Anions: | |||
| I | mg/L | 7.15 | 6.98 |
| Br | mg/L | 510 | 510 |
| Cl | mg/L | 48000 | 50000 |
| SO4 | mg/L | 340 | 320 |
| NO3 | mg/L | <0.2 | <0.2 |
| Sulfide | mg/L | - | 1.2 |
| Measurement | units | 2016 Before | 2016 After | 2013 | 2014 |
|---|---|---|---|---|---|
| EC | mS/cm | 102.6 | 102.9 | 103.3 1 | 102.0 1 |
| pH | 9.2 | 9.3 | 8.6 1 | 8.7 1 | |
| T | °C | 23.6 | 23.3 | 23.4 | 24.0 |
| O2 | mg/L | 0.05 | 0.04 | 0.03 | NM 2 |
| Sample ID | Depth (m) | Observed | Chao1 | ACE | Shannon | Inv. Simpson | Obs./Chao1 | Obs./ACE |
|---|---|---|---|---|---|---|---|---|
| Pyhäsalmi DNA | 2400 | 115 | 218 | 223 | 2.3 | 2.5 | 53% | 52% |
| Pyhäsalmi cDNA | 2400 | 115 | 202 | 234 | 2.4 | 2.5 | 57% | 49% |
| Otaniemi3 | 3203 | 1032 | 7440 | 20,614 | 3.7 | 15.9 | 14% | 5% |
| Otaniemi5 | 4203 | 714 | 5173 | 14,199 | 3.4 | 14.0 | 14% | 5% |
| Otaniemi4 | 4375 | 787 | 4718 | 8944 | 2.5 | 5.3 | 17% | 9% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purkamo, L.; Kietäväinen, R.; Nuppunen-Puputti, M.; Bomberg, M.; Cousins, C. Ultradeep Microbial Communities at 4.4 km within Crystalline Bedrock: Implications for Habitability in a Planetary Context. Life 2020, 10, 2. https://doi.org/10.3390/life10010002
Purkamo L, Kietäväinen R, Nuppunen-Puputti M, Bomberg M, Cousins C. Ultradeep Microbial Communities at 4.4 km within Crystalline Bedrock: Implications for Habitability in a Planetary Context. Life. 2020; 10(1):2. https://doi.org/10.3390/life10010002
Chicago/Turabian StylePurkamo, Lotta, Riikka Kietäväinen, Maija Nuppunen-Puputti, Malin Bomberg, and Claire Cousins. 2020. "Ultradeep Microbial Communities at 4.4 km within Crystalline Bedrock: Implications for Habitability in a Planetary Context" Life 10, no. 1: 2. https://doi.org/10.3390/life10010002
APA StylePurkamo, L., Kietäväinen, R., Nuppunen-Puputti, M., Bomberg, M., & Cousins, C. (2020). Ultradeep Microbial Communities at 4.4 km within Crystalline Bedrock: Implications for Habitability in a Planetary Context. Life, 10(1), 2. https://doi.org/10.3390/life10010002