Molecular Modeling of Interactions between N-(Carboxymethyl)-N-tetradecylglycine and Fluorapatite




Abstract
1. Introduction
2. Computational Details and Experimental Method
2.1. NCNT Characterization
2.2. FAp Bulk Structure Optimization
2.3. Surface Energy Calculation
2.4. Adsorption Energy Calculation
2.5. Flotation Experiment
3. Results
3.1. Structure and Property of NCNT
3.2. Simulation Parameter Screening and Rationality Verification of FAp Bulk
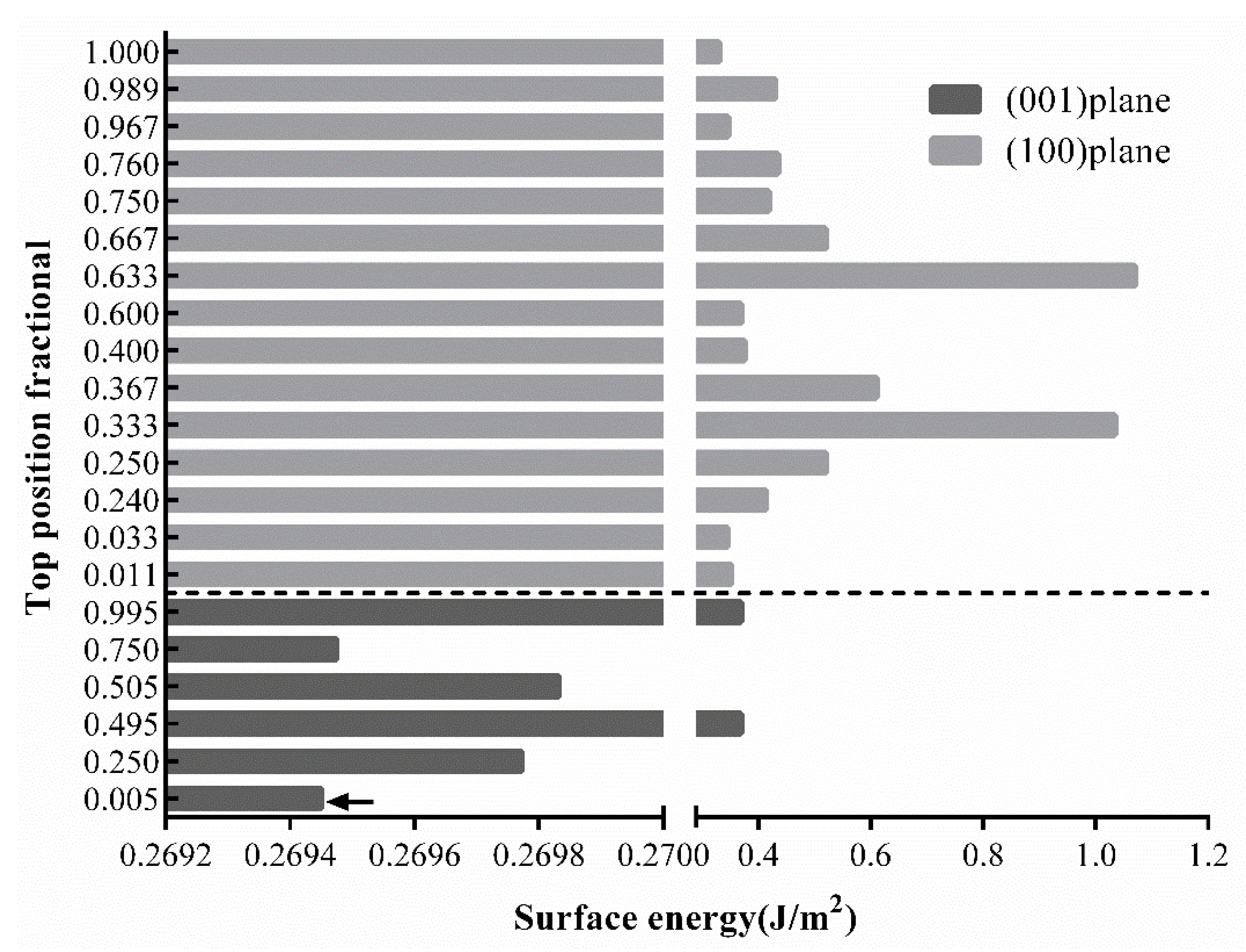
3.3. Structure Optimization of FAp Surface
3.4. Adsorption Site and Adsorption Configurations
3.5. Adsorption Energy
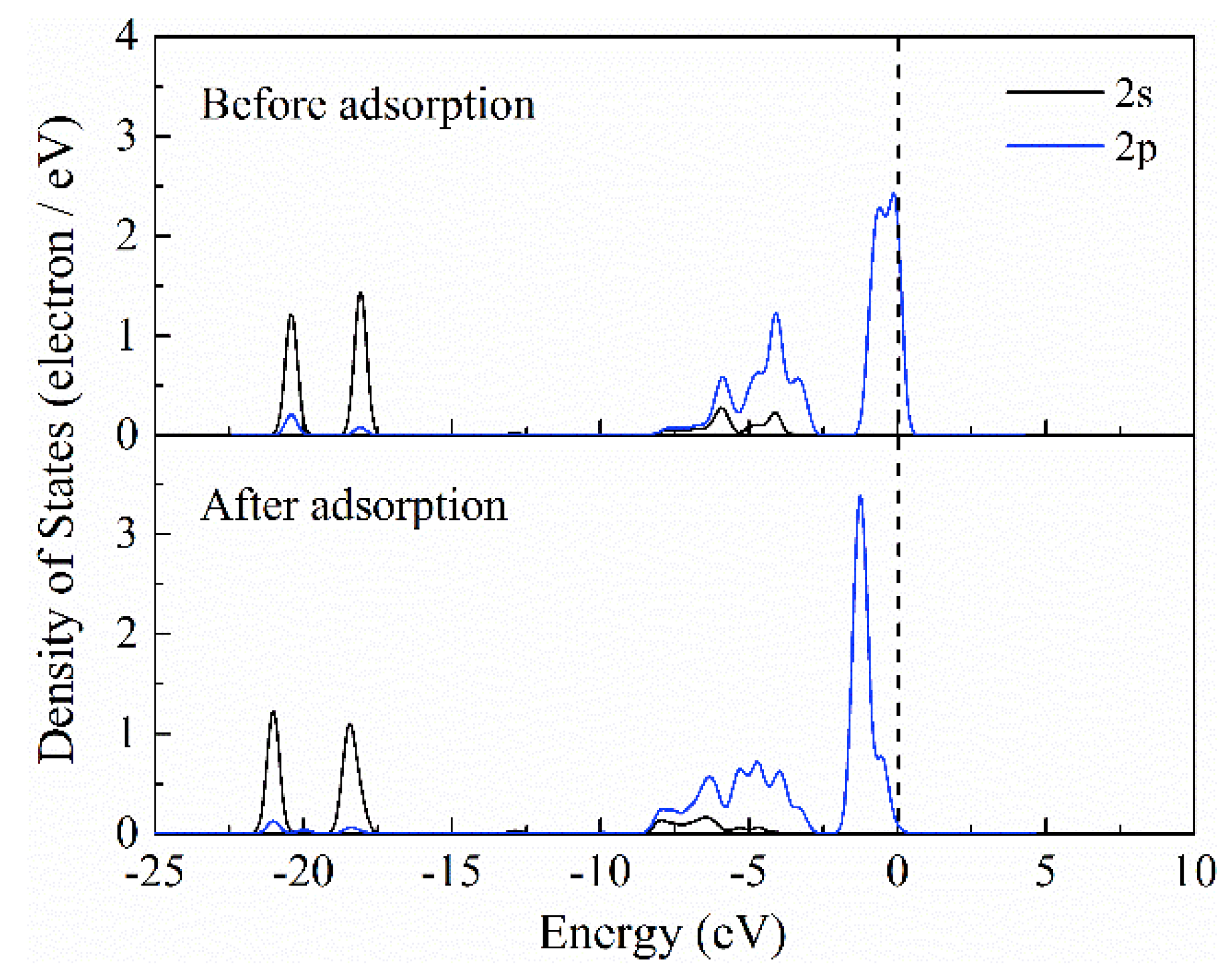
3.6. Adsorption Mechanism Study of NCNT2−
3.7. Flotation Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sharpley, A.; Rekolainen, S.; Tunney, H.; Carton, O. Phosphorus Loss from Soil to Water; CABI Publishing: Cambridge, UK, 1997. [Google Scholar]
- Sis, H.; Chander, S. Reagents used in the flotation of phosphate ores: A critical review. Miner. Eng. 2003, 16, 577–585. [Google Scholar] [CrossRef]
- Wills, B.A.; Napier-Munn, T. Mineral Processing Technology, 7th ed.; Elsevier Science & Technology Books: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Yu, J.; Ge, Y.Y.; Hou, J.T. Behavior and Mechanism of Collophane and Dolomite Separation Using Alkyl Hydroxamic Acid as a Flotation Collector. Physicochem. Probl. Mineral Pro. 2016, 52, 155–169. [Google Scholar]
- Borges, A.A.; da Luz, J.A.M.; Ferreira, E.E. Characterization of the magnetic phase from carbonatitic phosphate ore. Rem: Revista Escola de Minas 2008, 61, 29–34. [Google Scholar]
- Santos, E.P.; Dutra, A.J.B.; Oliveira, J.F. The effect of jojoba oil on the surface properties of calcite and apatite aiming at their selective flotation. Int. J. Miner. Process. 2015, 143, 34–38. [Google Scholar] [CrossRef]
- Su, F.W.; Rao, K.H.; Forssberg, K.S.E.; Samskog, P.O. The influence of temperature on the kinetics of apatite flotation from magnetite fines. Int. J. Miner. Process. 1998, 54, 131–145. [Google Scholar] [CrossRef]
- Cao, Q.B.; Cheng, J.H.; Wen, S.M.; Li, C.X.; Bai, S.J.; Liu, D. A mixed collector system for phosphate flotation. Miner. Eng. 2015, 78, 114–121. [Google Scholar] [CrossRef]
- Filippov, L.O.; Duverger, A.; Filippova, I.V.; Kasaini, H.; Thiry, J. Selective flotation of silicates and Ca-bearing minerals: The role of non-ionic reagent on cationic flotation. Miner. Eng. 2012, 36–38, 314–323. [Google Scholar] [CrossRef]
- Karlkvist, T.; Patra, A.; Rao, K.H.; Bordes, R.; Holmberg, K. Flotation selectivity of novel alkyl dicarboxylate reagents for apatite-calcite separation. J. Colloid Interface Sci. 2015, 445, 40–47. [Google Scholar] [CrossRef]
- Ahmed, E.; Salah, E.M.; Mohamed, A.K.; Nagui, A.K.; Ayman, E.M. Dolomite-apatite separation by amphoteric collector in presence of bacteria. J. Cent. South Univ. 2013, 20, 1645–1652. [Google Scholar] [CrossRef]
- Hu, Y.H.; Xu, Z.H. Interactions of amphoteric amino phosphoric acids with calcium-containing minerals and selective flotation. Int. J. Miner. Process. 2003, 72, 87–94. [Google Scholar] [CrossRef]
- Li, C.-X.; Duan, Y.-H.; Hu, W.-C. Electronic structure, elastic anisotropy, thermal conductivity and optical properties of calcium apatite Ca5(PO4)3X (X = F, Cl or Br). J. Alloy Compd. 2015, 619, 66–77. [Google Scholar] [CrossRef]
- Mkhonto, D.; de Leeuw, N.H. A computer modelling study of the effect of water on the surface structure and morphology of fluorapatite: Introducing a Ca10(PO4)6F2 potential model. J. Mater. Chem. 2002, 12, 2633–2642. [Google Scholar] [CrossRef]
- Pareek, A.; Torrelles, X.; Angermund, K.; Rius, J.; Magdans, U.; Gies, H. Structure of interfacial water on fluorapatite (100) surface. Langmuir 2008, 24, 2459–2464. [Google Scholar] [CrossRef]
- Pareek, A.; Torrelles, X.; Angermund, K.; Rius, J.; Magdans, U.; Gies, H. Competitive Adsorption of Glycine and Water on the Fluorapatite (100) Surface. Langmuir 2009, 25, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Schepers, T.; Brickmann, J.; Hochrein, O.; Zahn, D. Atomistic simulation study of calcium, phosphate and fluoride ion association to the teleopeptide-tails of collagen—Initial steps to biomineral formation. Zeitschrift für anorganische und allgemeine Chemie 2007, 633, 411–414. [Google Scholar] [CrossRef]
- Pradip; Rai, B.; Rao, T.K.; Krishnamurthy, S.; Vetrivel, R.; Mielczarski, J.; Cases, J.M. Molecular modeling of interactions of alkyl hydroxamates with calcium minerals. J. Colloid Interface Sci. 2002, 256, 106–113. [Google Scholar] [CrossRef]
- Pradip; Rai, B.; Rao, T.K.; Krishnamurthy, S.; Vetrivel, R.; Mielczarski, J.; Cases, J.M. Molecular modeling of interactions of diphosphonic acid based surfactants with calcium minerals. Langmuir 2002, 18, 932–940. [Google Scholar] [CrossRef]
- Filgueiras, M.R.T.; Mkhonto, D.; de Leeuw, N.H. Computer simulations of the adsorption of citric acid at hydroxyapatite surfaces. J. Cryst. Growth 2006, 294, 60–68. [Google Scholar] [CrossRef]
- Mkhonto, D.; Ngoepe, P.E.; Cooper, T.G.; de Leeuw, N.H. A computer modelling study of the interaction of organic adsorbates with fluorapatite surfaces. Phys. Chem. Miner. 2006, 33, 314–331. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Xie, X.-D.; Lu, D. Energy Band Theory of Solids; Fudan University Press: Shanghai, China, 1998. [Google Scholar]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Zeitschrift für Kristallographie-Crystalline Materials 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Mat. 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. Special points for Brillouin-zone integrations-a reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter. 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.M.; Luo, B.B.; Sun, C.Y.; Liu, J.; Sun, H.T.; Li, Y.J.; Han, Y.X. Density functional theory study of alpha-Bromolauric acid adsorption on the alpha-quartz (101) surface. Miner. Eng. 2016, 92, 72–77. [Google Scholar] [CrossRef]
- Sauer, J.; Sustmann, R. Mechanistic Aspects of Diels-Alder Reactions: A Critical Survey. Angew. Chem. Int. Ed. 1980, 19, 779–807. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Hendricks, S.B.; Jefferson, M.E.; Mosley, V.M. The Crystal Structures of some Natural and Synthetic Apatite-Like Substances. Zeitschrift für Kristallographie-Crystalline Materials 1932, 81, 352–369. [Google Scholar] [CrossRef]
- Deer, W.A.; Howie, R.A.; Zussman, J. An Introduction to the Rock-Forming Minerals; Longman Group: Harlow, UK, 1992. [Google Scholar]
- Zhu, H.; Qin, W.; Chen, C.; Liu, R. Interactions Between Sodium Oleate and Polyoxyethylene Ether and the Application in the Low-Temperature Flotation of Scheelite at 283 K. J. Surfactants Deterg. 2016, 19, 1289–1295. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbate | NCNT | NCNT2− | OA | OA− | H2O | OH− |
|---|---|---|---|---|---|---|
| EHOMO (eV) | −5.357 | 2.786 | −5.78 | 0.336 | −6.454 | 4.78 |
| ELUMO (eV) | −1.178 | 3.718 | −0.967 | 1.486 | −0.275 | 9.757 |
| EHOMO-LUMO (eV) | −4.179 | −0.932 | −4.813 | −1.15 | −6.179 | −4.977 |
| Data Resource | Functional | a/Å | b/Å | c/Å | Difference/% |
|---|---|---|---|---|---|
| Experimental | 9.370 | 9.370 | 6.880 | - | |
| Simulation | GGA-PBE | 9.305 | 9.305 | 6.858 | 0.593 |
| GGA-RPBE | 9.450 | 9.459 | 6.941 | 0.862 | |
| GGA-PW91 | 9.294 | 9.294 | 6.840 | 0.749 | |
| GGA-WC | 9.212 | 9.212 | 6.770 | 1.66 | |
| GGA-PBESOL | 9.199 | 9.199 | 6.785 | 1.71 |
| Slab depth (Å) | 6.858 | 10.287 | 13.716 | 17.145 | 20.574 |
| Surface energy (J∙m−2) | 0.3107 | 0.5483 | 0.5769 | 0.5856 | 0.6022 |
| Vacuum thickness (Å) | 10 | 12 | 14 | 16 | 18 |
| Surface energy (J∙m−2) | 0.3150 | 0.3167 | 0.3184 | 0.3186 | 0.3188 |
| Adsorbate | NCNT2− | OA− | H2O | OH− |
|---|---|---|---|---|
| Eads (J∙m−2) | −210.297 | −219.233 | −192.71 | −189.65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, N.; Zhu, Y.; Han, Y.; Liu, J. Molecular Modeling of Interactions between N-(Carboxymethyl)-N-tetradecylglycine and Fluorapatite. Minerals 2019, 9, 278. https://doi.org/10.3390/min9050278
Nan N, Zhu Y, Han Y, Liu J. Molecular Modeling of Interactions between N-(Carboxymethyl)-N-tetradecylglycine and Fluorapatite. Minerals. 2019; 9(5):278. https://doi.org/10.3390/min9050278
Chicago/Turabian StyleNan, Nan, Yimin Zhu, Yuexin Han, and Jie Liu. 2019. "Molecular Modeling of Interactions between N-(Carboxymethyl)-N-tetradecylglycine and Fluorapatite" Minerals 9, no. 5: 278. https://doi.org/10.3390/min9050278
APA StyleNan, N., Zhu, Y., Han, Y., & Liu, J. (2019). Molecular Modeling of Interactions between N-(Carboxymethyl)-N-tetradecylglycine and Fluorapatite. Minerals, 9(5), 278. https://doi.org/10.3390/min9050278