Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters


Abstract
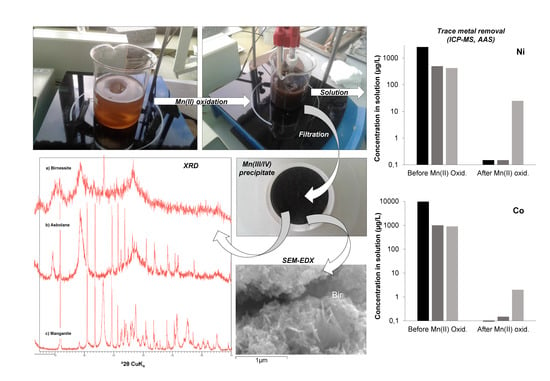
:
1. Introduction
2. Materials and Methods
2.1. Field Sampling and Preliminary Neutralization Stage
2.2. Batch Experiments of Mn(II) Oxidation
2.3. Mineralogical Identification and Analytical Methods
2.4. Geochemical Modeling
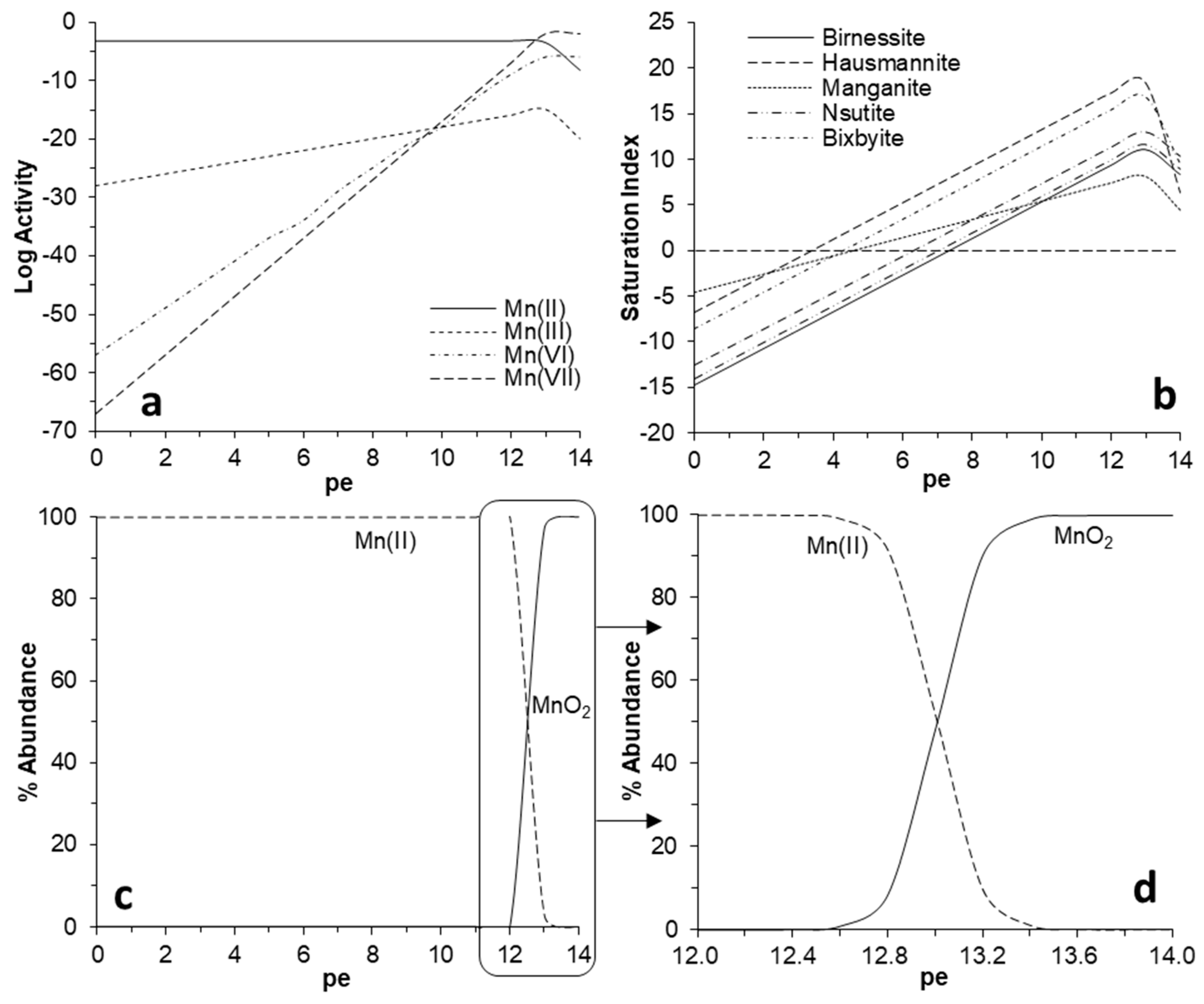
3. Results
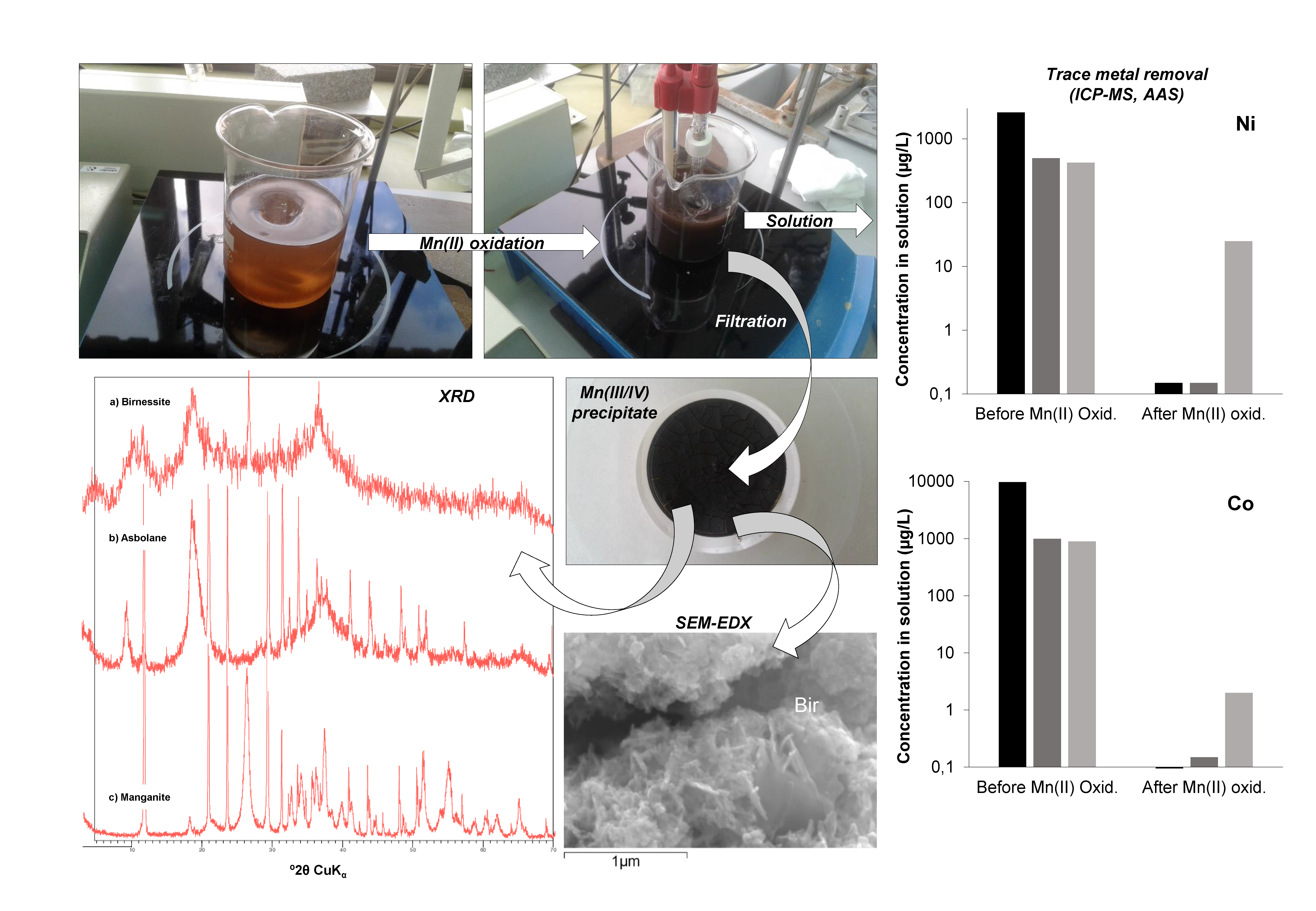
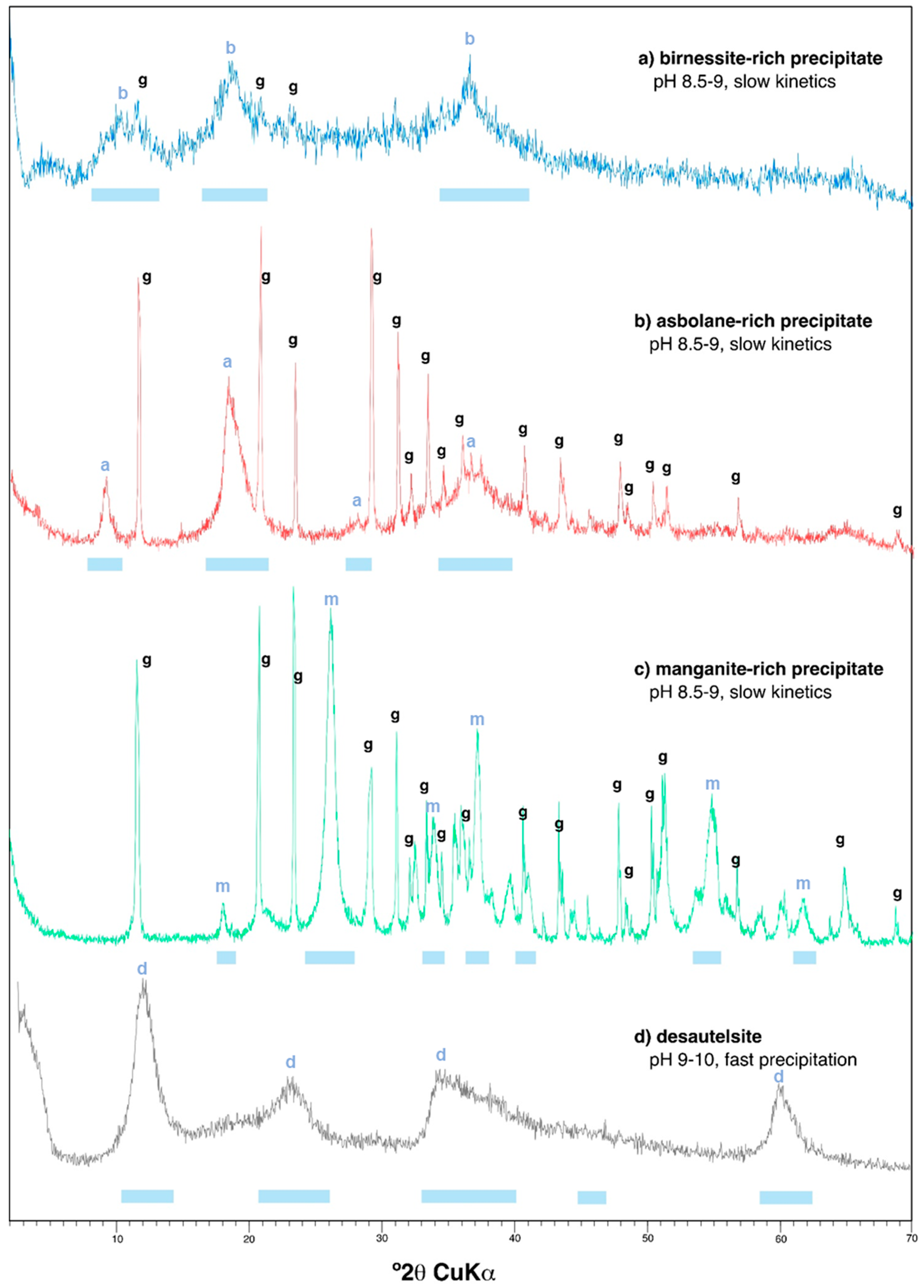
3.1. Mineralogy and Chemistry of the Manganese Oxide Precipitates
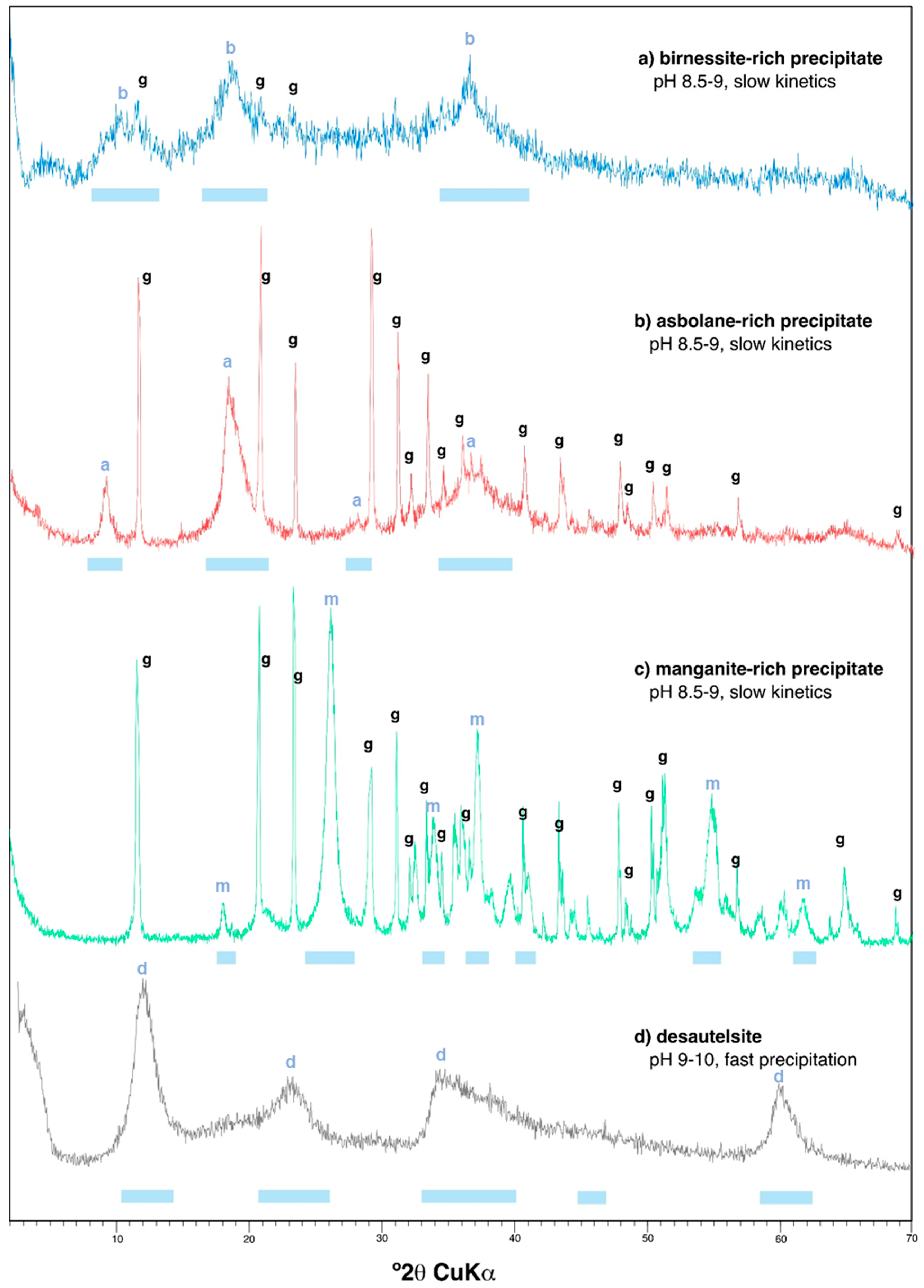
3.1.1. Mineral Composition Revealed by X-Ray Diffraction and Electron Microscopy
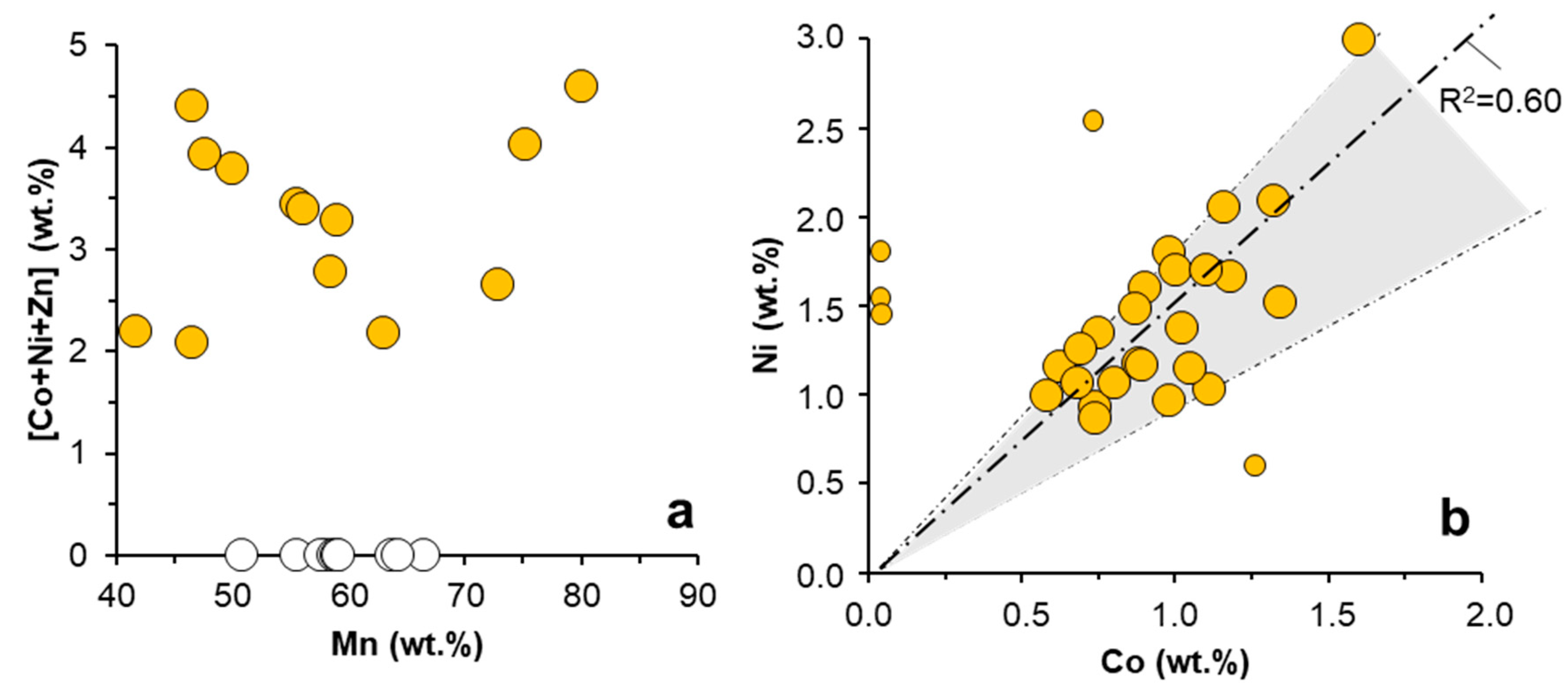
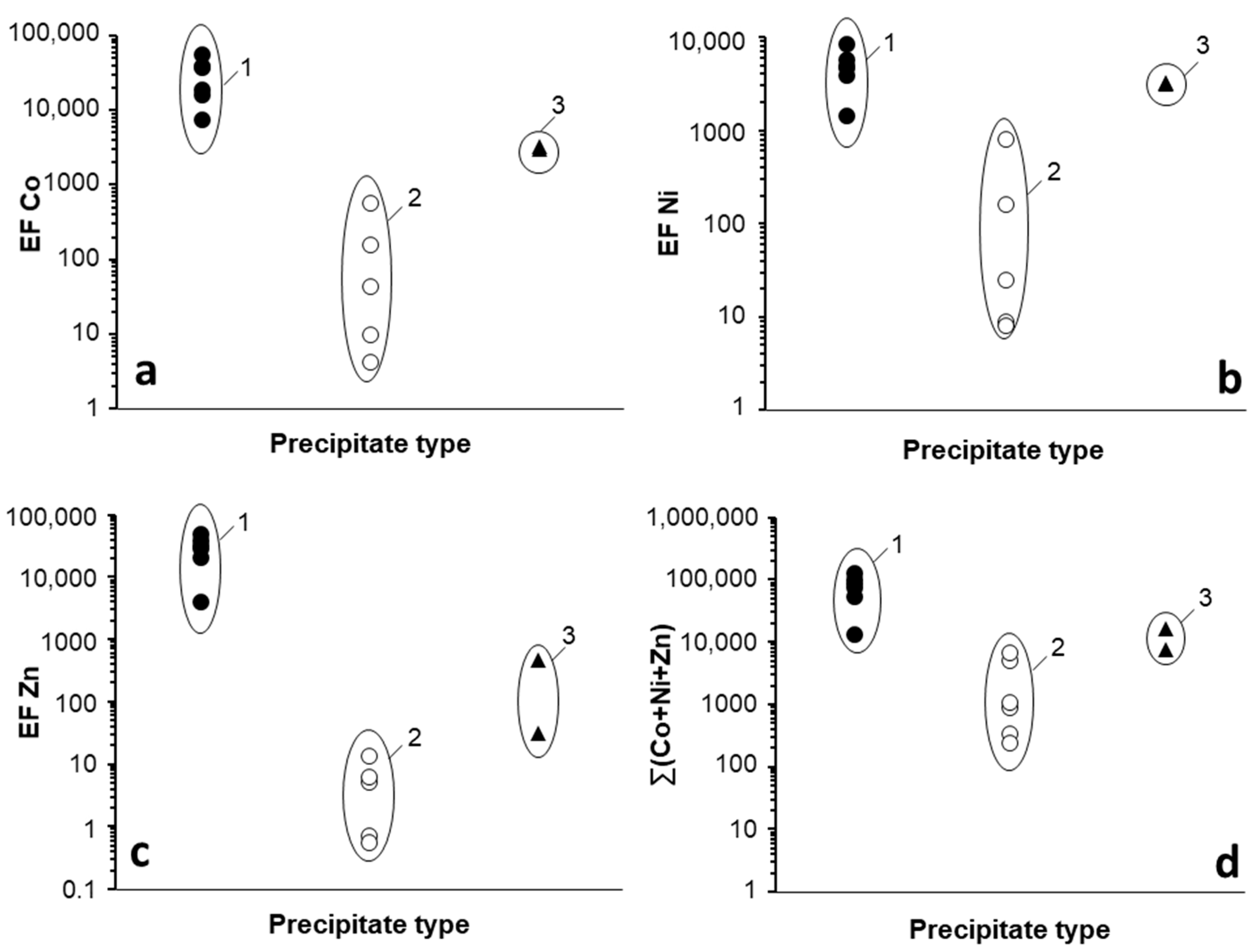
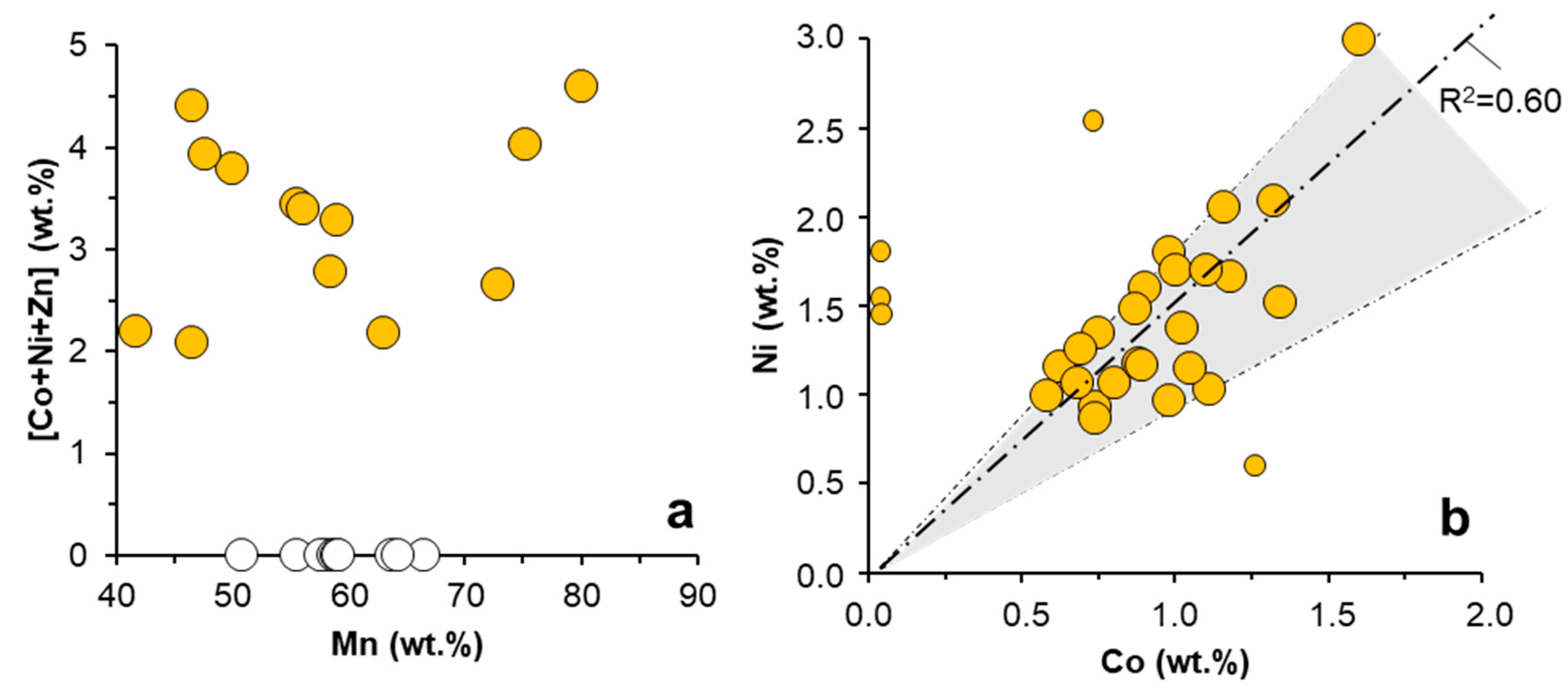
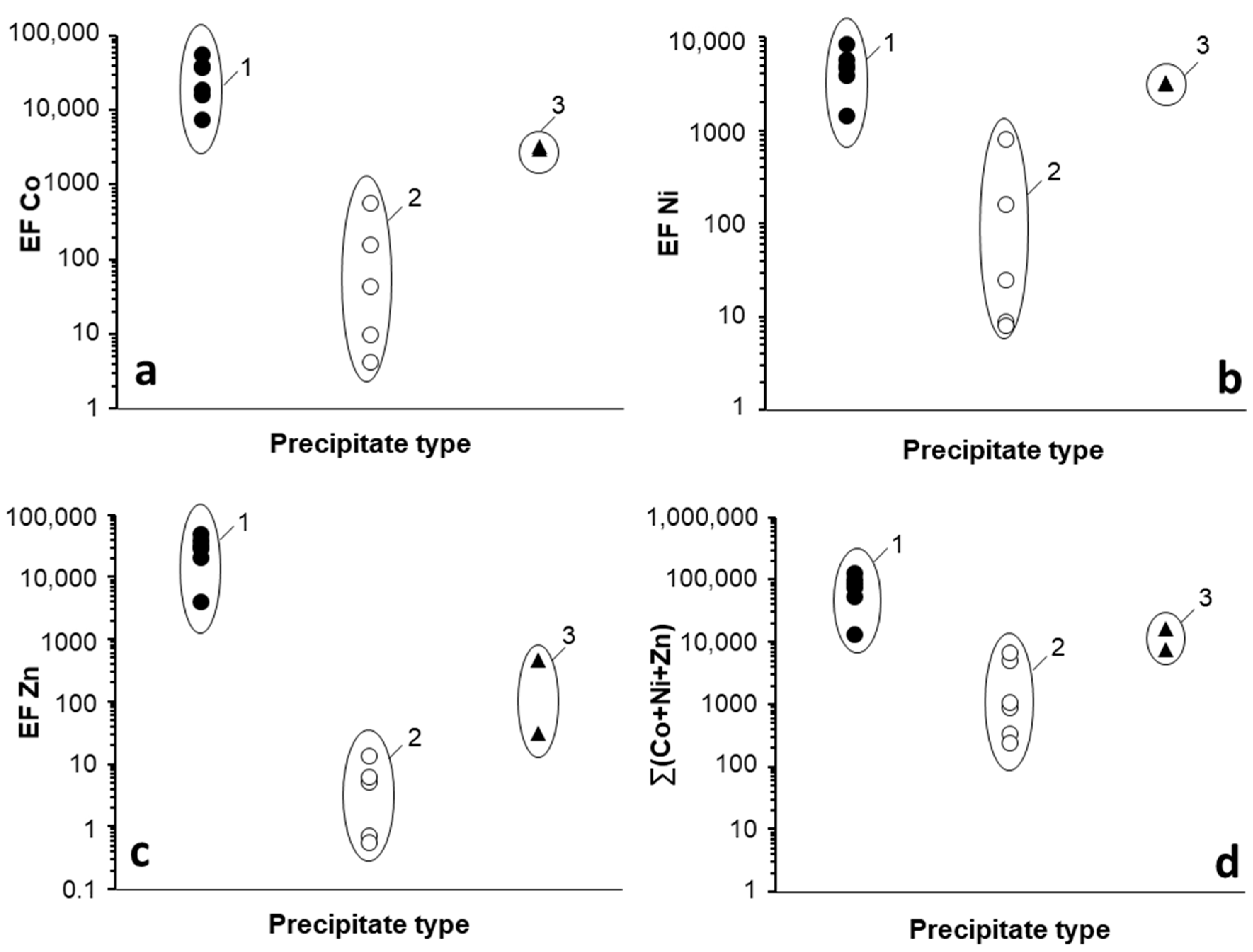
3.1.2. Trace Metal Content of the Mn Oxides
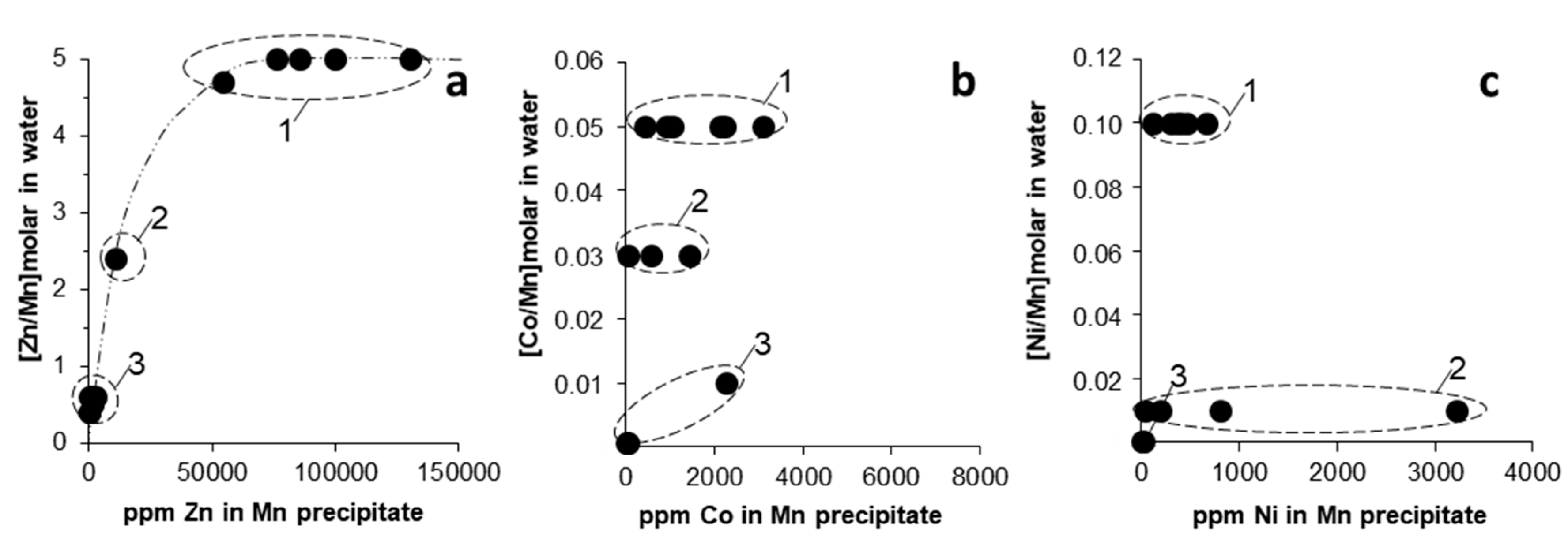
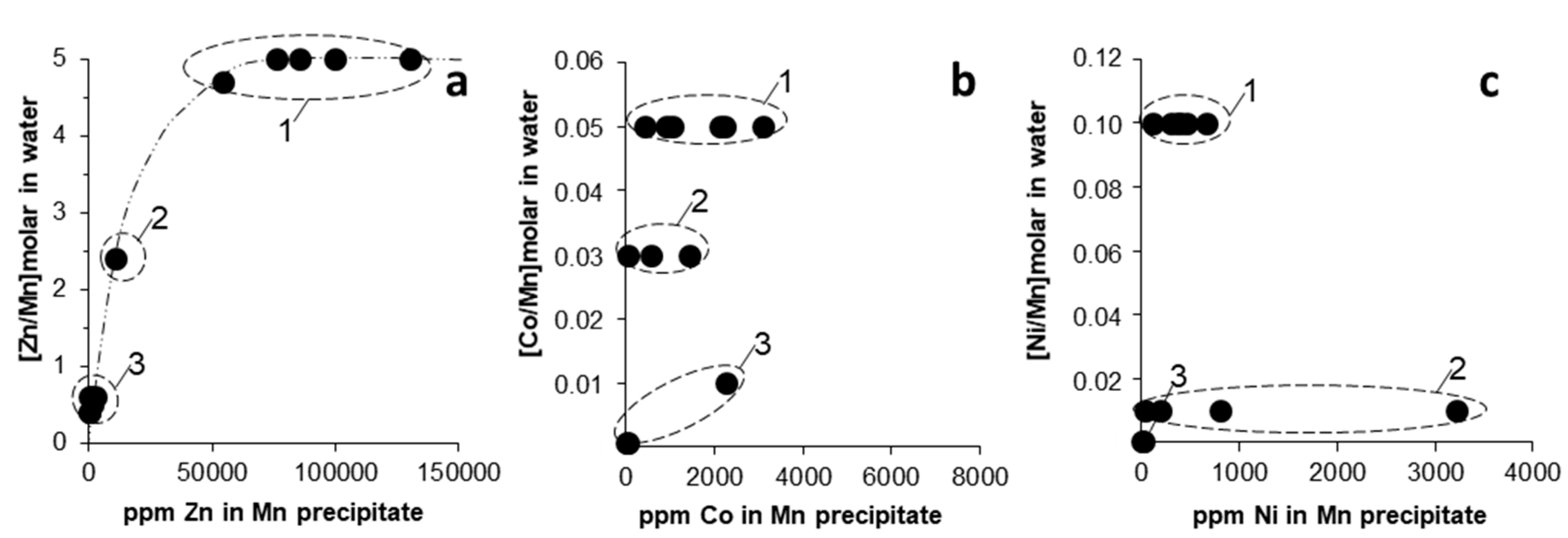
3.2. Trace Metal (Co, Ni, Zn) Partition between Parent Solutions and Mn Oxides
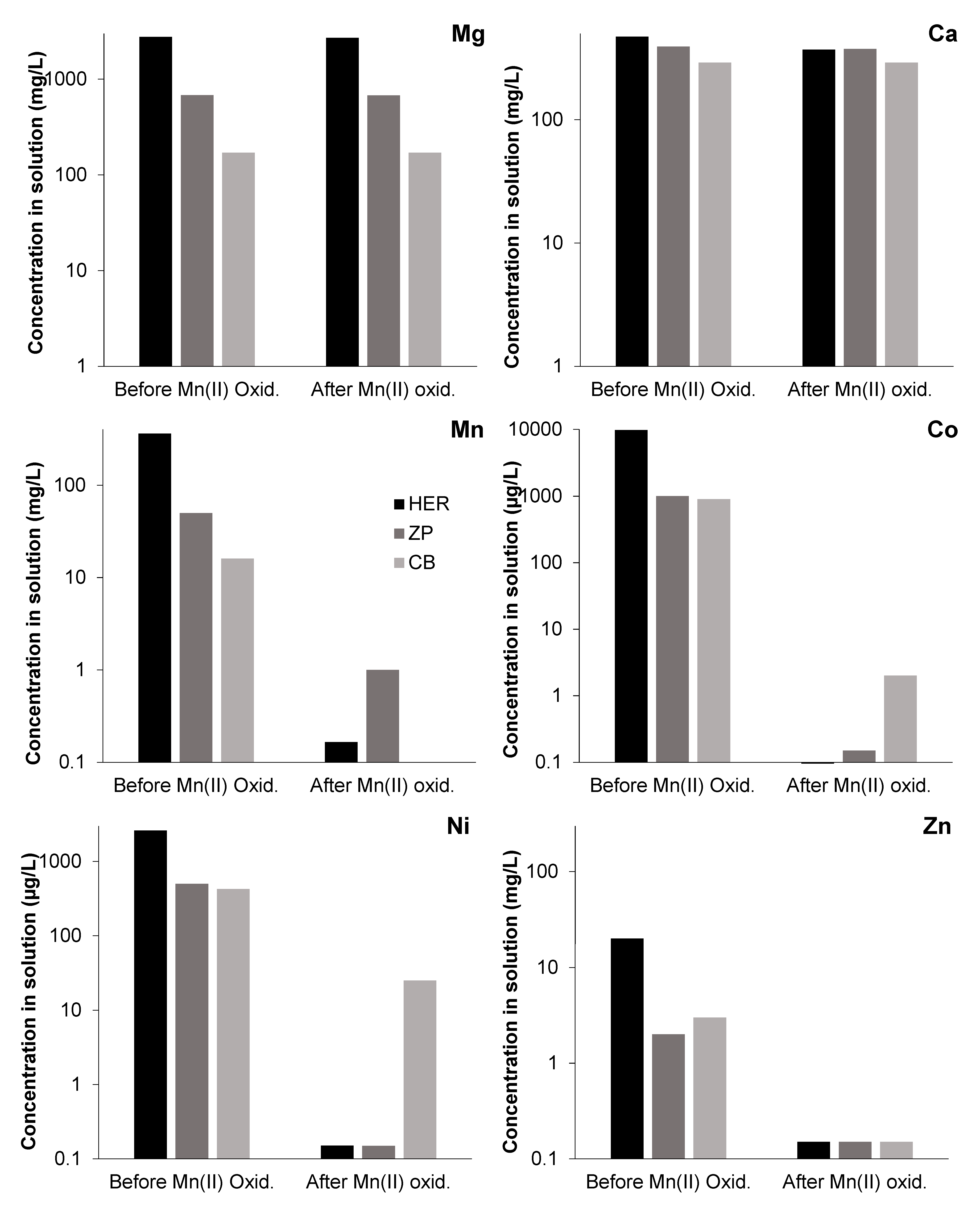
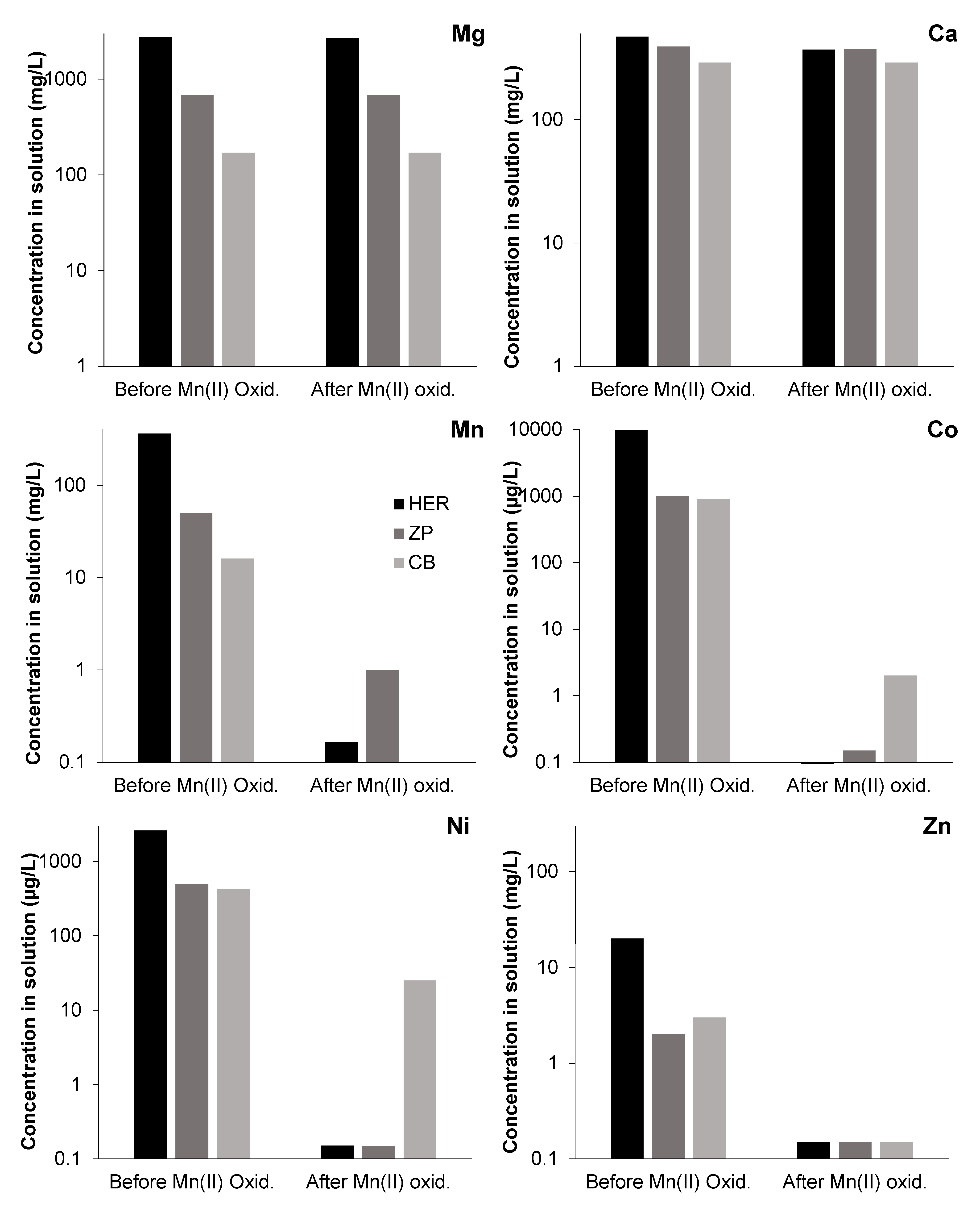
3.3. Effects of Mn(III/IV) Precipitation on Solution Chemistry
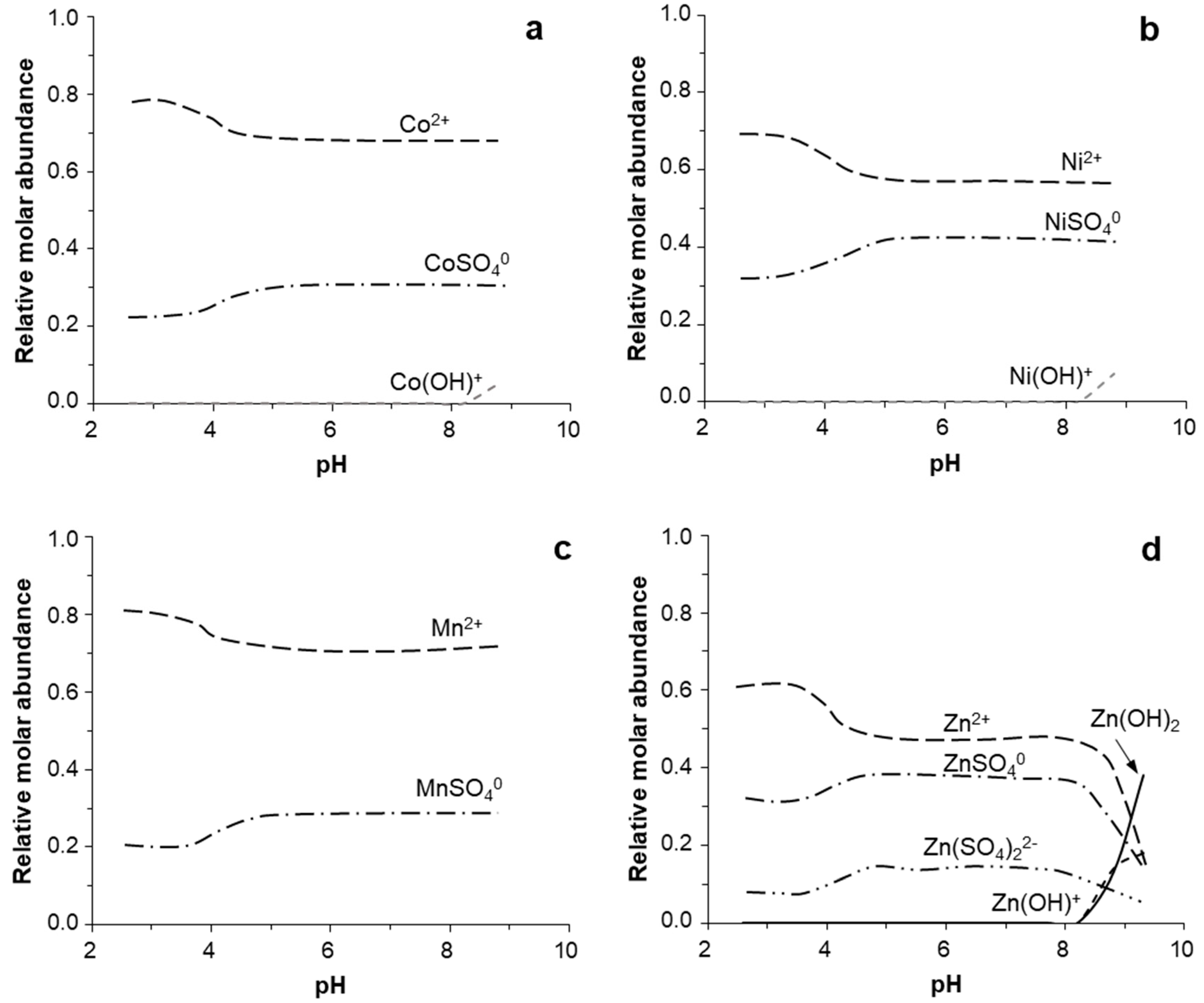
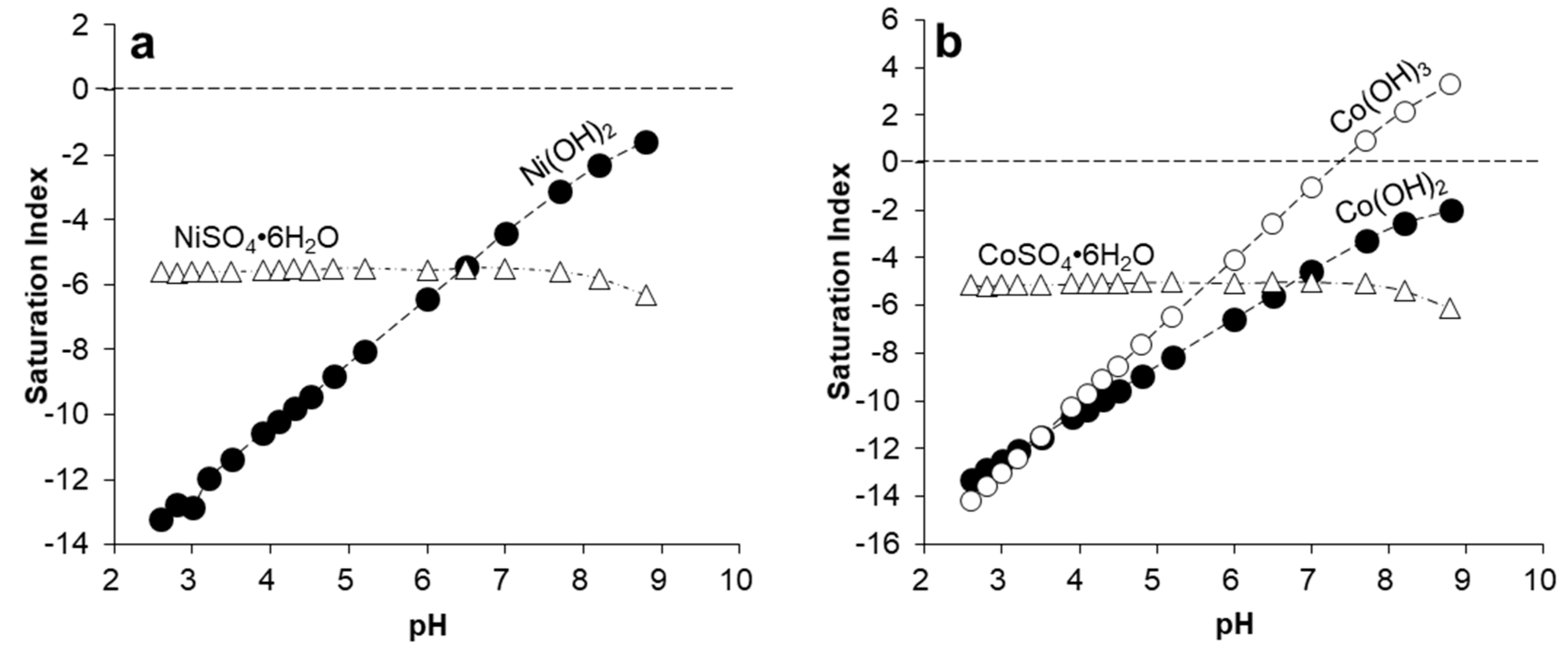
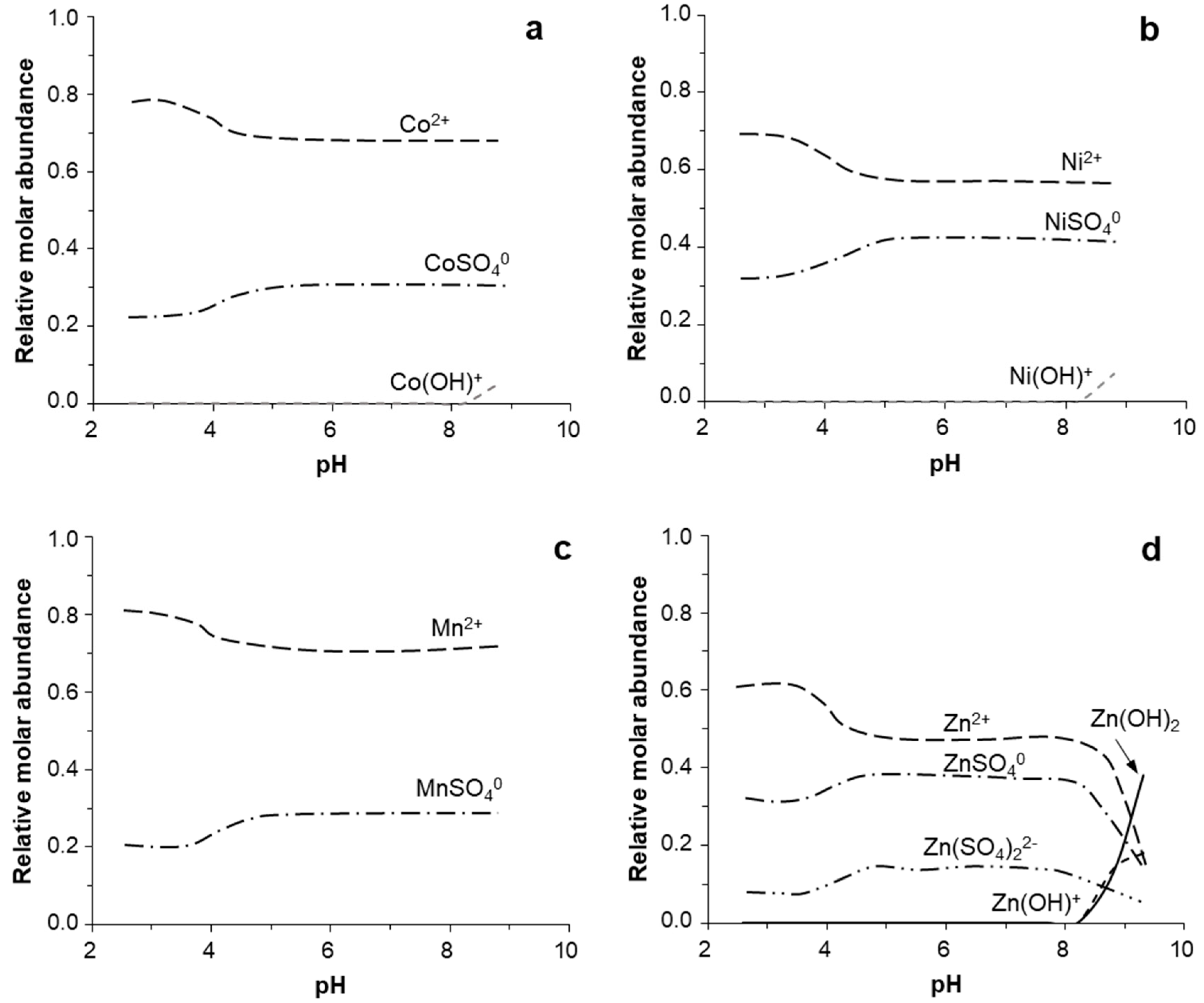
3.4. Geochemical Modeling
4. Discussion
4.1. Factors Controlling Mn(II) Oxidation and Mn Oxide Mineralogy in the Studied Mine Sites
4.2. Possible Implications for Remediation Technology and Metal Recovery
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Post, J.E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef]
- Tebo, B.M.; Johnson, H.A.; McCarthy, J.K.; Templeton, A.S. Geomicrobiology of Manganese(II) oxidation. Trends Microbiol. 2005, 13, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Tebo, B.M.; Bargar, J.R.; Clement, B.G.; Dick, G.J.; Murray, K.J.; Parker, D.; Verity, R.; Webb, S.M. Biogenic manganese oxides: Properties and Mechanisms of Formation. Annu. Rev. Earth Planet. Sci. 2004, 32, 287–328. [Google Scholar] [CrossRef]
- Tonkin, J.W.; Balistrieri, L.S.; Murray, J.W. Modeling sorption of divalent metal cations on hydrous manganese oxide using the diffuse double layer model. Appl. Geochem. 2004, 19, 29–53. [Google Scholar] [CrossRef]
- Yin, H.; Tan, W.; Zheng, L.; Cui, H.; Qiu, G.; Liu, F.; Feng, X. Characterization of Ni-rich hexagonal birnessite and its geochemical effects on aqueous Pb2+/Zn2+ and As(III). Geochim. Cosmochim. Acta 2012, 93, 47–62. [Google Scholar] [CrossRef]
- Yin, H.; Feng, X.; Qiu, G.; Tan, W.; Liu, F. Characterization of Co-doped birnessites and application for removal of lead and arsenite. J. Hazard. Mater. 2011, 188, 341–349. [Google Scholar] [CrossRef]
- Yin, H.; Liu, F.; Feng, X.; Liu, M.; Tan, W.; Qiu, G. Co2+-exchange mechanism of birnessite and its application for the removal of Pb2+ and As(III). J. Hazard. Mater. 2011, 196, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Tani, Y.; Naitou, H.; Miyata, N.; Seyama, H. Sequestration of Cd(II) and Ni(II) ions on fungal manganese oxides associated with Mn(II) oxidase activity. Appl. Geochem. 2014, 47, 198–208. [Google Scholar] [CrossRef]
- Wang, W.; Shao, Z.; Liu, Y.; Wang, G. Removal of multi-heavy metals using biogenic manganese oxides generated by a deep-sea sedimentary bacterium—Brachybacterium sp. Strain Mn32. Microbiology 2009, 155, 1989–1996. [Google Scholar] [CrossRef]
- Zhou, D.; Kim, D.-G.; Ko, S.O. Heavy metal adsorption with biogenic manganese oxides generated by Pseudomonas putida strain MnB1. J. Ind. Eng. Chem. 2015, 24, 132–139. [Google Scholar] [CrossRef]
- Manceau, A.; Llorca, S.; Calas, G. Crystal chemistry of cobalt and nickel in lithiophorite and asbolane from New Caledonia. Geochim. Cosmochim. Acta 1987, 51, 105–113. [Google Scholar] [CrossRef]
- Manceau, A.; Lanson, B.; Drits, V.A. Structure of heavy metal sorbed birnessite. Part III: Results from powder and polarized extended X-Ray absorption fine structure spectroscopy. Geochim. Cosmochim. Acta 2002, 66, 2639–2663. [Google Scholar] [CrossRef]
- Nicholson, K.; Eley, M. Geochemistry of manganese oxides: Metal adsorption in freshwater and marine environments. In Manganese Mineralization: Geochemistry and Mineralogy of Terrestrial and Marine Deposits; Nicholson, K., Hein, K., Bühn, J.R., Dasgupta, S., Eds.; Geological Society Special Publication No 119; Geological Society of London: London, UK, 1997; pp. 309–326. [Google Scholar] [CrossRef]
- Fuller, C.; Harvey, J. Reactive uptake of trace metals in the hyporheic zone of mining-contaminated stream, Pinal Creek, Arizona. Environ. Sci. Technol. 2000, 34, 1150–1155. [Google Scholar] [CrossRef]
- Kay, J.; Conklin, M.H.; Fuller, C.; O’Day, P.A. Processes of nickel and cobalt uptake by a manganese oxide forming sediment in Pinal Creek, Globe Mining District, Arizona. Environ. Sci. Technol. 2001, 35, 4719–4725. [Google Scholar] [CrossRef]
- Lee, G.; Bigham, J.M.; Faure, G. Removal of trace metals by coprecipitation with Fe, Al and Mn from natural waters contaminated with acid mine drainage in the Ducktown Mining District, Tennessee. Appl. Geochem. 2002, 17, 569–581. [Google Scholar] [CrossRef]
- Tani, Y.; Ohashi, M.; Miyata, N.; Seyama, H.; Iwahori, K.; Soma, M. Sorption of Co(II), Ni(II), and Zn (II) on biogenic manganese oxides produced by a Mn-oxidizing fungus, strain KR21-2. J. Environ. Sci. Health Part A 2004, 39, 2641–2660. [Google Scholar] [CrossRef]
- Tan, H.; Zhang, G.; Heaney, P.; Webb, S.; Burgos, W.D. Characterization of manganese oxide precipitates from Appalachian coal mine drainage treatment systems. Appl. Geochem. 2010, 25, 389–399. [Google Scholar] [CrossRef]
- Wetzel, R.G. Limnology: Lake and River Ecosystems, 3rd ed.; Elsevier-Academic Press: San Diego, CA, USA, 2001; p. 1001. [Google Scholar]
- Fosmire, G.J. Zinc toxicity. Am. J. Clin. Nutr. 1990, 51, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Leyssens, L.; Vinck, B.; Van Der Straeten, C.; Wuyts, F.; Maes, L. Cobalt toxicity in humans—A review of the potential sources and systemic health effects. Toxicology 2017, 387, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Das, K.K.; Das, S.N.; Dhundasi, S.A. Nickel, its adverse health effects & oxidative stress. Indian J. Med. Res. 2008, 128, 412–425. [Google Scholar] [PubMed]
- InfoMine. Available online: http://www.infomine.com (accessed on 21 January 2019).
- London Market Exchange. Available online: https://www.lme.com (accessed on 21 January 2019).
- Brant, D.L.; Ziemkiewicz, P.F. Passive removal of manganese from acid mine drainage. Proc. Am. Soc. Min. Reclam. 1997, 741–744. [Google Scholar] [CrossRef]
- Rose, A.W.; Shah, P.J.; Means, B. Case studies of limestone-bed passive systems for manganese removal from acid mine drainage. Proc. Am. Soc. Min. Reclam. 2003, 1059–1078. [Google Scholar] [CrossRef]
- Bamforth, S.M.; Manning, D.A.C.; Singleton, I.; Younger, P.L.; Johnson, K.L. Manganese removal from mine waters—Investigating the occurrende and importance of manganese carbonates. Appl. Geochem. 2006, 21, 1274–1287. [Google Scholar] [CrossRef]
- Teixeira, L.A.C.; Queiroz, J.P.; Marquez-Sarmiento, C. Oxidative precipitation of manganese from dilute waters. Mine Water Environ. 2017, 36, 452–456. [Google Scholar] [CrossRef]
- Gounot, A.M. Microbial oxidation and reduction of manganese: Consequences in groundwater and applications. FEMS Microbiol. Rev. 1994, 14, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.C.; Madgwick, J.C. Microbial formation of manganese oxides. App. Environ. Microbiol. 1991, 57, 1114–1120. [Google Scholar]
- Learman, D.R.; Wankel, S.D.; Webb, S.M.; Martínez, N.; Madden, A.S.; Hansel, C.M. Coupled biotic-abiotic Mn(II) oxidation pathway mediates the formation and structural evolution of biogenic oxides. Geochim. Cosmochim. Acta 2011, 75, 6048–6063. [Google Scholar] [CrossRef]
- Hallberg, K.B.; Johnson, D.B. Biological manganese removal from acid mine drainage in constructed wetlands and prototype bioreactors. Sci Total Environ. 2005, 338, 115–124. [Google Scholar] [CrossRef]
- López-Pamo, E.; Sánchez-España, J.; Santofimia, E.; Reyes, J.; Martín-Rubí, J.A. Limnología físico-química del lago formado durante la inundación de la Corta de Reocín, Cantabria (Marzo 2008–Febrero 2009); Unpublished Report; Instituto Geológico y Minero de España (IGME); Ministerio de Ciencia e Innovación: Madrid, Spain, 2009; p. 116.
- Diem, M.; Stumm, W. Is dissolved Mn2+ being oxidized by O2 in the absence of Mn-bacteria or surface catalysts? Geochim. Cosmochim. Acta 1984, 48, 1571–1573. [Google Scholar] [CrossRef]
- Morgan, A.J. Kinetics of reaction between O2 and Mn(II) species in aqueous solutions. Geochim. Csomochim. Acta 2005, 69, 35–48. [Google Scholar] [CrossRef]
- Hem, J.D. Chemical equilibria and rates of manganese oxidation. In Chemistry of Manganese in Natural Water; Water-Supply Paper 1667-A; U.S. Geological Survey: Washington, DC, USA, 1963. [Google Scholar]
- Martin, S.T. Precipitation and dissolution of iron and manganese oxides. In Environmental Catalysis; Grassian, V.H., Ed.; CRC Press: Boca Raton, FL, USA, 2005; Chapter 4; p. 701. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I. Low-crystallinity products of trace-metal precipitation in neutralized pit-lake waters without ferric and aluminous adsorbent: Geochemical modelling and mineralogical analyses. Mineral. Mag. 2015, 79, 781–798. [Google Scholar] [CrossRef]
- Parkurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Domensional Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; U.S. Geological Survey: Denver, CO, USA, 1999; p. 312.
- Allison, J.D.; Brown, D.S.; Novo-Gradac, J. MINTEQA2/PRODEAFA2, a Geochemical Assessment Model for Environmental Systems: User Manual Supplement for Version 4.0; USEPA: Washington, DC, USA; NERL: Athens, GA, USA, 1999.
- Chuchrov, F.V.; Gorshov, A.I.; Vitovskaya, I.V.; Drits, V.A.; Sivtsov, A.I.; Rudnitskaya, Y.S. Crystallochemical nature of Co-Ni asbolan. Int. Geol. Rev. 1982, 24, 598–604. [Google Scholar] [CrossRef]
- Garratt-Reed, A.; Bell, D.C. Energy-Dispersive X-Ray Analysis in the Electron Microscope; BIOS Scientific Publishers Limited: Oxford, UK, 2003; p. 160. [Google Scholar] [CrossRef]
- Santelli, C.M.; Webb, S.M.; Dohnalkova, A.C.; Hansel, C.M. Diversity of Mn oxides produced by Mn(II)-oxidizing fungi. Geochim. Cosmochim. Acta 2011, 74, 2762–2776. [Google Scholar] [CrossRef]
- Webb, S.M.; Tebo, B.M.; Bargar, J.R. Structural influences of sodium and calcium ions on the biogenic manganese oxides produced by the marine Bacillus Sp.; strain SG-1. Geomicrobiol. J. 2005, 22, 181–193. [Google Scholar] [CrossRef]
- Bohu, T.; Akob, D.M.; Abratis, M.; Lazar, C.S.; Küsel, K. Biological low-pH Mn(II) oxidation in a manganese deposit influenced by metal-rich groundwater. Appl. Environ. Microbiol. 2016, 82, 3009–3021. [Google Scholar] [CrossRef]
- White, W.B.; Scheetz, B.E.; Atkinson, S.D.; Ibberson, D.; Chess, C.A. Mineralogy of Rohrer’s Cave, Pennsylvania. NSS Bull. 1985, 47, 17–27. [Google Scholar]
- Yusta, I.; Castellano, A.; Aranburu, A.; Velasco, F. Adsorción de metales en espeleotemas de Mn-Al-Fe de la Cueva de Lazalday (Zarate-Alava). Macla 2009, 11, 203–204. [Google Scholar]
- Mindat.org. Available online: https://www.mindat.org (accessed on 30 January 2018).
- Mineralogy Database. Available online: https://www.webmineral.com (accessed on 30 November 2018).
- Sánchez-España, F.J.; López Pamo, E.; Santofimia, E.; Aduvire, O.; Reyes, J.; Barettino, D. Acid Mine Drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): Geochemistry, Mineralogy and Environmental Implications. Appl. Geochem. 2005, 20–27, 1320–1356. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Santofimia, E.; Reyes, J.; Martín Rubí, J.A. The natural attenuation of two acidic effluents in Tharsis and La Zarza-Perrunal mines (Iberian Pyrite Belt, Huelva, Spain). Environ. Geol. 2005, 49, 253–266. [Google Scholar] [CrossRef]
- Sánchez-España, J. The behavior of iron and aluminum in acid mine drainage: Speciation, mineralogy, and environmental significance. In Thermodynamics, Solubility and Environmental Issues; Letcher, T., Ed.; Elsevier: Amsterdam, The Netherlands; pp. 137–150.
- Sánchez-Espańa, J.; López-Pamo, E.; Santofimia, E.; Reyes, J.; Martín-Rubí, J.A. The impact of acid mine drainage on the water quality of the Odiel river (Huelva, Spain): Evolution of precipitate mineralogy and aqueous geochemistry along the Concepción-Tintillo segment. Water Air Soil Pollut. 2006, 173, 121–149. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Santofomia, E.; Reyes, J.; Martín-Rubí, J.A. The removal of dissolved metals by hydroxysulphate precipitates during oxidation and neutralization of acid mine waters, Iberian Pyrite Belt. Aquat. Geochem. 2006, 12–13, 269–298. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Reyes, J. Comparing Schwertmannite and Hydrobasaluminite Dissolution in Ammonium Oxalate (pH 3.0): Implications for Metal Speciation Studies by Sequential Extraction. Minerals 2019, 9, 57. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Santofimia, E.; Diez-Ercilla, M. The acidic mine pit lakes of the Iberian Pyrite Belt: An approach to their physical limnology and hydrogeochemistry. Appl. Geochem. 2008, 23, 1260–1287. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; Diez-Ercilla, M. Schwertmannite and hydrobasaluminite: A re-evaluation of therir solubility and control on the iron and aluminum concentration in acidic pit lakes. Appl. Geochem. 2011, 26, 1752–1774. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Diez, M.; Santofimia, E. Physico-chemical gradients and meromictic stratification in Cueva de la Mora and other acidic pit lakes of the Iberian Pyrite Belt. Mine Water Environ. 2009, 28, 15–19. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; López, G.A. Schwertmannite to jarosite conversion in the water column of an acidic mine pit lake. Mineral. Mag. 2012, 76, 2659–2682. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Diez, M.; Santofimia, E. Mine pit lakes of the Iberian Pyrite Belt: Some basic limnological, hydrogeochemical and microbiological con-siderations. In Acidic Pit Lakes: The Legacy of Coal and Metal Surface Mines; Geller, W., Schultze, M., Kleinmann, B., Wolkersdorfen, C., Eds.; Springer: Heidelberg, Germany, 2013; pp. 315–342. [Google Scholar]
- Sánchez-España, J.; Boehrer, B.; Yusta, I. Extreme carbon dioxide concentrations in acidic pit lakes provoked by water/rock interaction. Environ. Sci. Technol. 2014, 48, 4273–4281. [Google Scholar] [CrossRef]
- Balistrieri, L.S.; Murray, J.W.; Paul, B. The cycling of iron and manganese in the water column of Lake Sammamish, Washington. Limnol. Oceanogr. 1992, 37, 510–528. [Google Scholar] [CrossRef]
- Kawashima, M.; Takamatsu, T.; Koyama, M. Mechanisms of precipitation of manganese(II) in Lake Biwa, a fresh water lake. Water Resources 1988, 22, 613–618. [Google Scholar] [CrossRef]
- Davison, W.; Woof, C.; Rigg, E. The dynamics of iron and manganese in a seasonally anoxic lake: Direct measurements of fluxes using sediment traps. Limnol. Oceanogr. 1982, 27, 987–1003. [Google Scholar] [CrossRef]
- Douglas, G.B. Contaminant removal from acidic mine pit water via in situ hydrotalcite formation. Appl. Geochem. 2014, 51, 15–22. [Google Scholar] [CrossRef]
- Douglas, G.; Shackleton, M.; Woods, P. Hydrotalcite formation facilitates effective contaminant and radionuclide removal from acidic uranium mine barren lixiviant. Appl. Geochem. 2014, 42, 27–37. [Google Scholar] [CrossRef]
- Mubarok, M.Z.; Lieberto, J. Precipitation of nickel hydroxide from simulated and atmospheric-leach solution of nickel leterite ore. Proc. Earth Planet. Sci. 2013, 6, 457–464. [Google Scholar] [CrossRef]
- Kefeni, K.K.; Msagati, T.M.; Maree, J.P.; Mamba, B.B. Metals and sulphate removal from acid mine drainage in two steps via ferrite sludge and barium sulphate formation. Miner. Eng. 2015, 81, 79–87. [Google Scholar] [CrossRef]
- Cannon, W.F.; Kimball, B.E.; Corathers, L.A. Manganese. In Critical Mineral Resources of the United States-Economic and Environmental Geology and Prospects for Future Supply; Schulz, K.J., DeYoung, J.H., Seal, R.R., Bradley, D.C., Eds.; US Geological Survey Professional Paper 1802; U.S. Geological Survey: Reston, VA, USA, 2017; pp. L1–L28. [Google Scholar] [CrossRef]
- Singerling, S.A.; Tuck, C.A. Ferroalloys [Advanced Release]. In US Geological Survey Minerals Yearbook-2015, Volume I: Metals and Minerals; US Department of the Interior, US Geological Survey: Reston, VA, USA, May 2018; pp. 25.1–25.14. [Google Scholar]
- Mittal, N.K.; Sen, P.K. India’s first medium scale demonstration plant for treating polymetallic nodules. Miner. Eng. 2003, 16, 865–868. [Google Scholar] [CrossRef]
- Zubkov, M.V.; Plucinski, P.K.; Dartiguelongue, A.C.Y.; Lusty, P.A. Metal extraction from deep-ocean mineral deposits. Elements 2018, 14, 319–324. [Google Scholar] [CrossRef]
- Lusty, P.A.J.; Hein, J.R.; Josso, P. Formation and occurrence of ferromanganese crusts: Earth’s storehouse for critical metals. Elements 2018, 14, 313–318. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
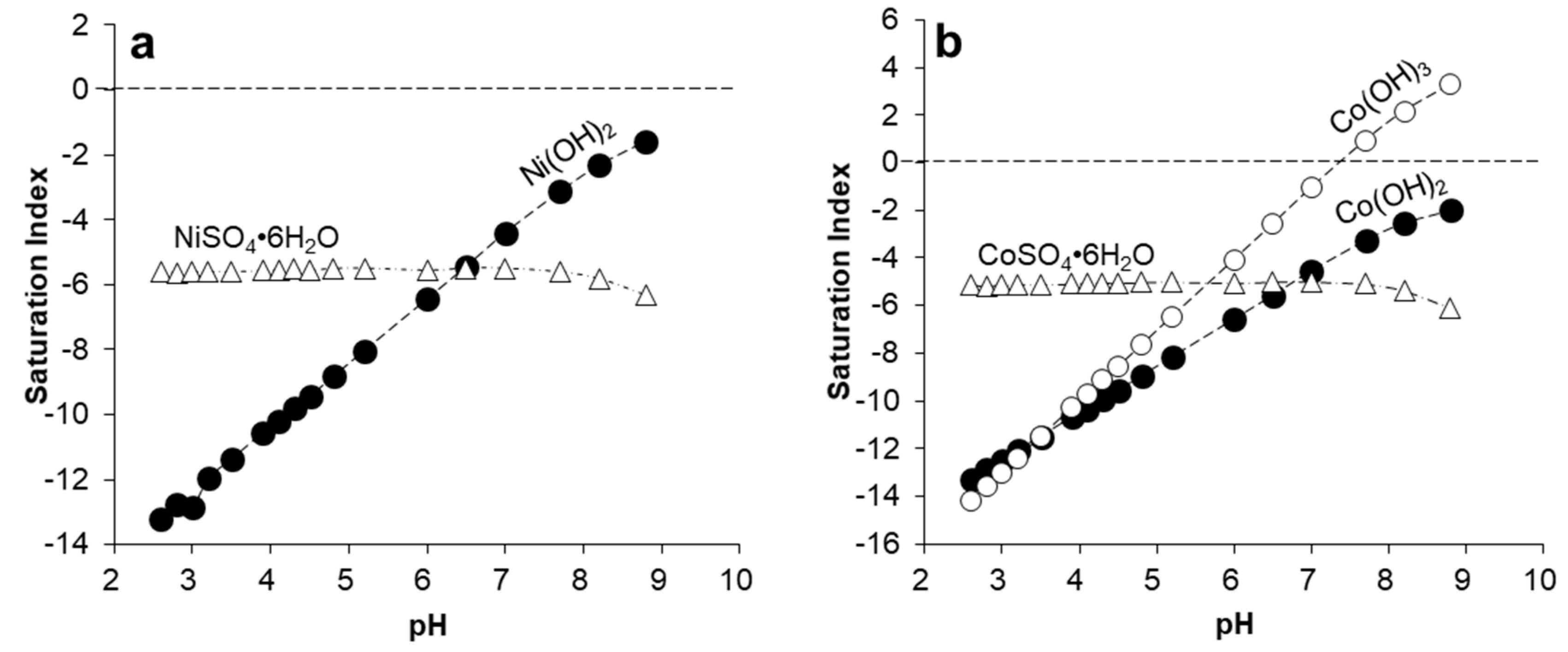{kind=link}
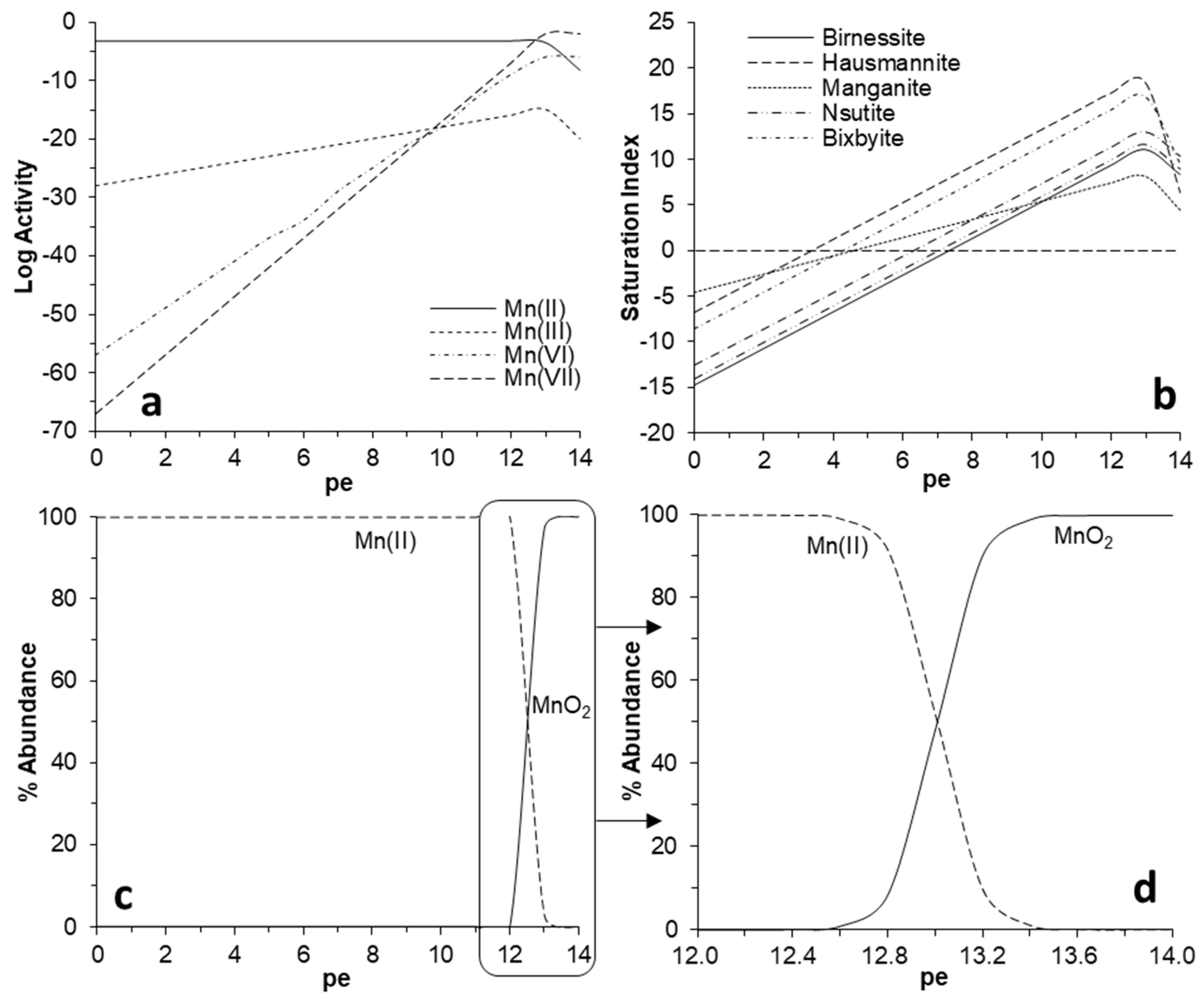{kind=link}
| Pit Lake | Depth | Na | K | Mg | Ca | SO4 | SiO2 | Fe | Al | Mn | Zn | Co | Ni |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | µg/L | µg/L | |
| La Zarza | 0 | 37 | 1 | 756 | 552 | 14,712 | 150 | 3901 | 876 | 317 | 182 | 4126 | 3994 |
| La Zarza | 30 | 39 | 9 | 1190 | 453 | 29,108 | 133 | 10,072 | 1870 | 489 | 523 | 7295 | 6877 |
| La Zarza | 70 | 50 | 16 | 1536 | 510 | 37,044 | 122 | 14,334 | 2178 | 647 | 624 | 11,081 | 10,394 |
| Guadiana | 0 | 48 | 1 | 666 | 530 | 5894 | 62 | 342 | 76 | 158 | 130 | 3379 | 2494 |
| Guadiana | 40 | 91 | 11 | 1394 | 472 | 14,459 | 43 | 3082 | 144 | 270 | 171 | 7117 | 4217 |
| Guadiana | 50 | 106 | 15 | 1834 | 477 | 18,664 | 13 | 4417 | 205 | 314 | 231 | 9456 | 5564 |
| Guadiana | 60 | 83 | 17 | 2351 | 472 | 20,971 | 10 | 4867 | 51 | 358 | 259 | 9724 | 5398 |
| Brunita | 0 | 590 | 2 | 5273 | 580 | 26,092 | 74 | 438 | 72 | 466 | 230 | 656 | 975 |
| Brunita | 12 | 565 | 12 | 5241 | 542 | 27,288 | 67 | 955 | 53 | 536 | 237 | 630 | 936 |
| Brunita | 15 | 344 | 16 | 5237 | 481 | 31,647 | 24 | 3661 | 23 | 814 | 436 | 704 | 863 |
| Brunita | 17 | 330 | 16 | 5184 | 470 | 30,542 | 21 | 4129 | 29 | 892 | 425 | 678 | 795 |
| Reocín | 0 | 42 | 17 | 210 | 556 | 1920 | 4.4 | 0.0 | 0.2 | 0.4 | 2.6 | 50 | 76 |
| Reocín | 8 | 44 | 18 | 211 | 561 | 1940 | 4.5 | 0.1 | 0.3 | 0.5 | 2.6 | 51 | 77 |
| Reocín | 13 | 45 | 18 | 204 | 559 | 1930 | 4.6 | 0.8 | 0.3 | 1.1 | 3.3 | 56 | 82 |
| Reocín | 50 | 41 | 18 | 198 | 571 | 1920 | 4.5 | 1.0 | 0.3 | 1.1 | 3.3 | 55 | 82 |
| Reocín | 90 | 41 | 18 | 199 | 557 | 1940 | 4.7 | 1.1 | 0.2 | 1.2 | 3.3 | 56 | 79 |
| Mineral | Formula |
|---|---|
| Manganite 1 | Mn3+O(OH) |
| Asbolane 1 | Ni0.3Co0.1Ca0.1Mn4+1.5O1.5(OH)2∙0.6(H2O) |
| Hausmannite 1,2 | Mn3+3O4 |
| Desautelsite 2 | Mg6Mn3+2(CO3)(OH)16∙4H2O |
| Birnessite 1,3 | (Na, Ca, K)x(Mn4+Mn3+)2-xO4∙1.5H2O |
| Todorokite 1,3 | (Na, Ca, K)2(Mn4+Mn3+)6O12∙3–4.5H2O |
| Pyrochroite 2,3 | Mn2+(OH)2 |
| Ranciecite 3 | (Ca, Mn2+)Mn4+4O9∙3H2O |
| Lithiophorite 3 | (Al, Li)Mn4+O2(OH)2 |
| Lake | Mineralogy | Conditions | Precip. Kinetics | pH | Ba | Cd | Co | Mo | Ni | Se | Pb | Zn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | |||||
| CB | Man | Lab oxidation | Slow (lab) | 8.5–9.0 | 333 | n.d. | 26 | n.d. | 25 | n.d. | n.d. | 279 |
| CB | Man | Lab oxidation | Slow (lab) | 8.5–9.0 | 445 | n.d. | 6 | n.d. | 8 | n.d. | n.d. | 225 |
| ZP | Asb | Lab oxidation | Slow (lab) | 8.5–9.0 | 532 | 153 | 2266 | n.d. | 3220 | n.d. | n.d. | 1101 |
| GUA | Bir | Lab oxidation | Slow (lab) | 8.5–9.0 | 2394 | 183 | 1424 | n.d. | 814 | n.d. | n.d. | 2713 |
| GUA | Bir | Lab oxidation | Slow (lab) | 8.5–9.0 | 2244 | n.d. | 38 | n.d. | 45 | n.d. | n.d. | 1010 |
| GUA | Tod | Lab oxidation | Slow (lab) | 8.5–9.0 | n.m. | n.d. | 560 | n.d. | 200 | n.d. | n.d. | 132 |
| CM | Des, Haus | Lab neutralization | Fast (lab) | 9.0–10.0 | n.m. | n.d. | 6566 | n.d. | 3095 | n.d. | n.d. | 6982 |
| ST | Des, Haus | Lab neutralization | Fast (lab) | 9.0–10.0 | n.m. | n.d. | 2981 | n.d. | 1601 | n.d. | n.d. | 3142 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 53 | 2125 | 123 | 675 | 228 | 177 | 10,533 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 65 | 2215 | 180 | 390 | 222 | 218 | 99,813 |
| Reo | Tod, Ran | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 74 | 3099 | 256 | 465 | 195 | 287 | 85,200 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 24 | 900 | 100 | 116 | 140 | 239 | 54,060 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 66 | 1042 | n.d. | 380 | 53 | 10 | 130,000 |
| Reo | Bir, Lit, Pyr | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 62 | 420 | 51 | 311 | 64 | 480 | 76,029 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-España, J.; Yusta, I. Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters. Minerals 2019, 9, 226. https://doi.org/10.3390/min9040226
Sánchez-España J, Yusta I. Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters. Minerals. 2019; 9(4):226. https://doi.org/10.3390/min9040226
Chicago/Turabian StyleSánchez-España, Javier, and Iñaki Yusta. 2019. "Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters" Minerals 9, no. 4: 226. https://doi.org/10.3390/min9040226
APA StyleSánchez-España, J., & Yusta, I. (2019). Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters. Minerals, 9(4), 226. https://doi.org/10.3390/min9040226