A Paris-Edinburgh Cell for High-Pressure and High-Temperature Structure Studies on Silicate Liquids Using Monochromatic Synchrotron Radiation

 , , , ,
, , , ,
Abstract
1. Introduction
2. Experimental Setup
2.1. The Paris-Edinburgh Press
2.2. The Multi-Channel Collimator
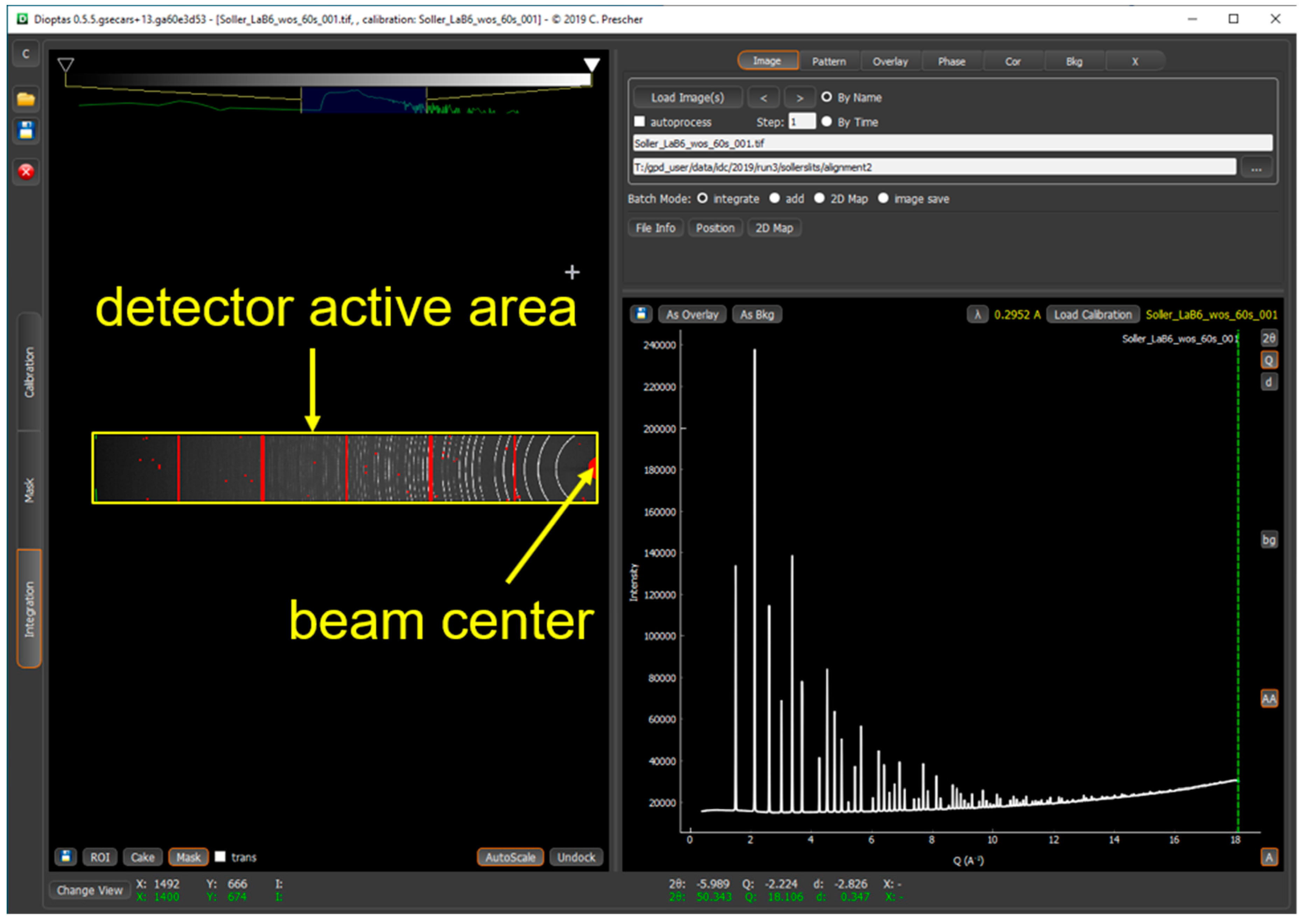
2.3. The Detector
3. Sample Environment and Preparation
3.1. The Paris-Edinburgh Cell Assembly
3.2. Sample Preparation
4. Data Collection and Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Funamori, N.; Yamamoto, S.; Yagi, T.; Kikegawa, T. Exploratory studies of silicate melt structure at high pressures and temperatures by in situ X-ray diffraction. J. Geophys. Res. 2004, 109, B03203. [Google Scholar] [CrossRef]
- McMillan, P. Structural studies of silicate glasses and melts—Applications and limitations of Raman spectroscopy. Am. Mineral. 1984, 69, 622–644. [Google Scholar]
- Stebbins, J.F.; Ellsworth, S.E. Temperature effects on structure and dynamics in borate and borosilicate liquids: High-resolution and high-temperature NMR results. J. Am. Ceram. Soc. 1996, 79, 2247–2256. [Google Scholar] [CrossRef]
- Angeli, F.; Villian, O.; Schuller, S.; Charpentier, T.; de Ligny, D.; Bressel, L.; Wondraczek, L. Effect of temperature and thermal history on borosilicate glass structure. Phys. Rev. B 2012, 85, 054110. [Google Scholar] [CrossRef]
- Susman, S.; Volin, K.J.; Price, D.L.; Grimsditch, M.; Rino, J.P.; Kalia, R.K.; Vashishta, P.; Gwanmesia, G.; Wang, Y.; Liebermann, R.C. Intermediate-range order in permanently densified vitreous SiO2: A neuton-diffraction and molecular-dynamics study. Phys. Rev. B 1991, 43, 1194–1197. [Google Scholar] [CrossRef]
- Zha, C.S.; Mao, H.K.; Hemley, R.J.; Duffy, T.S. Elasticity measurement and equation of state of MgO to 60 GPa. EOS. Trans. AGU 1997, 78, F752. [Google Scholar]
- Xu, M.; Jing, Z.; Chantel, J.; Jiang, P.; Yu, T.; Wang, Y. Ultrasonic velocity of diopside liquid at high pressure and temperature: Constraints on velocity reduction in the upper mantle due to partial melts. J. Geophys. Res. Sol. Earth 2018, 123, 8676–8690. [Google Scholar] [CrossRef]
- Jing, Z.; Karato, S. A new approach to the equation of state of silicate melts: An application of the theory of hard sphere mixtures. Geochim. Cosmochim. Acta 2011, 75, 6780–6802. [Google Scholar] [CrossRef]
- Wilding, M.C.; Benmore, C.J.; Weber, J.K.R. In situ diffraction studies of magnesium silicate liquids. J. Mater. Sci. 2008, 43, 4707–4713. [Google Scholar] [CrossRef][Green Version]
- Inoue, K.; Asada, T. Cubic anvil X-ray diffraction press up to 100 kbar and 1000 °C. Jpn. J. Appl. Phys. 1973, 12, 1786–1793. [Google Scholar] [CrossRef]
- Tsuji, K.; Yaoita, K.; Imai, M.; Shimomura, O.; Kikegawa, T. Measurements of X-ray diffraction for liquid metals under high pressure. Rev. Sci. Instrum. 1989, 60, 2425–2428. [Google Scholar] [CrossRef]
- Yamada, A.; Wang, Y.; Inoue, T.; Yang, W.; Park, C.; Yu, T.; Shen, G. High-pressure X-ray diffraction studies on the structure of liquid silicate using a Paris-Edinburgh type large volume press. Rev. Sci. Instrum. 2011, 82, 015103–015107. [Google Scholar] [CrossRef] [PubMed]
- Mezouar, M.; Faure, P.; Crichton, W.; Rambert, N.; Sitaud, N.; Bauchau, S.; Blattmann, G. Multichannel collimator for structural investigation of liquids and amorphous materials at high pressures and temperautures. Rev. Sci. Instrum. 2002, 73, 3570–3574. [Google Scholar] [CrossRef]
- Morard, G.; Mezouar, M.; Bauchau, S.; Alvarez-Murga, M.; Hodeau, J.L.; Garbarino, G. High efficiency multichannel collimator for structural studies of liquids and low-Z materials at high pressures and temperatures. Rev. Sci. Instrum. 2011, 82, 023904. [Google Scholar] [CrossRef]
- Besson, J.M.; Nelmes, R.J.; Hamel, G.; Loveday, J.S.; Weill, G.; Hull, S. Neutron powder diffraction above 10 GPa. Phys. B Condens. Matter 1992, 180, 907–910. [Google Scholar] [CrossRef]
- Klotz, S.; Besson, J.M.; Hammel, G.; Nelmes, R.J.; Loveday, J.S.; Marshall, W.G. High pressure neutron diffraction using the Paris-Edinburgh cell: Experimental possibilities and future prospects. High Press. Res. 1996, 14, 249–255. [Google Scholar] [CrossRef]
- Rivers, M.L.; Duffy, T.S.; Wang, Y.; Eng, P.J.; Sutton, S.R.; Shen, G. A new facility for high-pressure research at the Advanced Photon Source. In Properties of Earth and Planetary Materials at High Pressure and Temperature; Manghnani, M.H., Yagi, T., Eds.; AGU: Washington, DC, USA, 1998; pp. 79–88. [Google Scholar]
- Kono, Y.; Park, C.; Kenney-Benson, C.; Shen, G.; Wang, Y. Toward comprehensive studies of liquids at high pressures and high temperatures: Combined structure, elastic wave velocity, and viscosity measurements in the Paris-Edinburgh cell. Phys. Earth Planet. Int. 2014, 228, 269–280. [Google Scholar] [CrossRef]
- Yu, T.; Wang, Y.; Rivers, M.L. Imaging in 3D under pressure: A decade of high-pressure X-ray microtomography development at GSECARS. Prog. Earth Planet. Sci. 2016, 3, 17. [Google Scholar] [CrossRef]
- Brönnimann, C.; Trüb, P. Hybrid pixel photon counting X-ray detectors for synchrotron radiation. In Synchrotron Light Sources and Free-Electron Lasers; Jaeschke, E., Khan, S., Schneider, J., Hastings, J., Eds.; Springer: Cham, Switzerland, 2016; pp. 995–1027. [Google Scholar]
- Dewaele, A.; Fiquet, G.; Andrault, D.; Hausermann, D. P-V-T equation of state of periclase from synchrotron radiation measurements. J. Geophys. Res. 2000, 105, 2869–2877. [Google Scholar] [CrossRef]
- Prescher, C.; Prakapenka, V.B. DIOPTAS: A program for reduction of two-dimensional X-ray diffraction data and data exploration. High Press. Res. 2015, 35, 223–230. [Google Scholar] [CrossRef]
- Prescher, C. Glassure: An API and GUI program for analyzing angular dispersive total X-ray diffraction data. Zenodo 2017, 2017. [Google Scholar] [CrossRef]
- Wagner, C.N.J. Direct methods for the determination of atomic-scale structure of amorphous solids (X-ray, electron, and neutron scattering). J. Non Cryst. Solids 1978, 31, 1–40. [Google Scholar] [CrossRef]
- Parise, J.B.; Antao, S.M.; Michel, F.M.; Martin, C.D.; Chupas, P.J.; Shastri, S.D.; Lee, P.L. Quantitative high-pressure pair distribution function analysis. J. Synchrotron Radiat. 2005, 12, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Lorch, E. Neutron diffraction by Germania, silica and radiation-damaged silica glasses. J. Phys. C Solid State Phys. 1969, 2, 229–237. [Google Scholar] [CrossRef]
- Eggert, J.H.; Weck, G.; Loubeyre, P.; Mezouar, M. Quantitative structure factor and density measurements of high-pressrure fluids in diamond anvil cells by X-ray diffraction: Argon and water. Phys. Rev. B 2002, 65, 174105. [Google Scholar] [CrossRef]
- Misawa, M.; Price, D.L.; Suzuki, K. Short-range structure of alkali disilicate glasses by pulsed neutron total scattering. J. Non Cryst. Solids 1980, 37, 85–97. [Google Scholar] [CrossRef]
- Smith, W.; Greaves, G.N.; Gillan, M.J. Computer simulation of sodium disilicate glass. J. Chem. Phys. 1995, 103, 3031. [Google Scholar] [CrossRef]
- Huang, C.; Cormack, A.N. The structure of sodium silicate glass. J. Chem. Phys. 1990, 93, 8180–8186. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atomic Bond Lengths | This Study | Neutron Scattering [28] | MD Simulation [29] | MD Simulation [30] |
|---|---|---|---|---|
| Si–O | 1.61 ± 0.01 | 1.63 | 1.7 | 1.61 |
| Na–O | 2.20 ± 0.04 | - | 2.3 | 2.42 |
| O–O | 2.60 ± 0.02 | 2.66 | 2.6 | 2.62 |
| Si–Si | 3.14 ± 0.01 | - | 3.1 | 3.16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, T.; Prescher, C.; Ryu, Y.J.; Shi, F.; Greenberg, E.; Prakapenka, V.; Eng, P.; Stubbs, J.; Kono, Y.; Shen, G.; et al. A Paris-Edinburgh Cell for High-Pressure and High-Temperature Structure Studies on Silicate Liquids Using Monochromatic Synchrotron Radiation. Minerals 2019, 9, 715. https://doi.org/10.3390/min9110715
Yu T, Prescher C, Ryu YJ, Shi F, Greenberg E, Prakapenka V, Eng P, Stubbs J, Kono Y, Shen G, et al. A Paris-Edinburgh Cell for High-Pressure and High-Temperature Structure Studies on Silicate Liquids Using Monochromatic Synchrotron Radiation. Minerals. 2019; 9(11):715. https://doi.org/10.3390/min9110715
Chicago/Turabian StyleYu, Tony, Clemens Prescher, Young Jay Ryu, Feng Shi, Eran Greenberg, Vitali Prakapenka, Peter Eng, Joanne Stubbs, Yoshio Kono, Guoyin Shen, and et al. 2019. "A Paris-Edinburgh Cell for High-Pressure and High-Temperature Structure Studies on Silicate Liquids Using Monochromatic Synchrotron Radiation" Minerals 9, no. 11: 715. https://doi.org/10.3390/min9110715
APA StyleYu, T., Prescher, C., Ryu, Y. J., Shi, F., Greenberg, E., Prakapenka, V., Eng, P., Stubbs, J., Kono, Y., Shen, G., Watson, H., Rivers, M. L., Sutton, S. R., & Wang, Y. (2019). A Paris-Edinburgh Cell for High-Pressure and High-Temperature Structure Studies on Silicate Liquids Using Monochromatic Synchrotron Radiation. Minerals, 9(11), 715. https://doi.org/10.3390/min9110715